

Abstract
Whereas increased affinity enhances T cell competitiveness after immunization, the role of affinity in modulating the pathogenicity of self-reactive T cells is less established. To assess this, we generated two myelin-specific, class II MHC-restricted TCR that differ only in a buried hydroxymethyl that forms a common TRBV variant. The variation, predicted to increase TCR stability, resulted in a ~3log10 difference in TCR sensitivity with preserved fine specificity. The high affinity TCR markedly diminished T cell pathogenicity. T cells were not deleted, did not upregulate Foxp3, and barring disease induction were predominantly naïve. However, high affinity CD4+ T cells showed an altered cytokine profile characterized by the production of protective cytokines prior to experimental allergic encephalomyelitis induction and decreased effector cytokines after. Further, the high affinity TCR promoted the development of CD4−CD8− and CD8+ T cells that possessed low intrinsic pathogenicity, were protective even in small numbers when transferred into wild type mice and in mixed chimeras, and outcompete CD4+ T cells during disease development. Therefore TCR affinities exceeding an upper affinity threshold may impede the development of autoimmunity through altered development and functional maturation of T cells, including diminished intrinsic CD4+ T-cell pathogenicity and the development of CD4− Foxp3− regulatory populations.
Introduction
After infection or immunization, T cells with higher antigen (Ag) affinity outcompete lower affinity cells and come to dominate the immune response (1–4). The role of TCR affinity in determining pathogenicity among autoreactive T cells is less clear. This has been indirectly probed by examining T cell responses to altered peptide ligands (APL)(5). Antagonists or weak agonists can block or ameliorate autoimmune disease implying that low affinity cross-reactive interactions are fundamentally non-pathologic (6,7). Indeed, such APL have shown mixed results in clinical trials (8,9). Implicitly, higher affinity recognition is a prerequisite for pathogenicity. However, high affinity APL may also generate tolerance. Immunization with a proteolipid-protein (PLP)-derived APL stimulated T cells with a higher affinity for the APL than heteroclitic self-Ag. This led to the development of aTh0/Th2 response to self-Ag that inhibited disease development (10,11). Indeed, hyperstimulatory APL can also promote T cell apoptosis and tune down TCR responsiveness to cognate Ag and thereby confer protection (12,13). This suggests that T cells with either too low or high an affinity for stimulating Ag may be diverted into non-pathologic or even protective developmental pathways.
Whereas studies of APL have indicated that affinity thresholds may guide autoreactive T cell responses, few studies have directly assessed the role of affinity among T cells responding to cognate autoAg. NRP-A7-specific CD8+ T cells in NOD mice showed avidity-based competition during diabetes development, with higher avidity cells accumulating during disease progression (14). Likewise, 4E3 TCR Tg mice, which develop spontaneous fulminant EAE, bear a higher affinity TCR than 5B6 TCR Tg mice, which have a lower incidence of spontaneous disease (15). In contrast, in a direct assessment of the affinity of T cells responding in myelin oligodendrocyte glycoprotein-induced EAE (MOG-EAE) using 2D binding assays, the affinity range of responding cells varied by >100 fold (16). The majority of cells were low affinity, suggesting that high or moderate affinity cells were excluded from the response or otherwise tolerized. These low affinity cells demonstrated in vitro effector responses equivalent to those with higher affinity. In a separate study, higher affinity pancreatic Ag specific T cells were irreversibly tolerized whereas lower affinity cells persisted and were capable of initiating autoimmunity under stimulatory conditions (17). Two EAE studies also failed to correlate avidity with disease progression, one in which functional measurements of proteolipid protein (PLP)-specific T cell avidity was longitudinally followed (18), and a second exploring spontaneous EAE in retroviral transgenic (retrogenic) mice expressing a series of different TCR (19). These results imply that increased affinity is not necessarily associated with pathogenicity.
In prior analyses of the role of T cell affinity in autoimmunity, T cells expressed wholly distinct TCRs. How differences in fine specificity, degenerate recognition of additional Ags, or other recognition parameters influenced pathogenicity could not be dissected. To better characterize the role of affinity in T cell pathogenicity, we generated a mutant self-specific TCR with minimal structural differences compared to its parental receptor, but dramatically increased sensitivity for cognate autoAg. In a prior report we identified a variation in the use of G and S residues at position 107 in TRBV; TRBV13-2 possesses a G107 whereas most other TRBV have an S. This amino acid lies within the buried N-terminal core of the CDR3, with the side chain intercalating into the loop structure of the complementarity determining region (CDR) 3. We predicted based on structural modeling and molecular dynamics that substitution of the TRBV13-2 G with a S residue more typical of other TCR would position the S hydroxymethyl side chain to fill an internal gap within the CDR3 loop, and through increased van der Waals interactions and H-bonding stabilize it without substantially altering its structure (20). Indeed, a G107S substitution in the TRBV13-2+ IAb-MOG35–55-specific 1MOG9 TCR yielded a ~3log10 increase in cognate Ag sensitivity while preserving fine specificity.
We compare here the development and pathogenicity of T cells expressing these TCR. Either the high or low affinity MOG-specific T cells were well selected and predominantly naive in the absence of immunization. However, the high affinity TCR markedly diminished disease susceptibility. This resulted from a diminished pathogenicity of autoreactive CD4+ T cells. More prominently, the high affinity TCR led to the development of co-receptor independent, CD4−CD8− (double negative, DN) and CD8+ T cells that showed low intrinsic pathogenicity, outcompete CD4+ T cells, and inhibited disease after transfer to wild type (wt) mice. Therefore, high affinity autoAg specific T cells can suppress autoimmune responses through both altered lineage differentiation and functional maturation. Implicitly, the pathologic potential of an autoAg will depend on the affinity distribution of specific TCR; higher affinities clones may be protective.
Materials and Methods
Animals
C57BL/6 (B6), B6.129S7-Rag1tm1Mom/J (Rag1−/−), and B6.SJL-Ptprca Pepcb/BoyJ (CD45.1) mice were obtained from The Jackson Laboratory (Bar Harbor, ME). Mice were bred and maintained under specific pathogen-free conditions, including detectable strains of helicobacter. Experiments were performed under a protocol approved by and in accordance with Institutional Animal Care and Use Committee guidelines.
Peptides, antibodies, and flow cytometry
MOG35–55 peptide (MEVGWYRSPFSRVVHLYRNGK) was synthesized and HPLC purified by the St. Jude Hartwell Center for Biotechnology. Monoclonal antibodies specific for CD4 (clone H129.19), CD8 (clone 53-6.7), CD25 (clone 7D4), CD69 (cloneH1.2F3), CD45RB (clone 16A), CD44 (clone IM7) and TCR Vβ8.1, 8.2 (clone MR5-2) were from BD Biosciences (Franklin Lakes, NJ). CD62L (MEL-14) and the Foxp3 staining kit was from eBioscience (San Diego, CA). Flow cytometry was performed on a FACSCalibur (BD Biosciences), and flow cytometric sorting was performed on a MoFlo high-speed cell sorter (DakoCytomation, Fort Collins, CO).
TCR Constructs and Generation of retrogenic and chimeric mice
1MOG9 and G107S TCR α and β chains were isolated by PCR and cloned into the MSCV-I-GFP murine stem cell virus-based retroviral vector and retrogenic mice generated as described (19,20). Briefly, bone marrow cells were harvested from the femurs of Rag1−/− mice 48 h after the administration of 0.15 mg of 5-fluorouracil/g body weight. The pooled cells were cultured in complete Click’s medium (Invitrogen, Carlsbad, CA) containing 20% FCS, IL-3 (20 ng ml−1), IL-6 (50 ng ml−1), and SCF (50 ng ml−1) for 48 h at 37°C/5%CO2. The cells were then cocultured for an additional 48 h with1200-rad irradiated retroviral producer cells. The hematopoietic progenitor cells (HPC) were harvested, washed with PBS, transduction confirmed by flow cytometry for GFP, and injected i.v. into sublethally irradiated (450 rad) Rag1−/− mice at a ratio of two recipient mice per bone marrow donor. For the generation of chimeric mice, transduced HPC were admixed with B6 BM cells and transferred i.v. into 900-rad irradiated B6 recipients. Mice were assessed at the indicated times post-transplant.
EAE induction and clinical evaluation
EAE was induced by s.c. immunization with 100 μg of MOG35–55 peptide emulsified in complete Freunds adjuvant containing 0.4 mg (4mg/ml) Mycobacterium tuberculosis H37RA (Difco, Franklin Lakes, NJ). Two hundred ng of pertussis toxin (List Biological Laboratories, Campbell, CA) was administered i.v. on d 0 and 2. In some experiments, CD4+, CD8+, and DN TCRB+ cells were flow cytometrically isolated and transferred into recipient mice 1 day prior to immunization. Clinical scoring was as follows: 1, limp tail; 2, hind limb paresis or partial paralysis; 3, total hind limb paralysis; 4, hind limb paralysis and body/front limb paresis/paralysis; 5, moribund.
T cell proliferation
CD4+, CD8+ and DN TCRB+ T cells were isolated by flow cytometry or antibody-based magnetic bead selection (MACS; Miltenyi Biotec, Auburn, CA) per manufacturer’s instructions. These were either unimmunized or 7 d post-immunization with MOG35–55 peptide. Cells were cultured at 5×104 per well in 96-well plates with 3×105 irradiated B6 APCs and the indicated stimulus, pulsed with 1 μCi of [3H]thymidine after 72 h of culture, and then harvested for scintillation counting. Samples were analyzed in triplicate.
Cytokine analysis
CD4+, CD8+ and DN TCRB+ T cells were isolated and cultured as described above. Culture supernatants were collected at 48 h and analyzed for IL-2, IL-4, IL-10, IL-17 and IFNG using the Milliplex Map kit assay (Millipore, Billerica, MA).
Cytotoxicity assay and quantitative flow cytometry
Flow cytometrically purified CD4+, CD8+ and DN TCRB+ cells from CD45.1−CD45.2+ G107S mice were stimulated for 4 d with MOG35–55, then incubated at a 1:1 ratio with freshly purified congenic CD45.1+CD45.2− B cells or bone-marrow derived dendritic cell (DC) targets for 6 or 18 h with or without 100 μg/ml MOG35–55. Bone marrow (BM)-derived DCs were matured with GM-CSF and IL-4 (R&D Systems, Minneapolis, MN) as described (21). Samples were stained for the indicated target B-cell or DC population and then analyzed by quantitative flow cytometry as described (22). Viable cell counts are plotted for individual samples assayed in triplicate.
Results
Retrogenic mouse production and characterization
The parental MOG35–55-specific, IAb-restricted 1MOG9 and G017S TCR are identical except for the added hydroxymethyl group in the TRBV 107 side chain in the G107S receptor. This G/S variant residue is at the immediate junction of the TRBV and TRBJ, does not directly engage peptide-MHC, and is predicted to fill a buried gap in the 1MOG9 TRBV. The G107S TCR was previously observed in transduced hybridoma cells to possess an ~3log10 increase in Ag sensitivity (20).
The TCRA and B chains of the 1MOG9 and G107S TCR were subcloned into the MSCV-I-GFP retroviral vector separated by a viral 2A sequence to facilitate their stoichiometric expression. Retrovirus was transduced into Rag1−/− hematopoietic progenitor cells (HPC), which were transplanted into Rag1−/− recipients, thereby ensuring that only the retrogenic TCR was expressed.
G107S lineage infidelity
Skewed thymic development of CD4+ single positive T cells was seen in mice receiving HPC transduced either with the 1MOG9 or G107S TCR (Fig. 1a). Both 1MOG9 and G107S retrogenic mice also showed peripheral engraftment with the self-specific TCR (Fig. 1b). In each case, the CD4+ T cells were predominantly naïve, bearing a CD44lo, CD45Rbhi, CD62Lhi phenotype (Fig. 1c). Little expression of the activation and regulatory marker CD25 was apparent on either cell type, and despite the enhanced affinity for a self Agin the G107S mice, few Foxp3+ regulatory T cells were seen. In contrast with the virtually exclusively CD4+ T cell engraftment in mice expressing the wt 1MOG9 TCR, substantial numbers of peripheral CD8+ T cells were observed in G107S mice (Fig. 1b). These CD8+ cells, as the CD4+, were predominantly naïve (Fig. 1d). A smaller population of DN T cells was also seen in the G107S mice, whereas few were present in the 1MOG9 animals. These also displayed a largely naïve phenotype (Fig. 1e). Therefore the high affinity G107S TCR promotes lineage infidelity and T cell subset heterogeneity.
Figure 1. Immunophenotype of G107S and 1MOG9 retrogenic T cells.

Thymocytes, LN cells, and splenocytes were isolated from retrogenic mice ~8 wk following stem cell transfer or B6 controls. (A) CD4 and CD8 labeling of thymocytes is shown. (B) CD4 and CD8 expression on TCRB gated LN cells or splenocytes. (C) CD44, CD45Rb, and CD62L labeling was used to indicate the memory or naïve status of CD4+ TCRB+-gated lymphocytes. CD25 and Foxp3 were used as regulatory T cell markers.(D) Labeling of CD8+ TCRB+ T cells to evaluate the distribution of naïve and memory populations as in (C). (E) Naïve and memory cells among G107S DN TCRB+ T cells. Data is from representative animals.
Increased affinity for self is associated with the deletion of autoAg specific T cells (23). However, this was not apparent with the G107S TCR. To the contrary, 7–12 wk after stem cell transfer, the G107S mice showed significantly more splenic CD4+TCR+ T cells than matched wt 1MOG9 TCR retrogenic mice (p<0.0001; Fig. 2). Therefore, the G107S substitution does not adversely affect the development and engraftment of self-reactive T cells. Rather, it supports increased engraftment as well as production of CD8+ and smaller numbers of DN T cells. Notably, in a prior study of retrogenic mice expressing different MOG-specific TCR (19), we were unable to associate affinity with engraftment level, and the recognition parameters that are promoting this increased engraftment remain to be determined.
Figure 2. T cell subset engraftment in 1MOG9 and G107S mice.

Percent (A) and absolute numbers (B) of CD4+ (1MOG9 and G107S), CD8+ or DN (G107S only) T cell subsets from 7–12 wk 1MOG9 and G107S retrogenic mice are plotted. Circles indicate individual mice and lines population means. Substantial numbers of CD8+ and DN TCRB+ cells were not seen in the 1MOG9 mice. T-test comparison of CD4+ TCRB+ populations shows significantly increased relative (p=1×10−4) and absolute (p=2×10−4) engraftment in the G107S mice.
Heightened responsiveness of primary G107S T cells
Developmental tuning can dampen the responsiveness of high affinity T cells (24,25). To determine if the increased Ag sensitivity previously observed in G107S-TCR transduced cell lines was retained in primary T lymphocytes, we compared the proliferative response of isolated CD4+ T cells from 1MOG9 and G107S retrogenic mice. Similar to transduced cell lines, a ~3log10 increase in MOG sensitivity was seen in primary CD4+ T cells expressing the G107STCR (Fig. 3a). Isolated CD8+ and DNT cells present in the G107S mice also proliferated strongly in response to cognate Ag with response profiles similar to that of CD4+ T cells (Fig. 4a), verifying our prior observation in hybridomas that the G107S TCR is co-receptor independent. Therefore, a G107S substitution in a TRBV13-2 TCR is fully compatible with the development of functional T lymphocytes bearing markedly enhanced sensitivity for cognate Ag.
Figure 3. Proliferative response and cytokine profiles of 1MOG9 and G107S CD4+ T cells.

Splenic CD4+ T cells were isolated by magnetic bead selection from ~8 wk 1MOG9, G107S, or B6 splenocytes, and stimulated with the indicated concentration of Ag. (A) Proliferative response was determined by [3H]thymidine incorporation. (B) Stimulation-induced production of the indicated cytokines was measured using a bead multiplex assay. Mean ± 1 s.d. is plotted. Representative of 3 experiments.
Figure 4. Comparison of proliferative and cytokine profiles of CD4, CD8, and DN G107S T cells.

Analyses were performed as in figure 3, except CD4+, CD8+, and DN TCRB+ T cells were isolated by flow cytometric sorting from 16 wk G107S mice. Use of older mice proved necessary to acquire adequate numbers of DN cells for analysis. (A) Proliferative response of T cell subsets or control irradiated APCs. (B) Cytokine production by T cell populations. Note that the increased production of IFNG by cells from the older mice compared with younger G107S mice (plotted in Fig. 3) was consistently observed. Mean ± 1 s.d. is plotted. Representative of 2 experiments.
Distinct cytokine profiles of 1MOG9 and G107S T cells
To assess the differentiation potentials of 1MOG9 and G107S T cells, CD4+ T cells were bead isolated from 8 wk retrogenic mice and equal numbers stimulated in vitro with MOG35–55. IL-2, IL-4, IL-10, IFNG, and IL-17 were measured as representative Th0, Th2/Tr1, Th1 and Th17 cytokines. Either 1MOG9 or G107S CD4+ T cells showed good IL-2 production, though levels were greater with the G107S T cells (Fig. 3a). Both cell types also produced IFNG, associated with differentiation into Th1 cells that are pathologic in EAE (26,27), though here more was produced by the lower affinity 1MOG9T cells. G107S T cells produced modest amounts of IL-17, also associated with EAE pathogenicity (28), whereas none was seen with the 1MOG9 T cells. IL-4 is associated with disease protection (29). Little or no IL-4 was produced by the 1MOG9 T cells, while substantial quantities were produced by the G107S cells. The G107S T cells also exclusively demonstrated significant production of IL-10, a cytokine with pronounced regulatory activity in EAE (30–34). Therefore, CD4+ T cells from 1MOG9 mice showed a restricted cytokine production pattern characterized by IL-2 and IFN-γ production and little IL-4 and IL-10 synthesis. G107S T cells show a more degenerate and pronounced cytokine profile that included cytokines characteristic of Th1, Th2, and Th17 cells. IL10 and IL4 production, protective in EAE, was limited to the G107S cells.
CD8+ and DN T cells were similarly isolated, though from older 16 wk retrogenic mice, necessary to obtain adequate DN numbers by cell sorting. These demonstrated less IL-2 and more IFNG than similarly isolated CD4+ G107S T cells (Fig. 4b). Neither DN nor CD8+ T cells produced substantial quantities of IL-10. However, production of other cytokines diverged between the cell types. DN cells produced more IL-17, while CD8+ T cells produced little. CD8+ T cells also produced little IL-4 when compared with DN and CD4+ T cells. Therefore G107S T cell subsets possess distinct functional profiles when stimulated with Ag, with variability in the production of both inflammatory and protective cytokines.
Cytolytic activity of DN and CD8+ T cells
To determine if the T cells were able to kill MOG-expressing APCs, primary B cells or bone marrow-derived dendritic cells were pulsed with MOG35–55 and co-cultured for either 6 or 18 h with G107S T cells at a 1:1 effector:target ratio (Fig. 5). The DN and CD8+ T cells lysed the B cell populations at either time point. G107S (Fig. 5a) or 1MOG9 (not shown) CD4+ T cells were unable to kill these cells. At the 1:1 effector:target ratio, DCs proved insusceptible to CTL killing (Fig. 5b); such DC resistance to lysis has been previously reported (35). However, at a 4:1 ratio modest DC killing by the DN and CD8+ populations was evident (data not shown). Therefore, the CD8+ and DN but not CD4+ effector cells are capable of killing cells presenting Ag, though the extent of this is dependent on lineage sensitivity to lysis.
Figure 5. Ag specific cytolysis by G107S CD8+ and DN T cells.

Purified CD4+, CD8+ and DN G107S T cells were stimulated for 4 days with MOG35–55 Ag, isolated, and then co-cultured with purified B6 B lymphocytes (A) or BM-derived DCs (B) at a 1:1 E:T ratio in triplicate. Cultures were either pulsed with 100 μg/ml MOG35–55 or left without Ag. At 6 or 18 h numbers of residual viable target cells was quantitatively assessed by flow cytometry. The ratio of residual cells in Ag-pulsed/non-pulsed cultures is plotted. Mean + 1 s.d. is shown. Representative of 2 experiments.
Diminished EAE severity in G107S mice
We would predict that the altered differentiation profile of the higher affinity G107S T cells would influence EAE susceptibility. To test this, 1MOG9 and G107S mice were immunized with MOG35–55. The 1MOG9 mice developed fulminant disease, which proved nearly uniformly fatal by 2 weeks after immunization (Fig. 6a and Table I). In contrast, despite the increased T cell affinity for Ag, the G107S mice manifested milder disease which, though not delayed in onset relative to the 1MOG9 mice, failed to clinically progress. Mortality was absent among the G107S mice. Therefore a high affinity TCR in G107S retrogenic mice, despite promoting significantly enhanced T cell engraftment, led to markedly diminished disease severity.
Figure 6. Diminished EAE severity and altered subset expansion in G107S retrogenic mice.

(A) EAE was induced by immunization with MOG35–55. Clinical disease score was monitored longitudinally. Mean ± 1 s.e.m is plotted. Representative of 2 experiments. Additional data parameters are provided in Table I. (B-E) T lymphocytes were isolated from the spleen and CNS of mice 6 d after induction of EAE or spleen of control mice in which disease was not induced. At this time, the immunized 1MOG9 mice had disease scores ranging from 3–4 and the G107S mice all scores of 1. (B) Sample plots showing CD4 and CD8 labeling profiles of splenocytes (immunized and unimmunized) or CNS (immunized only) TCRB+ T cells. Absolute numbers of splenic T cell populations in 1MOG9 (C) or G107S (D) mice are plotted. Circles indicate individual animals and lines represent means. (E) Absolute numbers of isolated TCRB+ CNS T cell subsets are similarly plotted.
Table I. Actively induced EAE in 1MOG9 or G107S mice.
In experiments1 and 2 retrogenic mice were directly immunized. For experiments 3 and 4, 5×104 of the indicated flow cytometrically purified T cells were transferred into Rag1−/− recipients 1 d prior to immunization. For experiment 5, 105 of the indicated bead purified T cells were transferred 1 d prior to immunization. Onset time is from MOG immunization. AUC, area under the curve analysis using trapezoidal estimation. Longitudinal course of experiments 1 and 3 are plotted in figures 6 and 9 respectively.
| Expt | Follow-up (d) | Group | n | Incidence | Onset (d) | Max. Score | AUC | Mortality |
|---|---|---|---|---|---|---|---|---|
| 1 | 49 | 1MOG9 | 5 | 5/5 | 6.8±0.4 | 5.0±0 | 205.4±2.9 | 5/5 |
| G107S | 5 | 5/5 | 6.8±0.4 | 2.0±0 | 59.9±18.9 | 0/5 | ||
| 2 | 38 | 1MOG9 | 7 | 7/7 | 7.6±1.1 | 5.0±0 | 117.8±38.8 | 7/7 |
| G107S | 6 | 5/6 | 10.2±2.2 | 1.6±1.2 | 36.6±24.5 | 0/6 | ||
| 3 | 42 | 1MOG9, CD4 | 4 | 4/4 | 5.5±0.6 | 5.0±0 | 174.7±2.1 | 4/4 |
| G107S, CD4 | 4 | 4/4 | 6.2±0.9 | 4.2±0.9 | 131.6±31.3 | 2/4 | ||
| G107S, CD8 | 4 | 4/4 | 12.0±0.6 | 2.2±2.0 | 21.3±22.9 | 1/4 | ||
| G107S, DN | 5 | 1/5 | 12.0 | 0.2±0.4 | 1.0±2.1 | 0/5 | ||
| 4 | 27 | 1MOG9, CD4 | 5 | 5/5 | 8.2±0.4 | 5.0±0 | 87.7±2.8 | 5/5 |
| G107S, CD4 | 5 | 5/5 | 8.4±0.9 | 3.8±0.7 | 39.3±8.7 | 1/5 | ||
| G107S, CD8 | 4 | 4/4 | 16.0±6.0 | 2.0±0.7 | 19.7±12.0 | 0/4 | ||
| G107S, DN | 5 | 3/5 | 17.6±5.5 | 0.6±0.5 | 2.6±3.1 | 0/5 | ||
| 5 | 36 | 1MOG9, CD4 | 5 | 5/5 | 8.0±0.7 | 5.0±0 | 135.9±3.0 | 5/5 |
| G107S, CD4 | 5 | 5/5 | 7.2±0.4 | 3.0±1.2 | 79±14.5 | 1/5 | ||
| G107S, CD8 | 5 | 1/5 | 38.2±15.2 | 0.1±0.2 | 0.1±0.2 | 0/5 | ||
| 6 | 32 | 1MOG9, CD4 | 5 | 5/5 | 7.0±0.0 | 5.0±0 | 118.3±2.0 | 5/5 |
| G107S, CD4 | 5 | 5/5 | 8.2±0.4 | 3.0±1.4 | 58.8±29.1 | 2/5 | ||
| G107S, CD8 | 5 | 4/5 | 14.2±11.6 | 0.8±0.4 | 20.5±11.4 | 0/5 |
Distinct T lineage responses in G107S and 1MOG9 mice
To better characterize the responding cells in the different mice, we examined splenic and CNS cell numbers prior to and after immunization. As described above, CD4+ T cells were the predominant lymphoid constituent in pre-immune 1MOG9 mice, with only small numbers of CD8+ or DNT cells detected. After EAE induction, a dramatic expansion of CD4+ T cells was seen. These remained the dominant cell type in the spleen, and little expansion of CD8+ or DN T cells was apparent (Fig. 6b, c). In G107S mice, a majority of T cells were CD4+ in the absence of immunization, though a sizable population of CD8+ T cells and smaller numbers of DN cells were also present. After immunization all cell populations expanded, though not equally (Fig. 6b, d). The CD4+ population expanded less than in the 1MOG9 mice. CD8+ and DN T cells expanded to a much greater degree than CD4+ T cells, reversing the CD4+/CD8+ and CD4+/DN T cell ratios. Analysis of the CNS lymphoid infiltrate showed a predominance of CD4+ T cells in 1MOG9 mice, but CD8+ and DN significantly outnumbered CD4+ cells in G107S mice (Fig. 6e). Therefore, the autoimmune response in 1MOG9 and G107S mice is characterized by the differential expansion and CNS infiltration byCD4+, CD8+, and DN T cell subsets.
Functional potential of immune T cells
To functionally assess the responding cells, we analyzed the proliferative potential of purified T cell populations 7 days after immunization. In contrast to the distinct responses of pre-immune T cells, comparison of the T cell populations from mice with EAE demonstrated nearly equivalent in vitro proliferation of the 1MOG9 CD4+ T cells and G107S T cell subsets (Fig. 7a). This differed from results with pre-immune populations (Fig. 3). Indeed, direct comparison of the proliferative responses of purified CD4+ T cells isolated from immunized and unimmunized mice showed a corresponding enhanced responsiveness to Ag among immunized 1MOG9 but not G107S T cells (data not shown) compared with unimmunized controls. Therefore, prior activation differentially influences subsequent Ag sensitivity by G107S and 1MOG9 T cells, with only the lower affinity cells developing increased reactivity.
Figure 7. Functional profiles of 1MOG9 and G107S T cells in MOG-immunized mice.

T cell subsets were isolated by flow cytometry from 1MOG9 (CD4+) or G107S (CD4+, CD8+, DN) mice 7 d after MOG immunization. (A) Proliferation to the indicated concentration of MOG35–55 was measured by [3H] thymidine incorporation 72 h after stimulation. (B–F) Production of the indicated cytokines was measured by multiplex assay 48 h after stimulation. Responses of purified cells cultured without or with 100 μg/ml MOG35–55 is shown. Mean ± 1 s.d. is plotted. Representative of 2 experiments.
Cytokine profiles of the 1MOG9 and G107S T cells were further assessed at d 7, which though early after disease induction only shortly preceded the demise of most of the 1MOG9 mice(Fig. 7b–f). G107S CD4+, CD8+, and DN T cells showed significantly decreased IL-2, IFNG, and IL-17 compared with 1MOG9 cells. This indicates an overall diminished production of Th1 and Th17 cytokines relevant to EAE. Only modest IL-4 and IL-10 was produced by immune cells from either mouse type.
Therefore, an increased TCR affinity in G107S mice is associated with both attenuated disease course and diminished effector cytokine production. Notably, similar to the pre-immune mice (Fig. 1) few Foxp3+ Treg were detectable in either G107S or 1MOG9 mice after induction of EAE (data not shown).
Pathogenicity and protection by isolated G107S cellular subsets
Our results indicated that multiple T cell subsets responded in G107S EAE; the dominant T cell response was CD4−, and G107S T cells had diminished effector cytokine production. To examine the pathogenicity of isolated high affinity subsets, CD4+, CD8+ or DN T cells were purified from G107S mice and CD4+ T cells from 1MOG9 mice. These were transferred into Rag1−/− recipients and disease induced. CD4+ T cells from G107S mice manifested diminished disease relative to lower affinity 1MOG9 CD4+ T cells, indicating that increased TCR affinity decreases T cell pathogenicity in this population (Fig. 8 and Table I). In contrast to the G107S CD4+ T cells, mild or no disease was seen in recipients of CD8+ or DN G107S T cells. Therefore, pathogenicity is a function of both TCR affinity and cell lineage. DN and CD8+ T cells which show the greatest expansion in immunized G107S mice, have limited capacity to independently promote disease.
Figure 8. Pathologic potential of isolated T cell subsets.

Flow cytometrically isolated TCRB+ subsets from 1MOG9 (CD4+) or G107S (CD4+, CD8+, DN) were transferred i.v. into Rag1−/− recipients (5×104/mouse) and EAE was induced 1 d later. Clinical score was followed longitudinally. Mean ± 1 s.e.m is plotted. Representative of 2 experiments with flow cytometrically sorted T cells, and 2 with bead selected cells. Additional data is provided in Table I.
To determine whether G107S T cells could also suppress disease development and progression in a competitive environment, we analyzed their impact in B6 mice. Small numbers (1×105) of flow cytometrically purified CD4+, CD8+, or DN T cells were transferred into B6 recipients and 1 d later EAE was induced (Fig. 9a). Transfer of G107S CD4+ T cells diminished disease severity when compared with control treated mice. However, CD8+ T cells blocked G107S disease development and DN T cells markedly delayed time to disease onset as well as disease severity, indicating that these subsets are protective in wt mice.
Figure 9. Regulatory activity of G107S CD8+ and DN T cells in B6 mice and mixed chimeras.
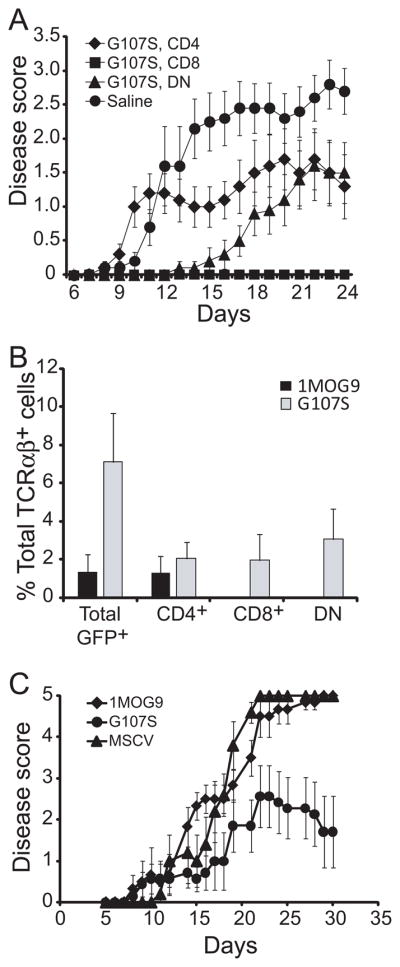
(A) 105 of the indicated flow cytometrically purified T cell subset or saline was transferred i.v. into B6 mice 1 d prior to EAE induction and disease course monitored. Data is pooled from 2 similar experiments. N=10 per group. Mean ± 1 s.e.m is plotted. (B) Engraftment of G107S or 1MOG9 T cell subsets in mixed chimeras. HPC transduced with the indicated TCR were admixed with wt B6 BM cells and transplanted into irradiated B6 recipients. The retrogenic T cells were identified in blood cells by co-expressed GFP. Mean + 1 s.d. is plotted. Representative of 2 experiments. (C) EAE was induced in mice in (B) and disease clinically monitored. N=7, G107S; N=6 other groups. Mean ± 1 s.e.m is plotted.
To determine if the MOG-specific CD4− T cells would develop not only in Rag−/− mice but in the context of a wt thymus, we diluted G107S, 1MOG9, or retroviral vector-transduced Rag−/− HPC with an excess of untransduced B6 bone marrow cells and transplanted these into B6 recipients. Only small numbers of retrogenic T cells, identifiable through their co-expression of GFP, were identified. They averaged fewer than 2% of TCRAB+ T cells in mice receiving 1MOG9-transduced progenitors, and were comprised virtually exclusively of CD4+ T cells (Fig. 9b). Approximately 7% of TCRAB+ cells in the G107S chimeras expressed the retrogenic TCR. Quantities of CD4+ retrogenic T cells were not significantly different from those in the 1MOG9 mice. However, CD8+ and DN G107S T cells were also observed, making up for the balance of cells. Therefore the high affinity MOG-specific TCR promotes the cross-differentiation of T cells into co-receptor independent CD8 and DN T cell subsets when developing as a minority population in wt mice.
To determine if the G107S T cells would protect against EAE development, disease was induced in these chimeric mice (Fig. 9c). Whereas mice receiving control MSCV vector- or 1MOG9 TCR transduced HPCs demonstrated similarly high levels of disease, mice receiving G107S-transduced cells demonstrated a milder disease course. Therefore, as with T cell transfer into wild type mice, G107S T cells developing in mixed stem cell chimeras diminish EAE severity.
Discussion
The presence of self-specific T cells is inadequate for autoimmunity. These cells must also be stimulated in a manner that triggers their maturation into destructive effector forms, promotes their expansion, and sustains their pathogenicity over time. To achieve this balance experimentally, specific induction regimens are often needed. In most experimental systems, relatively few epitopes have been identified against which T cell responses can provoke autoimmunity. For example, in B6 mice, MOG35–55 and the crossreactive NF-M18–30 epitope, are the only Ags identified able to induce EAE (36,37). The available T cell repertoire will determine the quality of the response to Ag, and hence susceptibility to autoimmunity. Thymic and peripheral T cell deletion may lead to specificity holes, and thereby tolerance. As described here, increased affinity can also lead to diminished disease susceptibility by altering differentiation potential.
TCR affinity plays a crucial role in conjunction with environmental signals in the functional maturation of T cells. Th subset maturation is avidity dependent. Thus, the response to pigeon cytochrome C is dominated by high affinity cells that following in vitro priming generates a Th1 biased response. Purging the high affinity cells leads to lower affinity repertoire that develop a Th2 bias (38). In a consistent manner low Ag dose or weak APL signaling leads to a Th2 responses in TCR Tg cells, whereas high dose promotes Th1 maturation (10,39). On the other hand, in matured T cell lines, it has also been observed that Th2 cells require greater signal strength on rechallenge than Th1 cells for cytokine production (40).
Affinity likewise is essential in determining cell survival and proliferation. In the Listeria-specific response, early stimulation activates both low and higher affinity cells. The lower affinity cells, however, exit the LNs earlier and have a more abbreviated expansion phase, potentially explaining the affinity maturation that is observed (41). In other conventional responses, affinity maturation is also present, and higher affinity cells dominate the secondary response. Despite these findings, in autoimmune systems strong agonists may be protective. Thus a myelin APL “superagonist” did not lead to EAE, but promoted the desensitization and AICD of autoAg specific T cells in a fas dependent manner(11,12).
Our results are consistent with a model in which T cell immunopathologic potential is bracketed by both low and high affinity thresholds, and describe new mechanisms for this. Importantly, the G107S TCR differs only in the presence of a buried hydroxymethyl group in the TRBV (20). The S107 residue supplying this moiety is present in the majority of expressed TCR; 20 of 23 mouse and 49 of 54 human TRBV possess a S whereas only 1 and 2 respectively incorporate a G, and therefore the substitution represents a natural variation at this site (20). The G leaves a gap at the base of the CDR3β which is filled by the S side chain. This minor structural perturbation influences affinity for MOG35–55 without substantially altering fine specificity. Yet it has several functionally significant effects. Most prominently, whereas the wt 1MOG9 TCR drives T cells into the CD4 T cell lineage, the G107S TCR leads to lineage infidelity, with CD4+, CD8+, and DN T cells identifiable. These CD4− populations also develop in mixed HPC chimeras in wt mice, indicating that they reflect the natural differentiation potential of this TCR.
Whether increased G107S TCR affinity for a self ligand is itself responsible for the lineage infidelity is uncertain from this study. In one other model, a high affinity self-specific TCR also guided T cell development into CD4−CD8− T cells (42), suggesting a role. Assessments of additional TCR series of varying affinity may help determine the recognition parameters controlling the fidelity of lineage assignments. Increased affinity is, however, essential for co-receptor independent antigen recognition. Studies of MHC class I-restricted TCR identified an affinity threshold in the μM range above which co-receptor is no longer required (43). The 1MOG9 TCR is class II MHC restricted, and threshold affinities for co-receptor independent class II MHC recognition are, to our knowledge, unavailable. Nevertheless, we previously demonstrated that CD4− hybridomas expressing the high affinity G107S TCR are co-receptor independent and remain class II MHC restricted, class II−/− APCs failing to stimulate them (19). This finding was replicated with the primary retrogenic T cells studied here (data not shown).
Isolated DN and CD8+ G107S T cells were only weakly pathogenic. Moreover, these populations outcompete CD4+ T cells during disease development. They expanded to form the majority of cells infiltrating the CNS, potentially explaining the dramatically reduced disease in G107S mice. After transfer into wt mice, very small numbers of the cells (105, Fig. 9 and as few as 4×104 (not shown)) were protective. This indicates a broader capacity to modulate autoimmune disease. Implicitly, in contrast to the competitive maturation to high affinity responses typical after immunization or infection, the low or moderate affinities often observed in autoimmunity may be necessary for the generation of pathologic responses.
The mechanism of protection of these CD4− cells in this model remains uncertain, though some clues are present. DN T cells have been observed more broadly to possess regulatory properties, as evidenced in other models of alloimmune and autoimmune diseases (44,45). It would seem likely here that the low pathologic potential of the DN cells coupled with their ability to outcompete more pathologic CD4+ T cells provides for their immunomodulatory activity. The source of these cells in natural circumstances has been debated. Small numbers of DN αβ T cells normally circulate, typically <2%, though these are increased in certain conditions such as ALPS (46,47). Defects in Fas signaling there suggest that alterations in this death signaling pathway may allow the accumulation of DN cells. Indeed, DN T cells survive chronic stimulation better than their coreceptor-positive peers (48). In some situations it is hypothesized that DN cells are generated through coreceptor downregulation with stimulation, which may occur genetically or epigentically (49–52). However other sources are possible (53), and the largely naïve state of the DN cells in unimmunized G107S mice and their distinct cytokine profile when compared with either CD4+ or CD8+ cells, would indicate that they are a primary population. Indeed, diversion of a large proportion of autoreactive T cells to DN cells was observed when a high affinity PLP-specific TCR was transgenically expressed on T cells in a disease resistant genetic background. Whereas DN TCRαβ T cells form a small portion of most immune responses, this evidence supports their formation from high affinity, self-specific cells through a common differentiation pathway (42). Further evaluation of these DN T cells and their role in EAE and other autoimmune diseases in umanipulated mice is warranted.
CD8+ T cells recognizing epitopes internal to MOG35–55 can modify MOG-EAE, and both pathologic and protective properties have been attributed to them (54–56). EAE in β2m−/− mice shows increased disease severity, suggesting that, overall, the class I restricted response is protective (57). In our analyses, the G107S CD8+ T cells recognized class II MHC restricted Ag. Such lineage crossover by co-receptor independent cells has been previously observed in immune responses (58), though the extent to which T cells are capable of crossing MHC restriction is unclear. Nevertheless, co-receptor independent signaling is adequate to activate T cells with high affinities (43). Further, the sheer magnitude of the degeneracy inherent in TCR recognition, where it is anticipated that a single TCR may recognize > 106 unique Ags, would suggest that some high affinity, co-receptor independent responses will exist and these may cross MHC class barriers (59).
To our knowledge, it has not been previously shown that high affinity cells specific for a class II restricted Ag may actually develop in situ into CD8+ T cells. To test the MHC requirements for this development, we generated G107S retrogenic mice using transduced β2m−/−Rag1−/− HPCs transferred into β2m−/−Rag1−/− recipients. Although the relative numbers of CD8+ T cells was diminished compared with Rag1−/− retrogenic mice, a significant population was nevertheless present, and DN cells were increased (Supp. Fig. S1). Therefore, both some CD8+ and DN G107S T cells can develop in a class I MHC-independent manner.
It is of interest, though not surprising, that the CD8+ and DN G107S T cells can kill Ag pulsed APCs, particularly B cells. Cytotoxicity is a characteristic of these populations, and it has been hypothesized that this can restrict autoreactive responses by reducing APC availability (54). The role of APC killing in the diminished pathogenicity and protection here will have to be established. However, DCs are a critical APC type in the EAE model system (60), and these possess mechanisms to resist lysis, including the expression of granzyme-inhibitory serpins (35). Indeed, at E:T ratios capable of easily killing B cells, DCs proved impervious to lytic attack.
The functional profile of the G107S and 1MOG9 T cells, defined here by their cytokine production patterns, differed significantly between the cells, and further explains the reduced pathogenicity of the higher affinity cells. Primary CD4+ G107S T cells produced increased IL-10 and IL-4, which are regulatory in EAE, compared with wt 1MOG9 cells. In mice with active disease, substantive IL-10 and IL-4 responses were not detected. Nevertheless, the G107S T cells produced less IFNG and IL-17, indicating a disruption in Th1/Th17 mediated immunopathogenesis. Further, isolated CD4+ G107S T cells were less potent than CD4+ 1MOG9 T cells in transfer models. Therefore, consistent with data on the use of “superagonistic” APL to stimulate autoreactive T cells, and the observed diminished pathogenicity of high affinity TCR transgenics, G107S CD4+ T cells possess an intrinsically reduced pathogenicity compared with paired 1MOG9 T cells. Further, lower affinity 1MOG9 but not G107S T cells demonstrated enhanced in vitro response to Ag after immunization, suggesting that prior stimulation differentially upregulates responsiveness and potentially supports expansion of the lower affinity cells upon Ag restimulation.
In summary, we demonstrate highly distinct T cell immunopathologic potential in two cohorts of mice expressing retrogenic TCR with minimal structural differences though more dramatically altered affinity for MOG35–55. Different sources are responsible, including cell intrinsic effects on cytokine patterns by pathologic CD4+ T cells as well as altered T cell differentiation and the induction of protective DN and CD8+ T cell responses. The presence of high affinity self-Ag specific T cells may therefore provide a barrier, protecting the host against pathologic autoimmune responses. Further analyses of the effect of TCR affinity on autoAg-specific T cell differentiation profiles is warranted to better define how T cell repertoire properties influence autoimmune susceptibility.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health Grant R01 AI056153 (to TLG) and by the American Lebanese Syrian Associated Charities (ALSAC)/St. Jude Children’s Research Hospital (to all authors).
We thank Richard Cross, Greig Lennon, Stephanie Morgan and the Immunology Core Flow Cytometry facility for assistance with flow cytometric sorting.
Reference List
- 1.Kedl RM, Kappler JW, Marrack P. Epitope dominance, competition and T cell affinity maturation. Curr Opin Immunol. 2003;15:120–127. doi: 10.1016/s0952-7915(02)00009-2. [DOI] [PubMed] [Google Scholar]
- 2.Malherbe L, Hausl C, Teyton L, McHeyzer-Williams M. Clonal selection of helper T cells is determined by an affinity threshold with no further skewing of TCR binding properties. Immunity. 2004;21:669–679. doi: 10.1016/j.immuni.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 3.Rees W, Bender J, Teague TK, Kedl RM, Crawford F, Marrack P, Kappler J. An inverse relationship between T cell receptor affinity and antigen dose during CD4(+) T cell responses in vivo and in vitro. Proc Natl Acad Sci U S A. 1999;96:9781–9786. doi: 10.1073/pnas.96.17.9781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Price DA, Brenchley JM, Ruff LE, Betts MR, Hill BJ, Roederer M, Koup RA, Migueles SA, Gostick E, Wooldridge L, Sewell AK, Connors M, Douek DC. Avidity for antigen shapes clonal dominance in CD8+ T cell populations specific for persistent DNA viruses. J Exp Med. 2005;202:1349–1361. doi: 10.1084/jem.20051357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nicholson LB, V, Kuchroo K. T cell recognition of self and altered self antigens. Crit Rev Immunol. 1997;17:449–462. [PubMed] [Google Scholar]
- 6.Kuchroo VK, Greer JM, Kaul D, Ishioka G, Franco A, Sette A, Sobel RA, Lees MB. A single TCR antagonist peptide inhibits experimental allergic encephalomyelitis mediated by a diverse T cell repertoire. J Immunol. 1994;153:3326–3336. [PubMed] [Google Scholar]
- 7.Smilek D, Wraith D, Hodgkinson S, Dwivedy S, Steinman L, McDevitt H. A single amino acid change in a a myelin basic protein peptide confers the capacity to prevent rather than induce experimental autoimmune encephalomyelitis. Proc Natl Acad Sci. 1991;88:9633–9637. doi: 10.1073/pnas.88.21.9633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Steinman L, Conlon P. Antigen specific immunotherapy of multiple sclerosis. J Clin Immunol. 2001;21:93–98. doi: 10.1023/a:1011020225433. [DOI] [PubMed] [Google Scholar]
- 9.Bielekova B, Goodwin B, Richert N, Cortese I, Kondo T, Afshar G, Gran B, Eaton J, Antel J, Frank JA, McFarland HF, Martin R. Encephalitogenic potential of the myelin basic protein peptide (amino acids 83–99) in multiple sclerosis: results of a phase II clinical trial with an altered peptide ligand. Nat Med. 2000;6:1167–1175. doi: 10.1038/80516. [DOI] [PubMed] [Google Scholar]
- 10.Nicholson LB, Greer JM, Sobel RA, Lees MB, Kuchroo VK. An altered peptide ligand mediates immune deviation and prevents autoimmune encephalomyelitis. Immunity. 1995;3:397–405. doi: 10.1016/1074-7613(95)90169-8. [DOI] [PubMed] [Google Scholar]
- 11.Nicholson LB, Anderson AC, Kuchroo VK. Tuning T cell activation threshold and effector function with cross-reactive peptide ligands. Int Immunol. 2000;12:205–213. doi: 10.1093/intimm/12.2.205. [DOI] [PubMed] [Google Scholar]
- 12.Munder M, Bettelli E, Monney L, Slavik JM, Nicholson LB, Kuchroo VK. Reduced self-reactivity of an autoreactive T cell after activation with cross-reactive non-self-ligand. J Exp Med. 2002;196:1151–1162. doi: 10.1084/jem.20020390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anderton SM, Radu CG, Lowrey PA, Ward ES, Wraith DC. Negative selection during the peripheral immune response to antigen. J Exp Med. 2001;193:1–11. doi: 10.1084/jem.193.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Amrani A, Verdaguer J, Serra P, Tafuro S, Tan R, Santamaria P. Progression of autoimmune diabetes driven by avidity maturation of a T-cell population. Nature. 2000;406:739–742. doi: 10.1038/35021081. [DOI] [PubMed] [Google Scholar]
- 15.Waldner H, Whitters MJ, Sobel RA, Collins M, Kuchroo VK. Fulminant spontaneous autoimmunity of the central nervous system in mice transgenic for the myelin proteolipid protein-specific T cell receptor. Proc Natl Acad Sci U S A. 2000;97:3412–3417. doi: 10.1073/pnas.97.7.3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sabatino JJ, Jr, Huang J, Zhu C, Evavold BD. High prevalence of low affinity peptide-MHC II tetramer-negative effectors during polyclonal CD4+ T cell responses. J Exp Med. 2011;208:81–90. doi: 10.1084/jem.20101574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zehn D, Bevan MJ. T cells with low avidity for a tissue-restricted antigen routinely evade central and peripheral tolerance and cause autoimmunity. Immunity. 2006;25:261–270. doi: 10.1016/j.immuni.2006.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hofstetter HH, Targoni OS, Karulin AY, Forsthuber TG, Tary-Lehmann M, Lehmann PV. Does the frequency and avidity spectrum of the neuroantigen-specific T cells in the blood mirror the autoimmune process in the central nervous system of mice undergoing experimental allergic encephalomyelitis? J Immunol. 2005;174:4598–4605. doi: 10.4049/jimmunol.174.8.4598. [DOI] [PubMed] [Google Scholar]
- 19.Alli R, Nguyen P, Geiger TL. Retrogenic modeling of experimental allergic encephalomyelitis associates T cell frequency but not TCR functional affinity with pathogenicity. J Immunol. 2008;181:136–145. doi: 10.4049/jimmunol.181.1.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alli R, Zhang ZM, Nguyen P, Zheng JJ, Geiger TL. Rational design of T cell receptors with enhanced sensitivity for antigen. PLoS ONE. 2011;6:e18027. doi: 10.1371/journal.pone.0018027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hikino H, Kasono K, Kanzaki M, Kai T, Konishi F, Kawakami M. Granulocyte/macrophage colony-stimulating factor and interleukin-4-induced dendritic cells. Anticancer Res. 2004;24:1609–1615. [PubMed] [Google Scholar]
- 22.Nguyen P, Geiger TL. Induction of B-cell immune tolerance by antigen-modified cytotoxic T lymphocytes. Transplantation. 2010;89:667–676. doi: 10.1097/TP.0b013e3181ca9048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Starr TK, Jameson SC, Hogquist KA. Positive and negative selection of T cells. Annu Rev Immunol. 2003;21:139–176. doi: 10.1146/annurev.immunol.21.120601.141107. [DOI] [PubMed] [Google Scholar]
- 24.Stephen TL, Tikhonova A, Riberdy JM, Laufer TM. The activation threshold of CD4+ T cells is defined by TCR/peptide-MHC class II interactions in the thymic medulla. J Immunol. 2009;183:5554–5562. doi: 10.4049/jimmunol.0901104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van den Berg HA, Rand DA. Quantitative theories of T-cell responsiveness. Immunol Rev. 2007;216:81–92. 81–92. doi: 10.1111/j.1600-065X.2006.00491.x. [DOI] [PubMed] [Google Scholar]
- 26.O’connor RA, Prendergast CT, Sabatos CA, Lau CW, Leech MD, Wraith DC, Anderton SM. Cutting edge: Th1 cells facilitate the entry of Th17 cells to the central nervous system during experimental autoimmune encephalomyelitis. J Immunol. 2008;181:3750–3754. doi: 10.4049/jimmunol.181.6.3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stromnes IM, Cerretti LM, Liggitt D, Harris RA, Goverman JM. Differential regulation of central nervous system autoimmunity by T(H)1 and T(H)17 cells. Nat Med. 2008;14:337–342. doi: 10.1038/nm1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Segal BM. Th17 cells in autoimmune demyelinating disease. Semin Immunopathol. 2010;32:71–77. doi: 10.1007/s00281-009-0186-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Whitacre CC, Song F, Wardrop RM, III, Campbell K, McClain M, Benson J, Guan Z, Gienapp I. Regulation of autoreactive T cell function by oral tolerance to self-antigens. Ann N Y Acad Sci. 2004;1029:172–9. 172–179. doi: 10.1196/annals.1309.033. [DOI] [PubMed] [Google Scholar]
- 30.Mekala DJ, Alli RS, Geiger TL. IL-10-dependent infectious tolerance after the treatment of experimental allergic encephalomyelitis with redirected CD4+CD25+ T lymphocytes. Proc Natl Acad Sci U S A. 2005;102:11817–11822. doi: 10.1073/pnas.0505445102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang X, Koldzic DN, Izikson L, Reddy J, Nazareno RF, Sakaguchi S, Kuchroo VK, Weiner HL. IL-10 is involved in the suppression of experimental autoimmune encephalomyelitis by CD25(+)CD4(+) regulatory T cells. Int Immunol. 2004;16:249–256. doi: 10.1093/intimm/dxh029. [DOI] [PubMed] [Google Scholar]
- 32.Mekala DJ, Alli RS, Geiger TL. IL-10-dependent suppression of experimental allergic encephalomyelitis by Th2-differentiated, anti-TCR redirected T lymphocytes. J Immunol. 2005;174:3789–3797. doi: 10.4049/jimmunol.174.6.3789. [DOI] [PubMed] [Google Scholar]
- 33.Spach KM, Nashold FE, Dittel BN, Hayes CE. IL-10 signaling is essential for 1,25-dihydroxyvitamin D3-mediated inhibition of experimental autoimmune encephalomyelitis. J Immunol. 2006;177:6030–6037. doi: 10.4049/jimmunol.177.9.6030. [DOI] [PubMed] [Google Scholar]
- 34.Gabrysova L, Wraith DC. Antigenic strength controls the generation of antigen-specific IL-10-secreting T regulatory cells. Eur J Immunol. 2010;40:1386–1395. doi: 10.1002/eji.200940151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Medema JP, Schuurhuis DH, Rea D, van TJ, de JJ, Bres SA, Laban S, Toes RE, Toebes M, Schumacher TN, Bladergroen BA, Ossendorp F, Kummer JA, Melief CJ, Offringa R. Expression of the serpin serine protease inhibitor 6 protects dendritic cells from cytotoxic T lymphocyte-induced apoptosis: differential modulation by T helper type 1 and type 2 cells. J Exp Med. 2001;194:657–667. doi: 10.1084/jem.194.5.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mendel I, Natarajan K, Ben-Nun A, Shevach EM. A novel protective model against experimental allergic encephalomyelitis in mice expressing a transgenic TCR-specific for myelin oligodendrocyte glycoprotein. J Neuroimmunol. 2004;149:10–21. doi: 10.1016/j.jneuroim.2003.12.007. [DOI] [PubMed] [Google Scholar]
- 37.Krishnamoorthy G, Saxena A, Mars LT, Domingues HS, Mentele R, Ben-Nun A, Lassmann H, Dornmair K, Kurschus FC, Liblau RS, Wekerle H. Myelin-specific T cells also recognize neuronal autoantigen in a transgenic mouse model of multiple sclerosis. Nat Med. 2009;15:626–632. doi: 10.1038/nm.1975. [DOI] [PubMed] [Google Scholar]
- 38.Milner JD, Fazilleau N, Heyzer-Williams M, Paul W. Cutting edge: lack of high affinity competition for peptide in polyclonal CD4+ responses unmasks IL-4 production. J Immunol. 2010;184:6569–6573. doi: 10.4049/jimmunol.1000674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Constant S, Pfeiffer C, Woodard A, Pasqualini T, Bottomly K. Extent of T cell receptor ligation can determine the functional differentiation of naive CD4+ T cells. J Exp Med. 1995;182:1591–1596. doi: 10.1084/jem.182.5.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holzer U, Kwok WW, Nepom GT, Buckner JH. Differential antigen sensitivity and costimulatory requirements in human Th1 and Th2 antigen-specific CD4+ cells with similar TCR avidity. J Immunol. 2003;170:1218–1223. doi: 10.4049/jimmunol.170.3.1218. [DOI] [PubMed] [Google Scholar]
- 41.Zehn D, Lee SY, Bevan MJ. Complete but curtailed T-cell response to very low-affinity antigen. Nature. 2009;458:211–214. doi: 10.1038/nature07657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Illes Z, Waldner H, Reddy J, Anderson AC, Sobel RA, Kuchroo VK. Modulation of CD4 co-receptor limits spontaneous autoimmunity when high-affinity transgenic TCR specific for self-antigen is expressed on a genetically resistant background. Int Immunol. 2007;19:1235–1248. doi: 10.1093/intimm/dxm094. [DOI] [PubMed] [Google Scholar]
- 43.Holler PD, Kranz DM. Quantitative analysis of the contribution of TCR/pepMHC affinity and CD8 to T cell activation. Immunity. 2003;18:255–264. doi: 10.1016/s1074-7613(03)00019-0. [DOI] [PubMed] [Google Scholar]
- 44.Thomson CW, Lee BP, Zhang L. Double-negative regulatory T cells: non-conventional regulators. Immunol Res. 2006;35:163–178. doi: 10.1385/IR:35:1:163. [DOI] [PubMed] [Google Scholar]
- 45.Voelkl S, Gary R, Mackensen A. Characterization of the immunoregulatory function of human TCR-alphabeta+ CD4-CD8-double-negative T cells. Eur J Immunol. 2011;41:739–748. doi: 10.1002/eji.201040982. [DOI] [PubMed] [Google Scholar]
- 46.Dowdell KC, Niemela JE, Price S, Davis J, Hornung RL, Oliveira JB, Puck JM, Jaffe ES, Pittaluga S, Cohen JI, Fleisher TA, Rao VK. Somatic FAS mutations are common in patients with genetically undefined autoimmune lymphoproliferative syndrome. Blood. 2010;115:5164–5169. doi: 10.1182/blood-2010-01-263145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Teachey DT, Manno CS, Axsom KM, Andrews T, Choi JK, Greenbaum BH, McMann JM, Sullivan KE, Travis SF, Grupp SA. Unmasking Evans syndrome: T-cell phenotype and apoptotic response reveal autoimmune lymphoproliferative syndrome (ALPS) Blood. 2005;105:2443–2448. doi: 10.1182/blood-2004-09-3542. [DOI] [PubMed] [Google Scholar]
- 48.Hamad AR, Srikrishnan A, Mirmonsef P, Broeren CP, June CH, Pardoll D, Schneck JP. Lack of coreceptor allows survival of chronically stimulated double-negative alpha/beta T cells: implications for autoimmunity. J Exp Med. 2001;193:1113–1121. doi: 10.1084/jem.193.10.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bilic I, Koesters C, Unger B, Sekimata M, Hertweck A, Maschek R, Wilson CB, Ellmeier W. Negative regulation of CD8 expression via Cd8 enhancer-mediated recruitment of the zinc finger protein MAZR. Nat Immunol. 2006;7:392–400. doi: 10.1038/ni1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pestano GA, Zhou Y, Trimble LA, Daley J, Weber GF, Cantor H. Inactivation of misselected CD8 T cells by CD8 gene methylation and cell death. Science. 1999;284:1187–1191. doi: 10.1126/science.284.5417.1187. [DOI] [PubMed] [Google Scholar]
- 51.Bristeau-Leprince A, Mateo V, Lim A, Magerus-Chatinet A, Solary E, Fischer A, Rieux-Laucat F, Gougeon ML. Human TCR alpha/beta+ CD4-CD8-double-negative T cells in patients with autoimmune lymphoproliferative syndrome express restricted Vbeta TCR diversity and are clonally related to CD8+ T cells. J Immunol. 2008;181:440–448. doi: 10.4049/jimmunol.181.1.440. [DOI] [PubMed] [Google Scholar]
- 52.Mehal WZ, I, Crispe N. TCR ligation on CD8+ T cells creates double-negative cells in vivo. J Immunol. 1998;161:1686–1693. [PubMed] [Google Scholar]
- 53.Zhang ZX, Young K, Zhang L. CD3+CD4-CD8-alphabeta-TCR+ T cell as immune regulatory cell. J Mol Med. 2001;79:419–427. doi: 10.1007/s001090100238. [DOI] [PubMed] [Google Scholar]
- 54.York NR, Mendoza JP, Ortega SB, Benagh A, Tyler AF, Firan M, Karandikar NJ. Immune regulatory CNS-reactive CD8+T cells in experimental autoimmune encephalomyelitis. J Autoimmun. 2010;35:33–44. doi: 10.1016/j.jaut.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bettini M, Rosenthal K, Evavold BD. Pathogenic MOG-reactive CD8+ T cells require MOG-reactive CD4+ T cells for sustained CNS inflammation during chronic EAE. J Neuroimmunol. 2009;213:60–68. doi: 10.1016/j.jneuroim.2009.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sun D, Zhang Y, Wei B, Peiper SC, Shao H, Kaplan HJ. Encephalitogenic activity of truncated myelin oligodendrocyte glycoprotein (MOG) peptides and their recognition by CD8+ MOG-specific T cells on oligomeric MHC class I molecules. Int Immunol. 2003;15:261–268. doi: 10.1093/intimm/dxg023. [DOI] [PubMed] [Google Scholar]
- 57.Linker RA, Rott E, Hofstetter HH, Hanke T, Toyka KV, Gold R. EAE in beta-2 microglobulin-deficient mice: axonal damage is not dependent on MHC-I restricted immune responses. Neurobiol Dis. 2005;19:218–228. doi: 10.1016/j.nbd.2004.12.017. [DOI] [PubMed] [Google Scholar]
- 58.Eshima K, Tachibana M, Suzuki H, Yamazaki S, Shinohara N. Co-receptor-independent signal transduction in a mismatched CD8+ major histocompatibility complex class II-specific allogeneic cytotoxic T lymphocyte. Eur J Immunol. 1997;27:55–61. doi: 10.1002/eji.1830270109. [DOI] [PubMed] [Google Scholar]
- 59.Wilson DB, Wilson DH, Schroder K, Pinilla C, Blondelle S, Houghten RA, Garcia KC. Specificity and degeneracy of T cells. Mol Immunol. 2004;40:1047–1055. doi: 10.1016/j.molimm.2003.11.022. [DOI] [PubMed] [Google Scholar]
- 60.Chastain EM, Duncan DS, Rodgers JM, Miller SD. The role of antigen presenting cellsin multiple sclerosis. Biochim Biophys Acta. 2011;1812:265–274. doi: 10.1016/j.bbadis.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.