

Abstract
Lymph node blood vessels play important roles in the support and trafficking of immune cells. The blood vasculature is a component of the vascular-stromal compartment that also includes the lymphatic vasculature and fibroblastic reticular cells (FRCs). During immune responses, as lymph nodes swell, the blood vasculature undergoes a rapid proliferative growth that is initially dependent on CD11c+ cells and VEGF but is independent of lymphocytes. The lymphatic vasculature grows with similar kinetics and VEGF dependence, suggesting co-regulation of blood and lymphatic vascular growth, but lymphatic growth has been shown to be B cell-dependent. Here we show that blood vascular, lymphatic, and FRC growth are coordinately regulated and identify two distinct phases of vascular-stromal growth—an initiation phase, characterized by upregulated vascular-stromal proliferation, and a subsequent expansion phase. The initiation phase is CD11c+ cell-dependent and T/B cell-independent while the expansion phase is dependent on B and T cells together. Using CCR7−/− mice and selective depletion of migratory skin dendritic cells, we show that endogenous skin-derived dendritic cells are not important during the initiation phase and uncover a modest regulatory role for CCR7. Finally, we show that FRC VEGF expression is upregulated during initiation and that dendritic cells can stimulate increased fibroblastic VEGF, suggesting the scenario that lymph node-resident CD11c+ cells orchestrate the initiation of blood and lymphatic vascular growth in part by stimulating FRCs to upregulate VEGF. These results illustrate how the lymph node microenvironment is shaped by the cells it supports.
Keywords: Spleen and lymph nodes, Endothelial cells, Dendritic cells, Stromal cells, Rodent
Introduction
Lymph nodes are sites of adaptive immune responses, allowing antigen-specific T and B cells to efficiently detect cognate antigen present in draining tissues and to interact with one another. Blood vessels are critical to lymph node function, as they support the metabolic needs of the nodes and deliver, via the high endothelial venules (HEVs), recirculating lymphocytes to the lymph node parenchyma. The blood vasculature is suspended along with the lymphatic vasculature within a reticular network of collagen-rich fibrils covered by fibroblastic reticular cells (FRCs). The lymphatic sinuses control entry of antigens and antigen presenting cells from the draining tissues and the egress of lymphocytes from the lymph nodes while the reticular network regulates lymphocyte localization, migration and homeostasis (1–4) During immune responses, when lymph nodes can swell to many times their original size, the different elements of the vascular-stromal compartment also grow. The feeding arteriole delivering arterial flow is remodeled and the HEVs and other portions of the microcirculation undergo a proliferative expansion, resulting in increased delivery and entry of blood-borne lymphocytes (5–10). The lymphatic vasculature also expands, allowing for increased migratory dendritic cell entry (11–13). The reticular network undergoes apparent expansion as well, presumably to accommodate the greatly increased cellularity in the stimulated lymph node (14). Understanding how the blood vascular growth is regulated and whether the growth is coordinately regulated with that of other cellular elements of the vascular-stromal compartment may help to understand how immune responses are orchestrated.
We have previously shown using flow cytometry that the total population of endothelial cells (consisting of a mix of 85% blood vascular endothelial cells and 15% lymphatic endothelial cells) in lymph nodes demonstrate upregulation of proliferation within 2 days after immunization, and that this proliferation is accompanied by marked expansion of endothelial cell numbers in subsequent days. The initial upregulation of proliferation is dependent on CD11c+ cells, as depletion of CD11c+ cells abrogates this upregulation. Injection of bone marrow- derived dendritic cells (BMDCs) can drive upregulation of proliferation at day 2 even in RAG1−/− mice, suggesting that dendritic cells can initiate vascular expansion in a manner independent of T and B cells (8). Studies focused on the lymphatic vasculature showed that lymphatic expansion is detectable by day 4 and that this expansion is dependent on B cells at this time point (11, 12), pointing to a role for lymphocytes either during initiation of lymphatic growth or during lymphatic expansion. Whether these studies together represent conflicting data on the role of lymphocytes in vascular growth, regulation that is unique to blood versus lymphatic endothelial cells, or whether they reflect an early lymphocyte-independent initiation phase followed by a lymphocyte-dependent expansion phase has not been fully addressed, but Liao and Ruddle showed that early phenotypic changes on HEVs are T and B cell-independent while later alterations to HEV are B cell-dependent (12), suggesting two phases of vascular regulation that could potentially also exist for the regulation of vascular growth.
The lymph node vasculature is intimately associated with FRCs, as the FRCs ensheathe the vessels as well as the network of collagen-rich fibrils that provide the infrastructure for the lymph nodes (15). The FRCs express chemokines and cytokines necessary for lymphocyte compartmentalization and homeostasis (16, 17), provide a substrate upon which lymphocytes migrate (18), and help to promote peripheral T cell tolerance (19). The reticular network may help regulate vascular activity, as the compartment between the FRCs and the fibrils and vessels they ensheathe comprise a conduit system that allows for the delivery of small molecules through the reticular network to the blood vessels. This conduit system receives afferent lymphatic flow, and molecules generated in the draining tissue or, potentially, by the FRCs of the reticular network can be delivered to the blood vessels (4, 15, 20). The FRCs are also likely to directly regulate vascular activity. The integrity of FRC organization around HEVs is associated with alterations in vascular permeability (21). GP38/podoplanin is highly expressed on lymphatic endothelial cells but, at lower levels, also marks FRC populations throughout the lymph node (17, 22–24). Analysis of VEGF reporter mice as well as of isolated FRCs showed that gp38+ FRCs, especially the ones immediately around and in the vicinity of blood and lymphatic vasculature, are the main expressers of VEGF within lymph nodes (9). VEGF regulates homeostatic endothelial cell proliferation, and is also required for the proliferative expansion of both the blood vascular and lymphatic vasculature after immunization (8, 9, 11, 13). VEGF protein levels are upregulated by 2-fold as early as day 1 after lymph node stimulation, and this increase is sensitive to CD11c+ cell depletion, suggesting that CD11c+ cells may mediate upregulation of endothelial cell proliferation at least in part by inducing upregulation of VEGF (8). GP38+ FRCs are the main expressers of VEGF mRNA both at homeostasis and at day 2 after immunization (9), but the source of the additional VEGF in the lymph nodes after immunization has been unclear. Immunochemical staining for VEGF localizes the protein to B cell areas after immunization and this localization is dependent on lymph node B cells, suggesting that B cells may be a potential source of or repository for VEGF (11). A role for B cells is further supported by increased blood and lymphatic vascular growth in transgenic mice expressing human VEGF in B cells (25). On the other hand, density of in-situ hybridization signals and real-time PCR for VEGF mRNA show no increase after immunization, leading to the idea that VEGF may be transported in from distal sources such as the inflamed skin (13). Understanding whether FRCs proliferate and expand along with endothelial cells and whether they upregulate VEGF expression after immunization would help to understand the regulation of lymph node vascular growth.
A further unresolved issue in the regulation of vascular growth in stimulated lymph nodes is the identity of CD11c+ cells that mediates the initial upregulation of endothelial cell proliferation (8). CD11chi MHCIImed cells appear to contribute in part, as their depletion attenuates upregulation of endothelial cell proliferation at day 2, but the majority of the effect is likely to be mediated by CD11cmed cells that include CD11cmedMHCIIhi skin-derived cells and CD11cmedMHCIImed cells (21). As mice were immunized in the footpad, a potential scenario is that skin dendritic cells are stimulated to migrate to the draining node to induce upregulation of endothelial cell proliferation. This scenario is also attractive because afferent lymphatic flow has been implicated to regulate HEV function (26–28). The requirement for skin dendritic cells or their migration to lymph nodes, however, has not been tested.
In this study, we first examine the role of T and B cells in lymph node blood vascular growth and show that the growth is comprised of a T/B cell-independent and CD11c+ cell-dependent initiation phase followed by a T and B cell-dependent expansion phase. Lymphatic endothelial cells and FRCs proliferate and expand coordinately with the blood vascular endothelial cells. We show that skin-derived dendritic or other CD11c+ cells do not mediate the upregulation of endothelial and FRC proliferation during the initiation phase, although our experiments uncover a regulatory role for CCR7-dependent radioresistant cells in controlling the magnitude of endothelial and FRC proliferation. Furthermore, we show here that FRCs upregulate VEGF expression during the initiation phase and that dendritic cells can stimulate fibroblastic cells to upregulate VEGF, supporting the thought that CD11c+ cells induce upregulation of blood vascular and lymphatic endothelial cell proliferation by stimulating FRCs to upregulate VEGF. These results extend our understanding of the regulation of lymph node vascular growth and underscore the coordinated regulation of the individual elements of the vascular-stromal compartment.
Materials and Methods
Mice
C57Bl/6 mice between 6–12 weeks of age were obtained from Jackson Laboratory (Bar Harbor, ME), Taconic Farms (Hudson, NY) or National Cancer Institute (Frederick, MD). RAG1−/− (29)and CD11c-DTR mice (30)were originally from Jackson Laboratory and bred in our facility. μMT and βδ TCR-deficient mice were from Jackson Laboratory. CCR7−/− mice (31) and Langerin-DTR mice (32) were as previously described. VEGF-lacZ mice were originally on a CD-1 background and crossed over 10 generations to B6 mice for our studies (9, 33). All animal procedures were performed in accordance with the regulations of the Institutional Animal Use and Care Committee of the Hospital for Special Surgery.
Mouse treatments and immunizations
For immunization with ovalbumin in complete Freund adjuvant (OVA/CFA), mice received hind footpad injection of 20ul of OVA/CFA as described (8) Mice receiving BMDCs were injected in hind footpads with 1×106 cells per footpad in 20ul of volume as described (8).
For proliferation studies, mice were given intraperitoneal injections of 2mg of BrdU at 18 hours and 1 hour prior to sacrifice and 0.8mg/ml BrdU in the drinking water in between injections as previously described (8, 21). CD11c-DTR mice express simian diphtheria toxin receptor that allows for depletion of CD11c+ cells upon diphtheria toxin (DT) administration (30). Depletion is as described previously (8, 21). Mice were injected in one footpad with indicated amount of DT (Calbiochem, San Diego, CA or List Biological Laboratory, Campbell, CA) in 10–15ul of volume. At 8 hours later, they were immunized with OVA/CFA in the same footpad. To control for the DT injection, control CD11c-DTR mice were injected with inactive mutant diphtheria toxin cross reactive material (CRM 197, Calbiochem).
For T cell depletion in μMT mice, 500ug of anti-CD4 (clone GK1.5) and of anti-CD8 (clone 53-6.72) or isotype controls (all from Bio × Cell, West Lebanon, NH) was injected intraperitoneally at days −5, −4, −3, 0 (day of immunization), and +3 (34). Depletion was verified by flow cytometry, with CD4+ T cells depleted by 97% and CD8+ T cells depleted by 92% in draining nodes.
BMDC preparation
BMDCs were generated as previously described (8). Briefly, bone marrow cells were cultured with 8–10% supernatant from GM-CSF-expressing J558L cells and matured with LPS on day 7. They were harvested on day 8, washed, and injected at 1×106 cells per hind footpad. BMDC preparations were depleted of contaminating Gr-1+ neutrophils using MACS magnetic selection (Miltenyi Biotec, Auburn, CA).
Flow cytometry analysis
For flow cytometric analysis of endothelial cell and FRC numbers and proliferation, we prepared single cell suspensions and stained cells as previously described (8). In brief, lymph nodes were digested with collagenase type II (Worthington, Lakewood, NJ) and stained with antibodies for flow cytometry. Antibodies used are against CD45 (BD Biosciences, San Jose, CA), CD31 (BD Biosciences), PNAd (BD Biosciences), gp38 (Biolegend, San Diego, CA or Developmental Studies Hybridoma Bank, Iowa City, IA), BrdU (Invitrogen, Carlsbad, CA). The unconjugated anti-PNAd is detected using fluorophor-conjugated anti-rat IgM (Jackson Immunoresearch, West Grove, PA). Cells were analyzed using a FACS CANTOS (BD Biosciences) and CellQuest Pro (BD Biosciences) software.
Flow cytometric analysis of ß-gal activity in VEGF-lacZ mice is as previously described (9). Briefly, the cells were stained for extracellular markers and then incubated with the ß-gal substrate fluorescein-di-beta-D-galactopyranoside (FDG)(Invitrogen) in water for 1 minute to allow entry of FDG and subsequent cleavage and release of fluorescein. The reaction was stopped by addition of 5-fold more media and placement on ice and samples were immediately run on the flow cytometer.
BMDC-3T3 cell co-cultures
NIH-3T3 cells were cultured in DMEM (Mediatech, Herndon, VA) containing 10% heat-inactivated calf serum (Hyclone, Logan, UT) as described previously (9). Cells were plated at 5χ10^3 cells per well in 24-well plates and, the next day, the media was changed and Gr-1-depleted BMDCs or the contaminating Gr-1+ fraction were added at 3×105/ well × 2 days. Supernatant was collected for VEGF ELISA, which was performed as described previously using a kit (R&D Systems, DuoSet-mouse VEGF)(9). For real-time PCR of the 3T3 cells, cells were trypsinized and 3T3 cells were isolated from adherent BMDCs by magnetic depletion of CD45+ cells, yielding >96–99% 3T3 cells. RNA was extracted and subjected to real-time PCR for VEGF and normalized to cyclophilin as described previously(9).
Results
Dependence on T and B cells separates initiation phase from subsequent phase of vascular expansion
We have shown previously that T and B cells are dispensable for the initial upregulation of endothelial cell proliferation at day 2 after lymph nodes are stimulated with footpad injection of BMDCs. After day 2, proliferation is accompanied by a marked expansion of endothelial cell numbers by day 5 (8, 21), and we asked whether T and B cells were also dispensable for this subsequent expansion. We examined endothelial cells as we have previously done, gating on CD45-CD31+ cells (Fig. 1A). At day 5 after BMDC injection, RAG1−/− lymph nodes showed no growth in cellularity (Fig. 1B) and total endothelial cell numbers showed no expansion (Fig. 1C) and a poor rate of proliferation (Fig. 1D), suggesting that T and/or B cells are critical for mediating the vascular expansion that occurs after day 2. This result combined with our previous results point to 2 separate phases of vascular growth—an early T and B cell-independent initiation phase, whereby endothelial cell proliferation is upregulated, followed by a T and/or B cell-dependent expansion phase.
Figure 1. Lymph node vascular growth is characterized by an initial T/B cell-independent initiation phase followed by a T/B cell-dependent expansion phase.

(A) Flow cytometry plots showing subsetting of CD45-CD31+ endothelial cells into PNAd+ HEV endothelial cells (HEV EC), PNAd-gp38− non-HEV blood vascular endothelial cells (non-HEV BEC), and PNAd-gp38+ lymphatic endothelial cells (LEC). (B–D) Wild-type (WT) or RAG1−/− mice were injected with 10^6 BMDCs in the hind footpads on day 0 and draining popliteal lymph nodes were examined on day 5. Unimmunized mice received either no footpad injection or were sham injected with PBS. (B) Lymph node cellularity as determined by count of lymph node cells. (C) Number of total CD45-CD31+ endothelial cells as determined by flow cytometric analysis. (D) Proliferation rate of CD45-CD31+ endothelial cells as determined by the percent of endothelial cells that are BrdU+. (E–J) WT or RAG1−/− mice were immunized with OVA/CFA on day 0 and draining popliteal nodes were examined on day 2 (E–G) or on day 5 (H–J). Unimmunized mice received either no footpad injection or were sham injected with PBS. (E, H) Lymph node cellularity. (F, I) Number of endothelial cells in each subset. (G, J) Proliferation rate of endothelial cell subsets. * indicates p value<.05 and ** indicates p value<.01 in comparison to the same cells in unimmunized controls of the same genotype using t-test; + indicates p-value<.05 and ++ indicates p-value<.01 in comparison to the same cells in WT mice receiving same treatment. For all studies except for the day 5 proliferation studies, n= at least 6 mice per group over at least 3 experiments. For day 5 proliferation studies, n=3 mice over 2 experiments for RAG1−/− PBS group, and n=at least 6 mice per group over at least 3 experiments for all other groups.
To confirm that these two phases were not specific to stimulation with BMDCs, we immunized wild-type and RAG1−/− mice with ovalbumin in complete Freund adjuvant (OVA/CFA). In these experiments, we also included additional markers of endothelial cells to examine the regulation of different subsets of endothelial cells in more detail. We have previously used peripheral node addressin (PNAd) expression to mark HEV endothelial cells within the CD45-CD31+ endothelial cell population(1, 9). Here, we identified HEV endothelial cells using PNAd again, and further divided the PNAd-population into gp38− blood vascular endothelial cells and gp38+ lymphatic endothelial cells (17, 22) (Fig. 1A). Note that the gp38 + lymphatic endothelial cells comprise only a small percentage (about 10–15%) of the total endothelial population in popliteal lymph nodes (Fig. 1A, 1F), while they comprise about 30–45% of the endothelial cells in brachial nodes (see Fig. 5D as an example). In this paper, we will refer to PNAd+ endothelial cells as “HEV endothelial cells,” PNAd− cells as a whole as “non-HEV mixed endothelial cells,” PNAD-gp38− cells as “non-HEV blood vascular endothelial cells,” and PNAD-gp38+ cells as “lymphatic endothelial cells.”
Figure 5. CCR7-dependent cells do not mediate upregulation of endothelial and FRC proliferation during the initiation phase.

Wild-type (WT) or CCR7−/− mice were immunized with OVA/CFA on day 0 and draining popliteal nodes and non-draining brachial nodes were examined on day 2. (A) Lymph node cellularity. (B) Number of dendritic cells per lymph node in the gates shown in (C). (C) Flow cytometry plot showing relative lack of CD11cmedMHCIIhi cells in CCR7−/− popliteal lymph nodes at day 2 after OVA/CFA. (D) Number of endothelial cells in all subsets. (E) Proliferation rate of endothelial cells. (F) Number of gp38+ FRCs. (G) Proliferation rate of gp38+ FRCs. Comparisons were made between WT and CCR7−/− samples using upaired t-test. * indicates p value<.05 and ** indicates p value<.01 in comparison to the equivalent cells in WT mice. n for proliferation determination= 5 mice per group over 2 experiments; n for lymph node, dendritic cell, endothelial cell, and FRC numbers=at least 7 mice per group over at least 3 experiments.
At day 2 after OVA/CFA in wild-type mice, lymph node cellularity was increased 4-fold from baseline (Fig. 1E). Total endothelial cell numbers were not significantly increased (Fig. 1F), but HEV endothelial cells doubled in numbers (Fig. 1F). The RAG1−/− mice showed a similar pattern of lymph node expansion (Fig. 1E) (wild-type=4.4-fold (+/−.8); KO=3.3-fold (+/−1.3); p=.05) and HEV expansion at day 2 (Fig. 1F). All endothelial cell types in both wild-type and RAG1−/− mice showed a marked increase in proliferation by day 2 (Fig. 1G). These results confirmed that the initiation of lymph node vascular growth is T and B cell-independent and also established that the T and B cell independence is consistent for all endothelial cell subsets examined.
By day 5 after OVA/CFA, lymph nodes were greatly enlarged (Fig. 1H) (18 –fold (+/−7.2)) and total endothelial cell numbers in wild-type mice expanded by nearly 4-fold, with the HEV subset expanding most dramatically and consequently accounting for about 50% of the total endothelial cells (Fig. 1I). In contrast to the unimportance of T and B cells at day 2 and similar to the findings in Fig. 1B, lymph nodes were only modestly expanded in size (Fig. 1H) (5-fold (+/−2.8); p<.01 compared to wild-type) and total endothelial cell numbers in RAG1−/− mice were not expanded, although HEV and lymphatic subsets had a low level of expansion (Fig. 1I), with the HEV endothelial cells demonstrating an expansion similar to what had been observed at day 2 (Fig. 1F). The proliferation rate in most endothelial cell subsets was upregulated at day 5 compared to unimmunized RAG1−/− mice, but the increase in proliferation was modest when compared to the increase in wild-type mice (Fig. 1J). Together, our results suggested that there are two distinct phases of vascular growth after immunization: a T and B cell-independent initiation phase between days 0–2 and a T and/or B cell-dependent vascular expansion phase after day 2 through at least day 5.
FRCs proliferate and expand with endothelial cells
We examined the extent to which the gp38+ FRCs proliferate and expand in number over time after OVA/CFA. We used the gating strategy shown in Fig. 2A. Similar to the pattern seen with endothelial cells, gp38+ FRCs in wild-type mice at day 2 after immunization had not yet expanded but showed upregulated proliferation (Fig. 2B, 2C). By day 5, the gp38+ FRCs in wild-type mice were expanded in number and continued to proliferate (Fig. 2D, 2E). In RAG1−/− mice at day 2, the upregulation of gp38+ FRC proliferation from baseline was preserved and even enhanced (Fig. 2C). GP38+ FRCs continued to proliferate at higher than baseline rates at day 5 in RAG1−/− mice, but the expansion in gp38+ FRC numbers was completely abrogated (Figure 2D, 2E), suggesting the possibility that proliferated cells were unable to survive in the absence of lymphocytes. Together, these results indicate that gp38+ FRCs proliferate and expand in parallel with endothelial cells in immune-stimulated lymph nodes and that, similar to endothelial cells, the initial proliferation is T and B cell-independent while the later expansion is T and/or B cell-dependent. Vascular growth, then, occurs in the context of a more generalized vascular-stromal proliferation and expansion.
Figure 2. FRCs proliferate and expand with the endothelial cells.

WT or RAG1y−/− mice were immunized with OVA/CFA on day 0 and draining popliteal nodes were examined on day 2 (B–C) or day 5 (D–E). (A) Gating scheme to identify gp38+ FRCs in flow cytometry plots. (B, D) Number of CD45-CD31-gp38+ FRCs at denoted time points. (C, E) FRC proliferation as measured by BrdU uptake at denoted time points. ** indicates p value<.01 in comparison to unimmunized controls of the same genotype using unpaired t-test; + indicates p-value<.05 in comparison to WT mice receiving same treatment. For all studies except for the day 5 proliferation studies, n= at least 6 mice per group over at least 3 experiments. For day 5 proliferation studies, n=3 mice over 2 experiments for RAG1−/− PBS group, and n=at least 6 mice per group over at least 3 experiments for all other groups.
B cells and T cells together mediate vascular-stromal expansion
To better understand the relative contributions of B and T cells to vascular-stromal expansion, we examined mice deficient in B or T cells. B cell-deficient uMT mice at day 5 showed an increase in lymph node cellularity (Fig. 3A) and in HEV and lymphatic endothelial cell numbers from baseline (Fig. 3B) that was only partial relative to that of wild-type mice but greater than the expansion found in RAG1−/− mice (Fig. 1I). Non-HEV blood vascular endothelial cells were reduced in number at baseline in uMT mice in comparison to that of wild-type mice (Fig. 3B), but the fold increase in expansion was the same as in wild-type mice (wild-type=2.8-fold (+/−1.3) and uMT=3.0-fold (+/−.5), p=.66 t-test), indicating that B cell absence did not affect immunization-induced expansion of this population. Proliferation rates of endothelial cells at day 5 in uMT mice were attenuated (Fig. 3C), but to a lesser extent than that seen in RAG1−/− mice (compare with Fig. 1J). FRCs also showed partial expansion in numbers in uMT mice (Fig. 3D), but, similar to what was observed in RAG1−/− mice (Fig. 2E), FRC proliferation rates were similar to that of wild-type mice (Fig. 3E). B cell deficiency, then, resulted in attenuated expansion and proliferation of HEV and lymphatic endothelial cells and in attenuated expansion but normal proliferation rate of FRCs.
Figure 3. B and T cells together mediate the expansion phase.

μMT mice (A–E) and βδ TCR−/− (F–J) and controls were immunized with OVA/CFA on Day 0 and draining popliteal nodes were examined on day 5. CD4 and CD8 T cell-depleted μMT mice (K–M) were immunized as above but examined on day 4. (A, F, K) Lymph node cellularity. (B, G, L) Number of endothelial cells in each subset. (C, H) Proliferation rate of endothelial cells in each subset. (D, I, M) Numbers of gp38+ FRCs. (E, J) Proliferation rate of gp38+ FRCs. For μMT mice and βδ TCR−/−, n=5–6 mice in each group over 3 experiments. For T cell-depleted μMT mice, n=5 mice in each group over 2 experiments. * indicates p value<.05 and ** indicates p value<.01 in comparison to the same cells in unimmunized controls of the same genotype using t-test; + indicates p-value<.05 and ++ indicates p-value<.01 in comparison to the same cells in WT mice receiving same treatment.
βδ TCR-deficient mice lacking αβ and γδ T cells had reduced lymph node cellularity and numbers of endothelial cells both at baseline and at day 5 after immunization when compared to wild-type mice (Fig. 3F–G). However, the fold increase in expansion in the numbers of HEV and nonHEV blood vascular endothelial cells were similar (for HEV, wild-type=12-fold (+/−8) and KO=12-fold (+/−9), p=.96; for nonHEV, wild-type =4-fold (+/−1.5) and KO=4-fold (+/−1.5), p=.87), suggesting that deficiency of T cells by themselves did not prevent expansion of these populations. The fold increase in lymphatic endothelial cell expansion was greater in βδ TCR-deficient mice (wild-type =7-fold (+/−5) and KO=17-fold (+/−8), p=.02) echoing the recent findings by Koh and colleagues (35). Curiously, despite equal or greater expansion, HEV and lymphatic endothelial cell proliferation rates in T cell-deficient mice were attenuated at day 5 (Fig. 3H), suggesting that T cells were required for full proliferation of these populations but this contribution was not required by itself for full expansion. Non-HEV blood vascular endothelial cell proliferation was reduced at baseline and after immunization (Fig. 3H), and showed a similar fold increase in wild-type and βδ TCR-deficient mice (wild-type=25-fold (+/−7.7) and KO=36-fold (+/− 11.7), p=.11), consistent with the idea that T cell deficiency by itself did not affect non-HEV blood endothelial cell proliferation. In contrast to the endothelial cells, FRC expansion and proliferation were both reduced (Fig. 3I–J). In summary, T cell deficiency resulted in attenuated HEV and lymphatic proliferation without attenuation of expansion, and in attenuated FRC proliferation and expansion.
As B cell-deficient mice had only a partial decrease in vascular-stromal expansion and T cell-deficient mice showed no decrease in vascular expansion, we considered that the nearly complete lack of vascular-stromal expansion in RAG1−/− mice might reflect partially redundant roles for B and T cells. We depleted CD4 and CD8 cells in uMT mice, and the resulting lack of both B and CD4/8 T cells led to small lymph nodes and complete abrogation of vascular-stromal expansion (Fig. 3K–M). Together, these results suggest that B and T cells together are required for vascular-stromal expansion.
CD11c+ cells are required for the blood vascular, lymphatic, and FRC proliferation during the initiation phase
We have previously shown that depletion of all CD11c+ subsets by footpad injection of 100ng of diphtheria toxin into CD11c-DTR mice greatly reduces the upregulation of endothelial cell proliferation normally seen at day 2 after immunization (8, 21). Here, we asked whether CD11c+ cells were important for the proliferation of both blood and lymphatic endothelial cells and also for the gp38+ FRCs. As we have previously shown, footpad administration of 100ng of DT resulted in relative depletion of all CD11c+ subsets, but especially of CD11chiMHCIImed and CD11cmedMHCIIhi subsets (Fig. 4A–B), and also resulted in reduced proliferation of total endothelial cells at day 2, with the HEV cells being less sensitive to and requiring greater levels of CD11c+ cell depletion than non-HEV mixed endothelial cells (Fig. 4C) (8, 21). Of the non-HEV mixed endothelial cells, both non-HEV blood vascular endothelial cell and lymphatic endothelial cell proliferation were sensitive to CD11c+ cell depletion (Fig. 4C). Upregulation of FRC proliferation was also attenuated upon CD11c+ cell depletion (Fig. 4D). These results indicated that the proliferation of blood vascular and lymphatic endothelial cells and FRCs during the initiation phase are coregulated and are suggestive of a scenario whereby CD11c+ cells orchestrate a program of vascular-stromal proliferation.
Figure 4. CD11c+ cells mediate upregulation of proliferation of blood and lymphatic endothelial cells and of FRCs during the initiation phase.
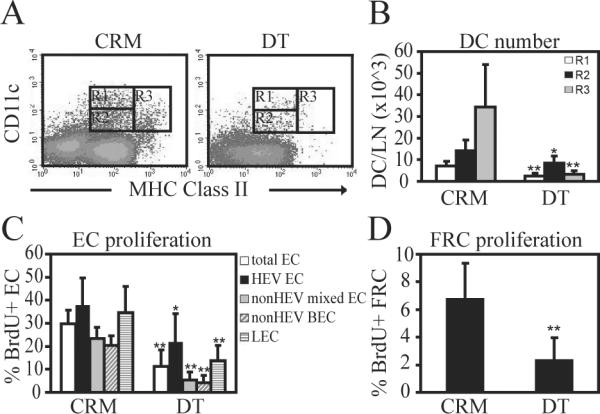
CD11c-DTR mice were injected with 100ng DT or control inactive mutant toxin (CRM) in the right footpad at 8 hours before OVA/CFA injection in the same footpad on day 0. Draining popliteal nodes were examined on day 2. (A) Flow cytometry plots showing relative depletion of all CD11c+ cell subsets with DT. (B) Number of cells per lymph node in the gates shown in (A). (C) Proliferation rate of endothelial cell subsets as measured by BrdU uptake. (D) Proliferation rate of gp38+ FRCs. * indicates p value<.05 and ** indicates p value<.01 in comparison to the same cells in CRM-treated mice using unpaired t-test. n=6 mice per condition over 4 experiments.
Skin-derived dendritic cells are not required for vascular-stromal proliferation during the initiation phase and CCR7 plays a modest regulatory role
The CD11c+ cells that mediate the vascular-stromal proliferation during the initiation phase could be resident within the lymph nodes or could derive from the draining skin. The CD11cmed MHCIIhi dendritic cells are a candidate population of CD11c+ cells (21) and include migratory skin-derived dendritic cells that require CCR7 to enter lymph nodes (31, 36, 37), so we asked about the importance of dendritic cell migration from the skin during the initiation phase by examining CCR7−/− mice. Consistent with the role of CCR7 in T and B cell entry into lymph nodes at homeostasis (31, 38), unstimulated brachial nodes were smaller in CCR7−/− mice than in wild-type mice. However, the cellularity of the draining popliteal lymph nodes at day 2 after OVA/CFA was similar to that of wild-type mice (Fig. 5A), suggesting that recruitment of lymphocytes after immunization was not CCR7-dependent. CD11cmedMHCIIhi dendritic cells were reduced in number as expected in CCR7−/− mice (Fig. 5B, 5C). CCR7−/− mice had more HEV endothelial cells in non-draining brachial nodes and more HEV and non-HEV blood vascular endothelial cells in the draining popliteal nodes (Fig. 5D), suggesting a regulatory role for CCR7-dependent cells. Endothelial cell proliferation rates in both non-draining and draining lymph nodes were equal to that of wild-type mice (Fig. 5E). GP38+ FRC numbers and proliferation were not affected in CCR7−/− mice (Fig. 5F, 5G). These results suggested that CCR7-dependent migration of dendritic cells was not required to mediate the upregulation of endothelial and FRC proliferation during the initiation phase and were, instead, suggestive of a regulatory role for CCR7-dependent cells.
We asked further whether any defect in vascular-stromal proliferation could be observed if we separated out the contributions of radiosensitive and radioresistant CCR7-dependent cells. Langerhans cells derived from the epidermis and a CD11b+ subpopulation of dermal dendritic cells are among the radioresistant dendritic cells in the skin (39, 40), while cells such as Langerin+ DEC205+ dermal dendritic cells are radiosensitive (41–43). We generated mixed bone marrow chimeras to examine the contribution of radiosensitive dendritic and other non-T non-B CCR7-dependent cells. Irradiated wild-type mice were reconstituted with 80% CCR7−/−RAG1−/− and 20% wild-type bone marrow. These chimeras had a less robust lymph node enlargement when compared to CCR7-sufficient chimeras at day 2 after OVA/CFA (Supplemental Fig. 1A), but had unchanged endothelial cell and FRC numbers and proliferation (Supplemental Fig. 1B–E), suggesting that radiosensitive CCR7-dependent non T-non B cells mediated the full extent of initial lymph node swelling but otherwise did not affect endothelial or FRC proliferation.
To test the potential role of radioresistant CCR7-dependent cells, we reconstituted CCR7−/− recipients with wild-type bone marrow. In these chimeras irrespective of the genotype, B cells and CD11chiMHCIImed cells were >99% donor-derived, indicating successful replacement of radiosensitive cells with donor cells. CD11cmedMHCIIhi cells were 91% donor-derived in the draining lymph nodes of wild-type→wild-type chimeras, reflecting a 9% contribution of the radioresistant Langerhans cells and dermal dendritic cell subset at day 2 after immunization. In the wild-type→CCR7 chimeras, the donor-derived cells were increased to >98%, likely reflecting the lack of migration to the lymph node of the radioresistant CCR7-dependent dendritic cells. The CD11cmedMHCIImed cells were 95% donor-derived in chimeras regardless of recipient genotype. WT→CCR7−/− chimeras had larger draining and non-draining nodes than wild-type→wild-type chimeras (Supplemental Fig. 1F). The WT→CCR7−/− chimeras had more HEV endothelial cells in the nondraining brachial nodes and more HEV and non-HEV blood endothelial cells in the draining popliteal nodes (Supplemental Fig. 1G), resembling the phenotype in the CCR7−/− mice. Proliferation of non-HEV blood vascular endothelial cells and of gp38+ FRCs was increased in the draining nodes of WT→CCR7−/− chimeras when compared to the nodes of WT→WT chimeras (Supplemental Fig. 1H, 1J). FRC numbers were unchanged (Supplemental Fig. 1I). These results suggested that radioresistant CCR7-dependent cells do not contribute to the upregulation of vascular-stromal proliferation but play a modest regulatory role, controlling the magnitude of lymph node cellularity, endothelial cell numbers, and non-HEV blood vascular endothelial cell and FRC proliferation.
Although CCR7-mediated migration of skin dendritic cells to lymph nodes was not necessary for initiation of lymph node vascular growth, cells in the skin can potentially regulate lymph node events by delivery of molecules to the lymph node via the afferent lymphatics (44, 45). We tested directly whether dendritic and other CD11c+ cells from the skin were required for the vascular-stromal proliferation seen during initiation. We administered a low dose (25ng) of DT locally to the footpad, reasoning that the DT should deplete local skin CD11c+ cells and, at this low dose, be less effective at reaching the lymph node to deplete CD11c+ cells located within the node. The DT resulted in a 70% reduction of CD11cmed MHCIIhi cells in the lymph node while other populations were preserved in number (Fig. 6A, 6B), consistent with the idea that we had mostly depleted skin-derived migratory dendritic cells. Endothelial cell proliferation was not affected (Fig. 6C), suggesting that the depleted skin-derived dendritic cells were not important in mediating the vascular proliferation during the initiation phase.
Figure 6. Depletion of skin-derived CD11c+ cells does not affect lymph endothelial cell proliferation during the initiation phase.

CD11c-DTR mice were injected with 25ng DT or CRM in the right footpad at 8 hours before OVA/CFA injection in the same footpad on day 0. Draining popliteal nodes were examined on day 2. (A) Flow cytometry plot showing depletion of CD11cmedMHCIIhi cells with DT. (B) Number of cells per lymph node in the gates shown in (A). (C) Endothelial cell proliferation. * indicates p value<.05 in comparison to same cells in CRM-treated mice using unpaired t-test. n =6 mice per group over 4 experiments.
Dendritic cells stimulate fibroblasts to upregulate VEGF expression
We asked whether FRCs upregulated VEGF expression after immunization by using VEGF-lacZ reporter mice to compare VEGF mRNA expression before and after immunization. FDG cleavage and release of fluorescein was used to measure beta-galactosidase activity in the gp38+ FRCs of VEGF-lacZ mice. As there is a low rate of continued FDG cleavage over the time required to run a sample on the flow cytometer, we ran the samples from the immunized mice prior to running the samples from unimmunized mice. This potentially underestimated any increase in beta-galactosidase activity upon immunization, but ensured that any increase seen in the samples from immunized mice was not an artifact of a longer incubation period. GP38+ FRCs at day 2 after OVA/CFA had greater beta-galactosidase activity than FRCs in unimmunized mice (Fig. 7A, 8B), and similar results were obtained at day 3 and with BMDC-stimulated mice (data not shown).
Figure 7. VEGF is upregulated in FRCs in vivo and in fibroblasts cultured with dendritic cells.

(A–B) VEGF-lacZ mice were immunized with OVA/CFA on day 0 and draining popliteal nodes were examined on day 2. Beta-galactosidase activity as an indicator of VEGF expression was measured as described in Methods. (A) Flow cytometry dot plots gated on CD45-CD31- cells showing beta-galactosidase activity of gp38+ FRCs from an immunized wild-type mouse, a sham-immunized VEGF-lacZ mouse, and an OVA/CFA-immunized VEGF-lacZ mouse. (B) Graph shows mean fluorescence intensity of gp38+ FRCs in the VEGF-lacZ mice. * indicates p value<.05 in comparison to unimmunized mice using paired t-test. Each symbol represents 1 mouse, with matched symbols in the unimmunized and OVA/CFA-immunized conditions representing a pair from a single experiment. Data was accumulated over 4 experiments. (C–D) 3T3 fibroblasts were cultured alone or with Gr-1 cell-depleted BMDCs (DC fraction) or the Gr1+ contaminating cells (Gr1 fraction) for 2 days. (C) VEGF levels in the culture supernatants. * p<.05 and ** p<.01 t-test. Each condition was plated in triplicate wells; bars show average value of the triplicate wells. Results representative of at least 5 similar experiments. (D) VEGF mRNA level in the 3T3 cells after culturing without or with DC fraction, as measured by real-time PCR. Each bar represents average values of two experiments, with each symbol representing the value obtained in a single experiment.
CD11c+ cells have been shown to be localized near or directly on FRCs (18, 21, 48, 49), and we have previously shown the CD11c+ cells are also enriched in regions rich in VEGF-expressing FRCs (9), raising the possibility that dendritic or other CD11c+ cells could interact with FRCs to promote VEGF upregulation. To test this possibility, we added BMDCs to cultures of 3T3 fibroblasts, which we have previously shown to share some of the characteristics of gp38+ FRCs, including gp38, lymphotoxin beta receptor, and VEGF expression (9). BMDCs were depleted of contaminating granulocytes using magnetic selection to yield a dendritic cell-rich fraction (DC fraction) that was 90% pure and a granulocyte-rich fraction (Gr fraction) that was relatively dendritic cell-poor, but still contained about 30% dendritic cells (data not shown). At baseline, the DC and Gr fractions both showed very low levels of VEGF expression while 3T3 cells expressed high levels of VEGF (Fig. 7C). Addition of the DC fraction to the 3T3 cells induced over a 2-fold rise in VEGF levels (Fig. 7C). This effect was specific for the DC fraction, as culturing of the fibroblasts with Gr fraction cells did not result in VEGF increases (Fig. 7C). The fibroblasts showed upregulated VEGF mRNA expression after exposure to the DC fraction (Fig. 7D). These results support the possibility that dendritic cell stimulation of FRCs may contribute to the increased VEGF levels found in stimulated lymph nodes.
Discussion
Our results presented here further our understanding of the how the vasculature grows after lymph node stimulation. Blood vascular and lymphatic endothelial cells proliferate and expand in a concerted manner with FRCs and there is a CD11c+ cell-dependent and T/B cell-independent initiation phase characterized by upregulation of proliferation that is followed by a T and B cell-dependent expansion phase that results in greater numbers of endothelial cells and FRCs. We further extend our understanding of how CD11c+ cells may orchestrate the initiation phase, finding that skin-derived dendritic cells are not the required CD11c+ cells that mediate the initiation phase, that gp38+ FRCs are stimulated to upregulate VEGF after immunization, and that dendritic cells can directly stimulate fibroblast-type cells to upregulate VEGF, suggesting the scenario that resident CD11c+ cells upregulate blood vascular and lymphatic endothelial cell proliferation in part by stimulating FRCs to produce more VEGF.
The T and B cell-independence during the initiation phase followed by T and B cell-dependence during the expansion phase mirrors the kinetics of innate and then adaptive immune cell activation during immune responses, and suggests a scenario whereby innate immunity begins to stimulate vascular-stromal growth that is only fully completed if there is antigen-specific activation of lymphocytes. Upon activation, the lymphocytes promote vascular-stromal expansion and the vascular-stromal expansion presumably ensures that the microenviroment can fully support the generation of effector and memory T and B cells. More experiments will be required to fully understand whether antigen-specific lymphocytes are necessary.
B cells have been shown to mediate lymphatic expansion after acute immunization (11, 12) and HEV expansion in a viral infection model (50), and our current findings showing a partial contribution by B cells are consistent with these studies. CD4 T cells are important for feeding arteriole remodeling (51) and suppress lymphatic expansion (35), and our findings of T cell contributions echo these findings. Here, our experiments reveal that B and T cells together contribute to mediate full vascular-stromal expansion and that there is partial redundancy or compensation between T and B cells. The normal vascular expansion in βδ TCR-deficient mice suggested that B cells can fully compensate for the lack of T cells while the partial defect in vascular expansion in B cell-deficient mice suggested that B cells possess unique properties for which T cells can only partially compensate. For FRCs, expansion was partially affected in both B and T cell-deficient mice, suggesting additive effects or partial redundancy of B and T cells.
The data were suggestive for a role for lymphocytes in mediating vascular-stromal expansion by actively promoting cell survival in addition to and somewhat independent of promoting cell proliferation. For example, although T cell-deficient mice had normal HEV and nonHEV blood vascular and FRC expansion, they had a partial proliferation defect, indicating that T cells make unique contributions to vascular-stromal proliferation. The normal expansion in the face of reduced proliferation may reflect the possibility that the proliferation defect was too mild to affect expansion, or it may reflect a role for B cells in compensating fully for the proliferation defect in part by promoting enhanced cell survival. Such a separation between cell proliferation and cell survival was also seen in the uMT mice, as FRCs showed reduced expansion but wild-type levels of proliferation. Similarly, in RAG1−/− mice, the initial drive for FRC proliferation can be seen to be intact even at day 5, but the expansion is completely prevented. Further work will be needed to test this idea, but cell proliferation and survival can be regulated by different sets of molecules (eg cyclins and Bcl-2 family members, respectively) and have been shown to be distinctly regulated. A recent example is in the case of mice deficient for p18, a negative modulator of cell cycling, where plasmablasts proliferate highly but do not survive (52). Also, in the dissection of the role of the anti-apoptotic molecule survivin in balloon-mediated arterial injury, survivin was required for PDGF-mediated cell survival but not for mitosis (53). It is possible that T and B cells can actively regulate both cell proliferation and cell survival to promote vascular-stromal expansion.
We have previously shown that CD11c+ cells are required for the initiation of lymph node vascular growth (8) and, here, we extend our findings to show that CD11c+ cells mediate upregulation of blood vascular as well as lymphatic endothelial cell and FRC proliferation. These results suggest that CD11c+ cells initiate the coordinated growth of the cellular elements of the vascular-stromal network. These findings may have implications for understanding the role of CD11c+ cells in the growth of other tissues. Recent studies have shown that, in inflamed airways, depletion of CD11c+ cells during the period of induced bronchus-associated lymphoid tissue (iBALT) growth did not affect the number of iBALTs but resulted in smaller iBALTs (54). This role for CD11c+ cells in mediating iBALT growth could potentially in part reflect the contribution of initiating CD11c+ cells to the growth of the vascular-stromal network in these lymph node-like structures. Furthermore, the beneficial effect of adding dendritic cells to a tissue-engineered lymph node (55, 56) may reflect increased dendritic cell-driven proliferation of the fibroblast-type cells and lymphatics as well as support blood vessel growth. Additionally, CD11c+ cells have been implicated in vascular expansion in inflamed non-lymphoid tissues and tumors (57–60), and our results suggest that CD11c+ cells may support the growth of these tissues beyond that of the blood vascular endothelial cells.
Here we have narrowed down the candidates for the CD11c+ cells that mediate the initiation phase. CD11cmedMHCIIhi cells include migratory dendritic cells that require CCR7 to migrate from skin and may also include monocyte-derived dendritic cells that enter lymph nodes via blood (31, 37, 61). Our results showed that neither preventing lymph node entry of CD11cmedMHCIIhi cells nor selectively depleting skin-derived CD11cmedMHCIIhi cells resulted in reduced vascular-stromal proliferation and suggest that skin-derived dendritic cells likely do not mediate the upregulation of vascular-stromal proliferation during the initiation phase. These results combined with our previous results showing only a partial role for CD11chiMHCIImed cells suggest that a subpopulation of CD11cmedMHCIImed cells residing in lymph nodes is important for mediating the events of the initiation phase; experiments are ongoing to test this hypothesis.
We show here that FRCs upregulate VEGF mRNA expression at day 2 and that, in vitro, dendritic cells can stimulate fibroblast-type cells to upregulate VEGF expression. Our results together suggest a scenario whereby, during the initiation phase, lymph node-resident CD11c+ cells stimulate FRCs to upregulate VEGF, which, in turn, induces upregulation of blood vascular and lymphatic endothelial cell proliferation. GP38+ FRCs can also express the lymphangiogenic molecule VEGF-C (62), and it is likely that CD11c+ cells alter FRC activity in additional ways to contribute to the initiation of vascular expansion. Dendritic cells are closely associated with the reticular network (18, 48, 49) and our co-culture experiments suggest that direct communication with FRCs could lead to VEGF upregulation. We have also shown a role for recruited blood-borne L-selectin-dependent cells in mediating a portion of the dendritic cell-driven endothelial cell proliferation (8), so it is possible that CD11c+ cells also promote VEGF upregulation in part by first promoting recruitment of L-selectin+ cells via increased blood delivery (5), increased vascular permeability (7), or altered HEV cell adhesion molecule or chemokine expression (12, 21, 63–65). Our results suggest that stimulation of blood and lymphatic vascular proliferation is mediated by at least a tripartite interaction among CD11c+ cells, FRCs, and the endothelial cells.
Together, our results add to the emerging concept that there is a constellation of vascular-stromal alterations that occur with the induction of an immune response in lymph nodes and highlight the coordinated regulation of vascular and stromal expansion by first innate and then adaptive immune cells. Lymph nodes are enlarged and the vascular-stromal compartment can potentially be altered in autoimmune and lymphoproliferative diseases. Understanding the extent of vascular-stromal alterations in stimulated lymph nodes, how these alterations are regulated and function in immunity may lead to the targeting of the vascular-stromal compartment in the development of novel therapeutics.
Supplementary Material
Acknowledgements
We thank all the members of the Lu lab for helpful discussions, Dr. Sanjiv Luther for critical reading of the manuscript, Drs. Miriam Merad and Bernard Malissen for providing Langerin-DTR mice and Dr. Sergio Lira for providing CCR7 breeders.
Funding support: This work was funded by R01-AI069800, R01 AI079178, and a Lupus Research Institute award (TL),
Abbreviations
- FRC
fibroblastic reticular cell
- HEV
high endothelial venule
- VEGF
vascular endothelial growth factor
References
- 1.von Andrian UH, Mempel TR. Homing and cellular traffic in lymph nodes. Nat Rev Immunol. 2003;3 doi: 10.1038/nri1222. [DOI] [PubMed] [Google Scholar]
- 2.Mueller SN, Germain RN. Stromal cell contributions to the homeostasis and functionality of the immune system. 2009;9 doi: 10.1038/nri2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lammermann T, Sixt M. The microanatomy of T-cell responses. Immunol Rev. 2008;221 doi: 10.1111/j.1600-065X.2008.00592.x. [DOI] [PubMed] [Google Scholar]
- 4.Junt T, Scandella E, Ludewig B. Form follows function: lymphoid tissue microarchitecture in antimicrobial immune defence. Nat Rev Immunol. 2008;8 doi: 10.1038/nri2414. [DOI] [PubMed] [Google Scholar]
- 5.Soderberg KA, Payne GW, Sato A, Medzhitov R, Segal SS, Iwasaki A. Innate control of adaptive immunity via remodeling of lymph node feed arteriole. Proc Natl Acad Sci U S A. 2005;102 doi: 10.1073/pnas.0506190102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herman PG, Yamamoto I, Mellins HZ. Blood microcirculation in the lymph node during the primary immune response. J Exp Med. 1972;136 doi: 10.1084/jem.136.4.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anderson ND, Anderson AO, Wyllie RG. Microvascular changes in lymph nodes draining skin allografts. Am J Pathol. 1975;81 [PMC free article] [PubMed] [Google Scholar]
- 8.Webster B, Ekland EH, Agle LM, Chyou S, Ruggieri R, Lu TT. Regulation of lymph node vascular growth by dendritic cells. J Exp Med. 2006;203 doi: 10.1084/jem.20052272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chyou S, Ekland EH, Carpenter AC, Tzeng T-CJ, Tian S, Michaud M, Madri JA, Lu TT. Fibroblast-Type Reticular Stromal Cells Regulate the Lymph Node Vasculature. J Immunol. 2008;181 doi: 10.4049/jimmunol.181.6.3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mebius RE, Breve J, Duijvestijn AM, Kraal G. The function of high endothelial venules in mouse lymph nodes stimulated by oxazolone. Immunology. 1990;71 [PMC free article] [PubMed] [Google Scholar]
- 11.Angeli V, Ginhoux F, Llodra J, Quemeneur L, Frenette PS, Skobe M, Jessberger R, Merad M, Randolph GJ. B cell-driven lymphangiogenesis in inflamed lymph nodes enhances dendritic cell mobilization. Immunity. 2006;24 doi: 10.1016/j.immuni.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 12.Liao S, Ruddle NH. Synchrony of High Endothelial Venules and Lymphatic Vessels Revealed by Immunization. J Immunol. 2006;177 doi: 10.4049/jimmunol.177.5.3369. [DOI] [PubMed] [Google Scholar]
- 13.Halin C, Tobler NE, Vigl B, Brown LF, Detmar M. VEGF-A produced by chronically inflamed tissue induces lymphangiogenesis in draining lymph nodes. Blood. 2007;110 doi: 10.1182/blood-2007-01-066811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Katakai T, Hara T, Sugai M, Gonda H, Shimizu A. Lymph Node Fibroblastic Reticular Cells Construct the Stromal Reticulum via Contact with Lymphocytes. J. Exp. Med. 2004;200 doi: 10.1084/jem.20040254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gretz JE, Anderson AO, Shaw S. Cords, channels, corridors and conduits: critical architectural elements facilitating cell interactions in the lymph node cortex. Immunol Rev. 1997;156 doi: 10.1111/j.1600-065x.1997.tb00955.x. [DOI] [PubMed] [Google Scholar]
- 16.Cyster JG. Chemokines, sphingosine-1-phosphate, and cell migration in secondary lymphoid organs. Annu Rev Immunol. 2005;23 doi: 10.1146/annurev.immunol.23.021704.115628. [DOI] [PubMed] [Google Scholar]
- 17.Link A, Vogt TK, Favre S, Britschgi MR, Acha-Orbea H, Hinz B, Cyster JG, Luther SA. Fibroblastic reticular cells in lymph nodes regulate the homeostasis of naive T cells. Nat Immunol. 2007;8 doi: 10.1038/ni1513. [DOI] [PubMed] [Google Scholar]
- 18.Bajenoff M, Egen JG, Koo LY, Laugier JP, Brau F, Glaichenhaus N, Germain RN. Stromal cell networks regulate lymphocyte entry, migration, and territoriality in lymph nodes. Immunity. 2006;25 doi: 10.1016/j.immuni.2006.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee J-W, Epardaud M, Sun J, Becker JE, Cheng AC, Yonekura A.-r., Heath JK, Turley SJ. Peripheral antigen display by lymph node stroma promotes T cell tolerance to intestinal self. 2007;8 doi: 10.1038/ni1427. [DOI] [PubMed] [Google Scholar]
- 20.Gretz JE, Norbury CC, Anderson AO, Proudfoot AE, Shaw S. Lymph-borne chemokines and other low molecular weight molecules reach high endothelial venules via specialized conduits while a functional barrier limits access to the lymphocyte microenvironments in lymph node cortex. J Exp Med. 2000;192 doi: 10.1084/jem.192.10.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tzeng TC, Chyou S, Tian S, Webster B, Carpenter AC, Guaiquil VH, Lu TT. CD11chi dendritic cells regulate the re-establishment of vascular quiescence and stabilization after immune stimulation of lymph nodes. J Immunol. 2010;184 doi: 10.4049/jimmunol.0902914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Breiteneder-Geleff S, Soleiman A, Kowalski H, Horvat R, Amann G, Kriehuber E, Diem K, Weninger W, Tschachler E, Alitalo K, Kerjaschki D. Angiosarcomas express mixed endothelial phenotypes of blood and lymphatic capillaries: podoplanin as a specific marker for lymphatic endothelium. Am J Pathol. 1999;154 doi: 10.1016/S0002-9440(10)65285-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Farr A, Berry M, Kim A, Nelson A, Welch M, Aruffo A. Characterization and cloning of a novel glycoprotein expressed by stromal cells in T-dependent areas of peripheral lymphoid tissues. J. Exp. Med. 1992;176 doi: 10.1084/jem.176.5.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roozendaal R, Mempel TR, Pitcher LA, Gonzalez SF, Verschoor A, Mebius RE, von Andrian UH, Carroll MC. Conduits mediate transport of low-molecular-weight antigen to lymph node follicles. Immunity. 2009;30 doi: 10.1016/j.immuni.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shrestha B, Hashiguchi T, Ito T, Miura N, Takenouchi K, Oyama Y, Kawahara K, Tancharoen S, Ki IY, Arimura N, Yoshinaga N, Noma S, Shrestha C, Nitanda T, Kitajima S, Arimura K, Sato M, Sakamoto T, Maruyama I. B cell-derived vascular endothelial growth factor A promotes lymphangiogenesis and high endothelial venule expansion in lymph nodes. J Immunol. 2010;184 doi: 10.4049/jimmunol.0903063. [DOI] [PubMed] [Google Scholar]
- 26.Drayson MT, Ford WL. Afferent lymph and lymph borne cells: their influence on lymph node function. Immunobiology. 1984;168 doi: 10.1016/S0171-2985(84)80123-0. [DOI] [PubMed] [Google Scholar]
- 27.Hendriks HR, Eestermans IL, Hoefsmit EC. Depletion of macrophages and disappearance of postcapillary high endothelial venules in lymph nodes deprived of afferent lymphatic vessels. Cell Tissue Res. 1980;211 doi: 10.1007/BF00234394. [DOI] [PubMed] [Google Scholar]
- 28.Mebius R, Streeter P, Breve J, Duijvestijn A, Kraal G. The influence of afferent lymphatic vessel interruption on vascular addressin expression. J. Cell Biol. 1991;115 doi: 10.1083/jcb.115.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1-deficient mice have no mature B and T lymphocytes. Cell. 1992;68 doi: 10.1016/0092-8674(92)90030-g. [DOI] [PubMed] [Google Scholar]
- 30.Jung S, Unutmaz D, Wong P, Sano G, De los Santos K, Sparwasser T, Wu S, Vuthoori S, Ko K, Zavala F, Pamer EG, Littman DR, Lang RA. In vivo depletion of CD11c(+) dendritic cells abrogates priming of CD8(+) T cells by exogenous cell-associated antigens. Immunity. 2002;17 doi: 10.1016/s1074-7613(02)00365-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Forster R, Schubel A, Breitfeld D, Kremmer E, Renner-Muller I, Wolf E, Lipp M. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell. 1999;99 doi: 10.1016/s0092-8674(00)80059-8. [DOI] [PubMed] [Google Scholar]
- 32.Kissenpfennig A, Henri S, Dubois B, Laplace-Builhe C, Perrin P, Romani N, Tripp CH, Douillard P, Leserman L, Kaiserlian D, Saeland S, Davoust J, Malissen B. Dynamics and Function of Langerhans Cells In Vivo Dermal Dendritic Cells Colonize Lymph Node AreasDistinct from Slower Migrating Langerhans Cells. Immunity. 2005;22 doi: 10.1016/j.immuni.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 33.Miquerol L, Gertsenstein M, Harpal K, Rossant J, Nagy A. Multiple developmental roles of VEGF suggested by a LacZ-tagged allele. Dev Biol. 1999;212 doi: 10.1006/dbio.1999.9355. [DOI] [PubMed] [Google Scholar]
- 34.Kruisbeek AM. UNIT 4.1 In Vivo Depletion of CD4− and CD8-Specific T Cells. John Wiley & Sons, Inc.; 2001. [DOI] [PubMed] [Google Scholar]
- 35.Kataru RP, Kim H, Jang C, Choi DK, Koh BI, Kim M, Gollamudi S, Kim YK, Lee SH, Koh GY. T lymphocytes negatively regulate lymph node lymphatic vessel formation. Immunity. 2011;34 doi: 10.1016/j.immuni.2010.12.016. [DOI] [PubMed] [Google Scholar]
- 36.Lopez-Bravo M, Ardavin C. In vivo induction of immune responses to pathogens by conventional dendritic cells. Immunity. 2008;29 doi: 10.1016/j.immuni.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 37.Ohl L, Mohaupt M, Czeloth N, Hintzen G, Kiafard Z, Zwirner J, Blankenstein T, Henning G, Forster R. CCR7 governs skin dendritic cell migration under inflammatory and steady-state conditions. Immunity. 2004;21 doi: 10.1016/j.immuni.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 38.Okada T, Ngo VN, Ekland EH, Forster R, Lipp M, Littman DR, Cyster JG. Chemokine requirements for B cell entry to lymph nodes and Peyer's patches. J Exp Med. 2002;196 doi: 10.1084/jem.20020201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bogunovic M, Ginhoux F, Wagers A, Loubeau M, Isola LM, Lubrano L, Najfeld V, Phelps RG, Grosskreutz C, Scigliano E, Frenette PS, Merad M. Identification of a radio-resistant and cycling dermal dendritic cell population in mice and men. J Exp Med. 2006;203 doi: 10.1084/jem.20060667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Merad M, Manz MG, Karsunky H, Wagers A, Peters W, Charo I, Weissman IL, Cyster JG, Engleman EG. Langerhans cells renew in the skin throughout life under steady-state conditions. Nat Immunol. 2002;3 doi: 10.1038/ni852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ginhoux F, Collin MP, Bogunovic M, Abel M, Leboeuf M, Helft J, Ochando J, Kissenpfennig A, Malissen B, Grisotto M, Snoeck H, Randolph G, Merad M. Blood-derived dermal langerin+ dendritic cells survey the skin in the steady state. J Exp Med. 2007;204 doi: 10.1084/jem.20071733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bursch LS, Wang L, Igyarto B, Kissenpfennig A, Malissen B, Kaplan DH, Hogquist KA. Identification of a novel population of Langerin+ dendritic cells. J Exp Med. 2007;204 doi: 10.1084/jem.20071966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Poulin LF, Henri S, de Bovis B, Devilard E, Kissenpfennig A, Malissen B. The dermis contains langerin+ dendritic cells that develop and function independently of epidermal Langerhans cells. J Exp Med. 2007;204 doi: 10.1084/jem.20071724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Palframan RT, Jung S, Cheng G, Weninger W, Luo Y, Dorf M, Littman DR, Rollins BJ, Zweerink H, Rot A, von Andrian UH. Inflammatory chemokine transport and presentation in HEV: a remote control mechanism for monocyte recruitment to lymph nodes in inflamed tissues. J Exp Med. 2001;194 doi: 10.1084/jem.194.9.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kunder CA, St John AL, Li G, Leong KW, Berwin B, Staats HF, Abraham SN. Mast cell-derived particles deliver peripheral signals to remote lymph nodes. J Exp Med. 2009;206 doi: 10.1084/jem.20090805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martin-Fontecha A, Sebastiani S, Hopken UE, Uguccioni M, Lipp M, Lanzavecchia A, Sallusto F. Regulation of dendritic cell migration to the draining lymph node: impact on T lymphocyte traffic and priming. J Exp Med. 2003;198 doi: 10.1084/jem.20030448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Braun A, Worbs T, Moschovakis GL, Halle S, Hoffmann K, Bolter J, Munk A, Forster R. Afferent lymph-derived T cells and DCs use different chemokine receptor CCR7-dependent routes for entry into the lymph node and intranodal migration. Nat Immunol. 2011;12 doi: 10.1038/ni.2085. [DOI] [PubMed] [Google Scholar]
- 48.Luther SA, Tang HL, Hyman PL, Farr AG, Cyster JG. Coexpression of the chemokines ELC and SLC by T zone stromal cells and deletion of the ELC gene in the plt/plt mouse. Proc Natl Acad Sci U S A. 2000;97 doi: 10.1073/pnas.97.23.12694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sixt M, Kanazawa N, Selg M, Samson T, Roos G, Reinhardt DP, Pabst R, Lutz MB, Sorokin L. The conduit system transports soluble antigens from the afferent lymph to resident dendritic cells in the T cell area of the lymph node. Immunity. 2005;22 doi: 10.1016/j.immuni.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 50.Kumar V, Scandella E, Danuser R, Onder L, Nitschke M, Fukui Y, Halin C, Ludewig B, Stein JV. Global lymphoid tissue remodeling during a viral infection is orchestrated by a B cell-lymphotoxin-dependent pathway. Blood. 2010;115 doi: 10.1182/blood-2009-10-250118. [DOI] [PubMed] [Google Scholar]
- 51.Kumamoto Y, Mattei LM, Sellers S, Payne GW, Iwasaki A. CD4+ T cells support cytotoxic T lymphocyte priming by controlling lymph node input. Proc Natl Acad Sci U S A. 2011;108 doi: 10.1073/pnas.1100567108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bretz J, Garcia J, Huang X, Kang L, Zhang Y, Toellner K-M, Chen-Kiang S. Noxa mediates p18INK4c cell-cycle control of homeostasis in B cells and plasma cell precursors. Blood. 2011;117 doi: 10.1182/blood-2010-06-288027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Blanc-Brude OP, Yu J, Simosa H, Conte MS, Sessa WC, Altieri DC. Inhibitor of apoptosis protein survivin regulates vascular injury. Nat Med. 2002;8 doi: 10.1038/nm750. [DOI] [PubMed] [Google Scholar]
- 54.Halle S, Dujardin HC, Bakocevic N, Fleige H, Danzer H, Willenzon S, Suezer Y, Hammerling G, Garbi N, Sutter G, Worbs T, Forster R. Induced bronchus-associated lymphoid tissue serves as a general priming site for T cells and is maintained by dendritic cells. J Exp Med. 2009;206 doi: 10.1084/jem.20091472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Suematsu S, Watanabe T. Generation of a synthetic lymphoid tissue-like organoid in mice. Nat Biotechnol. 2004;22 doi: 10.1038/nbt1039. [DOI] [PubMed] [Google Scholar]
- 56.Okamoto N, Chihara R, Shimizu C, Nishimoto S, Watanabe T. Artificial lymph nodes induce potent secondary immune responses in naive and immunodeficient mice. J Clin Invest. 2007;117 doi: 10.1172/JCI30379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fainaru O, Almog N, Yung CW, Nakai K, Montoya-Zavala M, Abdollahi A, D'Amato R, Ingber DE. Tumor growth and angiogenesis are dependent on the presence of immature dendritic cells. Faseb J. 2010;24 doi: 10.1096/fj.09-147025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fainaru O, Adini A, Benny O, Adini I, Short S, Bazinet L, Nakai K, Pravda E, Hornstein MD, D'Amato RJ, Folkman J. Dendritic cells support angiogenesis and promote lesion growth in a murine model of endometriosis. Faseb J. 2008;22 doi: 10.1096/fj.07-9034com. [DOI] [PubMed] [Google Scholar]
- 59.Nakai K, Fainaru O, Bazinet L, Pakneshan P, Benny O, Pravda E, Folkman J, D'Amato RJ. Dendritic cells augment choroidal neovascularization. Invest Ophthalmol Vis Sci. 2008;49 doi: 10.1167/iovs.07-1640. [DOI] [PubMed] [Google Scholar]
- 60.Conejo-Garcia JR, Benencia F, Courreges MC, Kang E, Mohamed-Hadley A, Buckanovich RJ, Holtz DO, Jenkins A, Na H, Zhang L, Wagner DS, Katsaros D, Caroll R, Coukos G. Tumor-infiltrating dendritic cell precursors recruited by a beta-defensin contribute to vasculogenesis under the influence of Vegf-A. Nat Med. 2004;10 doi: 10.1038/nm1097. [DOI] [PubMed] [Google Scholar]
- 61.Cheong C, Matos I, Choi JH, Dandamudi DB, Shrestha E, Longhi MP, Jeffrey KL, Anthony RM, Kluger C, Nchinda G, Koh H, Rodriguez A, Idoyaga J, Pack M, Velinzon K, Park CG, Steinman RM. Microbial stimulation fully differentiates monocytes to DC-SIGN/CD209(+) dendritic cells for immune T cell areas. Cell. 2010;143 doi: 10.1016/j.cell.2010.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Peduto L, Dulauroy S, Lochner M, Spath GF, Morales MA, Cumano A, Eberl G. Inflammation Recapitulates the Ontogeny of Lymphoid Stromal Cells. J Immunol. 2009;182 doi: 10.4049/jimmunol.0803974. [DOI] [PubMed] [Google Scholar]
- 63.Chen Q, Fisher DT, Clancy KA, Gauguet JM, Wang WC, Unger E, Rose-John S, von Andrian UH, Baumann H, Evans SS. Fever-range thermal stress promotes lymphocyte trafficking across high endothelial venules via an interleukin 6 trans-signaling mechanism. Nat Immunol. 2006;7 doi: 10.1038/ni1406. [DOI] [PubMed] [Google Scholar]
- 64.Guarda G, Hons M, Soriano SF, Huang AY, Polley R, Martin-Fontecha A, Stein JV, Germain RN, Lanzavecchia A, Sallusto F. L-selectin-negative CCR7- effector and memory CD8+ T cells enter reactive lymph nodes and kill dendritic cells. Nat Immunol. 2007;8 doi: 10.1038/ni1469. [DOI] [PubMed] [Google Scholar]
- 65.Martin-Fontecha A, Baumjohann D, Guarda G, Reboldi A, Hons M, Lanzavecchia A, Sallusto F. CD40L+ CD4+ memory T cells migrate in a CD62P-dependent fashion into reactive lymph nodes and license dendritic cells for T cell priming. J Exp Med. 2008;205 doi: 10.1084/jem.20081212. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.