

Abstract
Cardiac fibroblasts are exposed to both cyclic strain and interstitial fluid flow in the myocardium. The balance of these stimuli is affected by fibrotic scarring, during which the fibroblasts transition to a myofibroblast phenotype. The present study investigates the mechanisms by which cardiac fibroblasts seeded in three-dimensional (3D) collagen gels differentiate between strain and fluid flow. Neonatal cardiac fibroblast-seeded 3D collagen gels were exposed to interstitial flow and/or cyclic strain and message levels of collagens type I and III, transforming growth factor β1 (TGF-β1), and α-smooth muscle actin (α-SMA) were assessed. Flow was found to significantly increase and strain to decrease expression of myofibroblast markers. Corresponding immunofluorescence indicated that flow and strain differentially regulated α-SMA protein expression. The effect of flow was inhibited by exposure to losartan, an angiotensin II type 1 receptor (AT1R) blocker, and by introduction of shRNA constructs limiting AT1R expression. Blocking of TGF-β also inhibited the myofibroblast transition, suggesting that flow-mediated cell signaling involved both AT1R and TGF-β1. Reduced smad2 phosphorylation in response to cyclic strain suggested that TGF-β is part of the mechanism by which cardiac fibroblasts differentiate between strain-induced and flow-induced mechanical stress. Our experiments show that fluid flow and mechanical deformation have distinct effects on cardiac fibroblast phenotype. Our data suggest a mechanism in which fluid flow directly acts on AT1R and causes increased TGF-β1 expression, whereas cyclic strain reduces activation of smad proteins. These results have relevance to the pathogenesis and treatment of heart failure.
Keywords: Cardiac fibroblast, collagen hydrogel, angiotensin II type 1 receptor, mechanotransduction, interstitial flow
Introduction
Fibrotic scars in the myocardium are characterized by dense extracellular matrix deposition, a marked reduction in the number of functioning cardiomyocytes, and a transition of resident fibroblasts to a contractile myofibroblast phenotype. The mechanisms by which fibrotic scars develop and the role of myofibroblasts in the progression to heart failure are poorly understood, though a number of putative mechanisms have been proposed [1–3]. Recent studies suggest adverse fibrotic remodeling by resident myofibroblasts may both affect and be affected by the altered mechanical microenvironment within a scar [4, 5]. The myofibroblast phenotype is characterized by increased expression of α-smooth muscle actin (SMA) [6], secretion of extracellular matrix proteins [7], increased contractility, and an increased prevalence of stress fibers in the cytoskeleton [8, 9]. A number of factors including mechanical stress [10], transforming growth factor β1 (TGF-β1) and its downstream effectors smad2/3 [11–14], as well as hypoxia [15] can initiate the myofibroblast transition. There is cross-talk between the TGF-β and the angiotensin II signaling pathways and the angiotensin II type 1 receptor (AT1R) is activated by flow-induced shear even in the absence of a biochemical agonist in monolayers of cardiomyocytes [16–18] and endothelial cells [19]. However, the role of AT1R in mechanotransduction of the cardiac myofibroblast transition, and the relationship between mechanical induction of this transition and signal transduction during scar formation are unclear.
Fibroblasts in the myocardium are exposed to a combination of interstitial fluid flow caused by perfusion from the vasculature and cyclic strain from the contraction of surrounding cardiomyocytes. A recent study established a connection between intravascular flow in the heart and interstitial fluid in the myocardium [20]. Fluid flow in the heart can also be produced by edema in the response to injury [21]. Early studies demonstrated that cyclic, uniaxial strain increased collagen type I secretion [22] and α-SMA expression [23, 24] in cardiac fibroblasts plated on two-dimensional elastic substrates, and that these effects are mediated through canonical integrin-mediated pathways such as p38, ERK, and JNK [25, 26]. However, less is known about the response of cardiac fibroblasts to static or cyclic strain in a three-dimensional (3D) environment, which is more representative of the in vivo milieu. Initial studies using fibroblasts embedded in 3D collagen type I hydrogels have demonstrated that α-SMA levels are correlated with the stiffness of the 3D environment [4], and that stiffness also affects cardiac fibroblast morphology [8].
The effects of interstitial flow on the phenotype of a variety of mesenchymal cell types have been investigated. These studies have provided valuable insight into how fluid flow-induced shear stress affects cell function [27–30]. For example, interstitial flow stimulates signaling via α1β1 integrins that influences the orientation of dermal fibroblasts in collagen matrices [31], and also has been shown to act through heparin sulfate proteoglycans and activation of the FAK-ERK signaling pathway [32]. In other studies, TGF-β played a key role in the dermal fibroblast response to interstitial flow, while matrix metalloproteinase 1 (MMP1) mediated the motility of fibroblasts in response to interstitial flow [10, 33]. A complete picture of the signaling pathways activated in response to interstitial fluid flow and their impact on the fibroblast to myofibroblast transition has yet to emerge, though a recent study linked flow to α-SMA expression in smooth muscle cells [34]. While cardiac mechanical strain levels are relatively well characterized, the rates of interstitial fluid flow in the myocardium are more difficult to determine, with only rudimentary estimates generated so far [20]. Based on the information available, flow velocities on the order of microns per second appear to mimic in vivo interstitial flows [10, 33].
Our primary goals in the present study were to simulate the mechanical stimuli experienced by cardiac fibroblasts, to characterize the morphological and functional outcomes of these stimuli, and to identify receptor(s) and signaling pathways involved in transducing mechanical signals into cellular responses. We cultured fibroblasts in a 3D collagen matrix and applied cyclic mechanical strain and interstitial fluid flow, both alone and in combination. We used this model to characterize the myofibroblast transition in response to mechanical cues in 3D, and to query selected signaling mechanisms involved in the phenotype shift. Our findings suggest that the mechanical microenvironment of a fibrotic scar modulates the myofibroblast transition, and may therefore be a factor in the subsequent development of the scar region. This study provides fundamental insights into the fibrotic response that is a key contributor to the progression to heart failure, and may provide a basis for the development of future treatments to modify fibrosis and associated cardiac pathologies.
Methods
A detailed description of the following methods section is provided in the online supplemental material.
Cell Culture
Cardiac fibroblasts were isolated from the ventricles of 2- to 3-day-old Sprague-Dawley rats following euthanasia by means of decapitation [35]. This procedure was approved by the Institutional Animal Use and Care Committee at the University of Michigan and was in compliance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health. Cells were cultured for one week in M199 media supplemented with 5% fetal bovine serum (FBS) and antibiotics.
Mechanical and Biochemical Stimulation
Fibroblast-seeded gels were polymerized in polydimethylsiloxane (PDMS) wells capable of 5% cyclic strain and/or perfusion with 10 µL/min cross flow, which were developed and characterized in a previous study [36]. All gels were cultured at 37C with 100% humidity and 5% CO2 for 24 hours prior to further testing. Gels digested after the 24 hour incubation served as initial controls, while the remaining gels were transferred to the testing apparatus or to an oxygen-controlled incubator set at 2.5% O2 for an additional 48 hours. Gels transferred to the bath but not exposed to any loading served as controls (denoted 48 h control). The remaining unloaded gels were transferred from the bath into the incubator for another 48 hours (denoted the 96 h control). The loading conditions included flow, strain, and a combination of strain and flow with and without inhibitors of angiotensin type II receptor and TGF-β.
qRT-PCR
A guanidium thiocyanate-phenol-chloroform extraction protocol (TRIzol, Invitrogen, Carlsbad, CA) was used to isolate mRNA from the cells. Briefly, homogenized gels seeded with fibroblasts were dissolved in TRIzol, buffer, and reverse transcription of mRNA was performed with a high-capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA) and a C-1000 Thermocycler (Bio-Rad, Hercules, CA). The quantitative PCR protocol is described in a previous publication [4].
Immunofluorescence
Gels were fixed, permeabilized, and stained using previously described protocols [4]. Immunostains included fluorescent DAPI, phalloidin conjugated to Texas Red, and monoclonal mouse anti-α-SMA conjugated with FITC. Cells were visualized using confocal microscopy. Projection image Z-stacks of approximately 30 µm were collected using a confocal microscope (Olympus America, Inc., Center Valley, PA). To quantify the phosphorylation of smad2/3, a rabbit polyclonal antibody and secondary Texas Red-tagged goat anti-rabbit Ab were utilized to quantify smad2/3 phosphorylation. The intensity of the fluorescence within the nucleus of the cell (determined by DAPI staining) was normalized to the intensity within the outline of the cell body (determined by phalloidin staining) for three cells from three images for each condition using the open source software, ImageJ.
Immunoblotting
Gels were homogenized in Garner buffer containing protease and phosphatase inhibitors for cellular protein separation. Tubes were then boiled in a water bath for 3 minutes, sonicated for 10 minutes, and centrifugated 3 times at 2000 g for 15 seconds to isolate cellular protein, before quantifying the protein with a BCA assay. For gels treated with control and AT1R lentivirus particles, trypsinized cells washed in PBS were lysed in ice-cold sample buffer, and then stored at −20C. Protein separation was performed as previously described [37]. Blots were blocked, probed, and visualized using standard protocols.
Lentiviral knockdown
Cardiac fibroblasts isolated from neonatal rats were grown to 50% confluency prior to lentiviral-mediated gene transfer. Viral particles were added to M199 media containing 5 µg/mL Polybrene using an MOI of 1. After viral transduction, cells were cultured in puromycin-containing M199 media (2.5 µg/mL) to deplete non-infected cardiac fibroblasts from the cultures.
Proliferation Assays
DNA quantity was assessed using a PicoGreen assay. Cell-seeded collagen gels were washed thoroughly with PBS, frozen at −80 C, then lyophilized overnight before being incubated in a proteinase K solution at 55 C for 12–16 hours. DNA quantity was normalized to the initial time points.
Statistical Analysis
Results show sample sizes of three to six separate experiments, with data expressed as mean±SD. An unpaired Student's t test was used to determine statistical significance for comparisons of two groups, with P<0.05 considered significantly different. An analysis of variance (ANOVA) followed by Tukey's multiple comparison test (P<.05) was used to compare multiple groups using the open source statistical package R [38].
Results
The Effect of Mechanical Stimuli on Expression of Myofibroblast Markers
The present study demonstrates that mechanical stimuli modulate the fibroblast to myofibroblast transition, though the mechanisms involved result in distinct transduction of fluid-induced and stretch-induced stresses. In a previous study, we showed that fibroblasts in collagen gels created in standard culture plates and cultured for 48 and 96 h transitioned to a myofibroblast phenotype over time, marked by significant elevations in the mRNA message levels for collagen type III and α-SMA, and a reduction in TGF-β1 expression [4]. This previous study indicated that TGF-β levels peaked at 24 hours (the initial time point used in the present study), which was likely an initiating event for the subsequent increases in α-SMA and collagen type III levels. This baseline pattern was repeated in gels polymerized in the PDMS wells in the present study, as indicated in Figure 1. Collagens type I and III serve as markers for matrix deposition in the myocardium, and α-SMA as a marker for the myofibroblast phenotype. Therefore, polymerizing cardiac fibroblast-seeded collagen gels in PDMS wells did not alter the baseline shift to a myofibroblast phenotype.
Figure 1. Control message levels as a function of time.
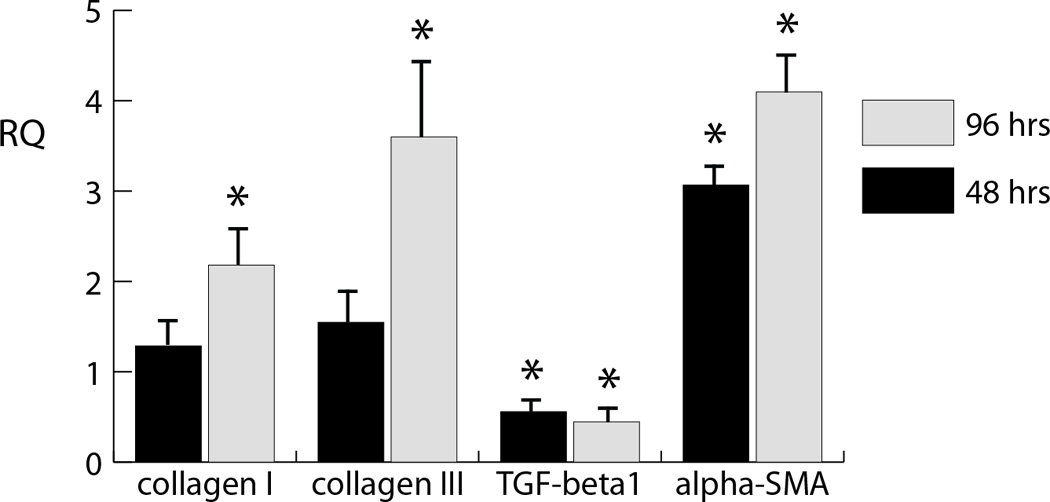
Message expression of the 48 h and 96 h control samples levels showing collagen type I (A), collagen type III (B), TGF-β (C), and α-SMA (D) after. Levels are normalized to initial (t=0) levels.
mRNA expression of myofibroblast markers (collagen I and III, α-SMA, and TGF-β1) was measured and immunolocalization of α-SMA were performed in response to a 48 h exposure to combinations of interstitial flow and cyclic strain, as well as to a 2.5% oxygen environment. As shown in Figure 2a, neither mechanical stimulation (by cyclic stretch or cross-flow) nor hypoxia significantly affected collagen type I message levels. However, cross flow significantly increased collagen type III levels over control levels, both in the presence and absence of cyclic strain, whereas cyclic strain alone did not significantly affect message levels (Fig. 2b). Similarly, cross flow both with and without cyclic strain prevented the drop in TGF-β1 expression observed in the control samples and in cyclic strain conditions (Fig. 2c). Cross flow alone produced a marked increase in α-SMA message levels compared with both the initial control level and that of samples exposed to cyclic strain. The competing effects of cross flow and cyclic strain were most apparent in the samples exposed to a combination of both fluid flow and cyclic strain, which resulted in no significant change from the 48 h controls (Fig. 2d). Hypoxia caused a significant increase in collagen type III and α-SMA message, but not in TGF-β1 levels. Assessment of DNA content in these constructs showed that cell numbers were constant across samples, and therefore the observed effects were not a result of changes in proliferation or cell death (Figure 3).
Figure 2. Message levels in response to strain, flow, and hypoxia.
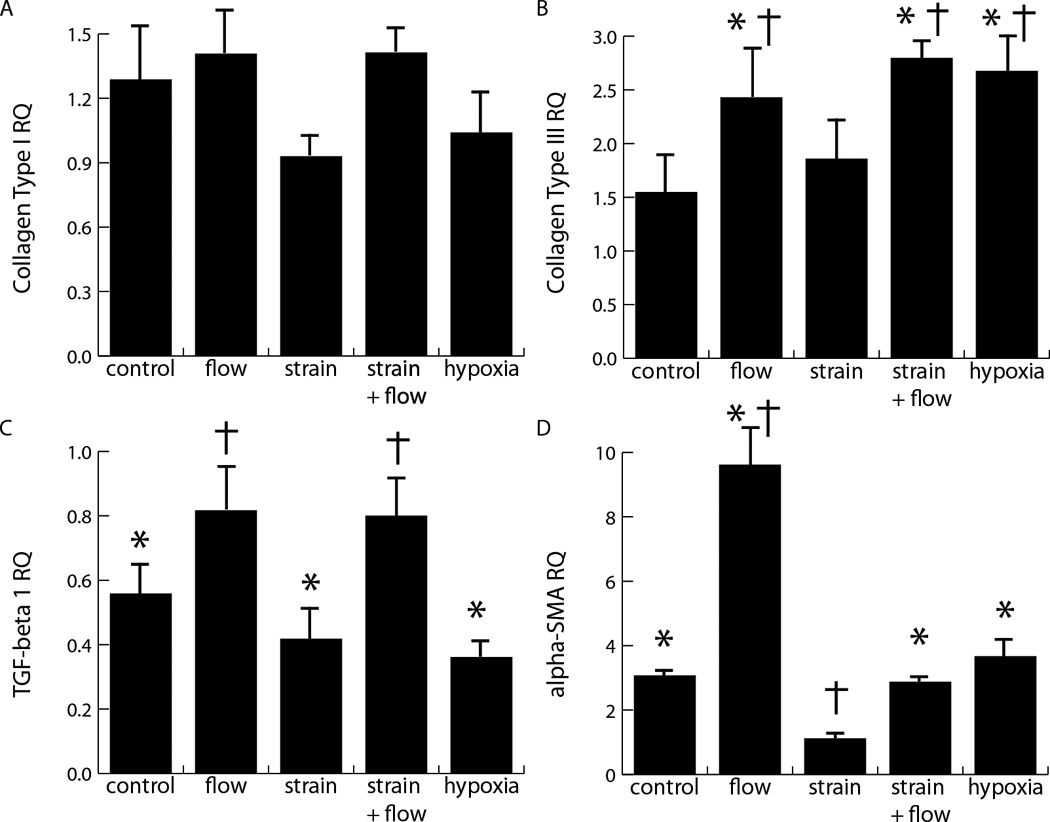
Influence of hypoxia and mechanical stimuli produced by cross flow and cyclic strain on message levels of collagen type I (A), collagen type III (B), TGF-β (C), and α-SMA (D) after 48 h of stimulation, indicating significant differences compared to initial control levels (*: p<0.05) and showing significant differences compared to 48 h controls (†; p<0.05). Relative quantity (RQ) is defined as 2−ΔΔCT, where CT is the cycles to threshold, double normalized to both GAPDH and the initial control levels.
Figure 3. The effect of mechanical and biochemical stimulation on DNA content.
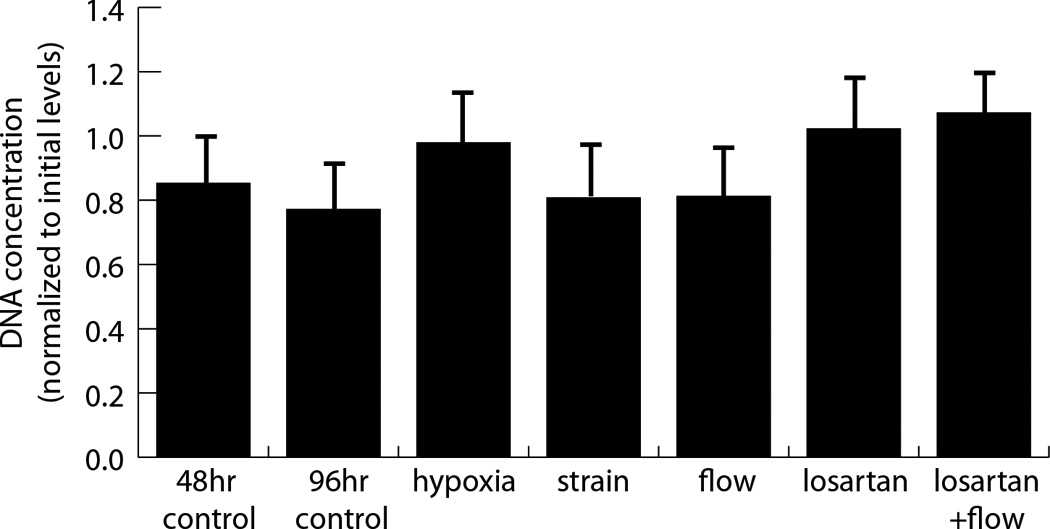
DNA concentrations quantified using the picoGreen assay were normalized to initial levels for many of the testing conditions. No significant differences were found.
Immunohistochemistry was performed to provide insight into the mechanical influences on cell phenotype (Fig. 4). Previous studies indicated that the myofibroblast transition is associated with a distinct morphology characterized by prominent stress fibers and a less stellate shape. The observed increase in α-SMA gene expression was corroborated by the presence of distinct stress fibers that stained for α-SMA. This effect was evident in control samples, which exhibited increased positive staining for α-SMA over 96 h (Fig. 4a–c). Cells exposed to cross flow exhibited prominent stress fibers, a more stellate morphology, and markedly increased α-SMA staining compared to the other conditions (Fig. 4d). Cells exposed to cyclic strain, both with and without the presence of cross flow, showed decreased expression of α-SMA compared to the cross flow samples (Fig. 4e, f). Moreover, the cells exposed to strain appeared to align with the direction of strain, though they exhibited less prominent stress fibers compared to the 96 h controls and cells exposed to flow alone. These results support the qRT-PCR data for α-SMA message levels, further implicating that cross flow stimulates and cyclic strain attenuates the myofibroblast transition.
Figure 4. Immunofluorescence of cells stimulated by strain and flow.

Immunofluorescence images of embedded fibroblasts for initial, 48 h, and 96 h controls (panels A–C) and for flow (panel D), strain (panel E), and strain + flow (panel F). Blue indicates DAPI staining of the nucleus, red shows Texas red phalloidin staining of fibrillar actin, and green represents FITC-stained anti-α-SMA. Note scale bars and higher magnification in panels C and E, compared to A, B, D, F.
The role of the AT1R and TGF-β signaling pathways in cardiac fibroblast mechanotransduction
Since the response of cardiac fibroblasts exposed to cross flow and cyclic strain revealed distinct and opposing effects of these two types of mechanical signal, we performed experiments to further examine signal transduction in these cells. In particular, we blocked TGF-β and AT1R-mediated signaling to determine the roles of these pathways in the cellular response, as shown in Figure 5. These data complement our initial findings in Figures 2 and 4, and show that blocking of TGF-β to inhibit autocrine and paracrine signaling by the cells through this growth factor did not affect collagen type I message levels, both in the presence or absence of cross flow (Fig. 5a). However, inhibiting TGF-β attenuated the cross flow-induced increase in both collagen type III and TGF-β1 expression (Fig. 5b–c). The presence of the antibody did not affect levels of either marker in the absence of flow. However, blocking TGF-β not only prevented the cross flow-induced increase in α-SMA expression, but also reduced the message levels to initial control levels both with and without cross flow (Fig. 5d), suggesting that cross flow might be activating a separate pathway.
Figure 5. Inhibition of AT1R and TGF-β.
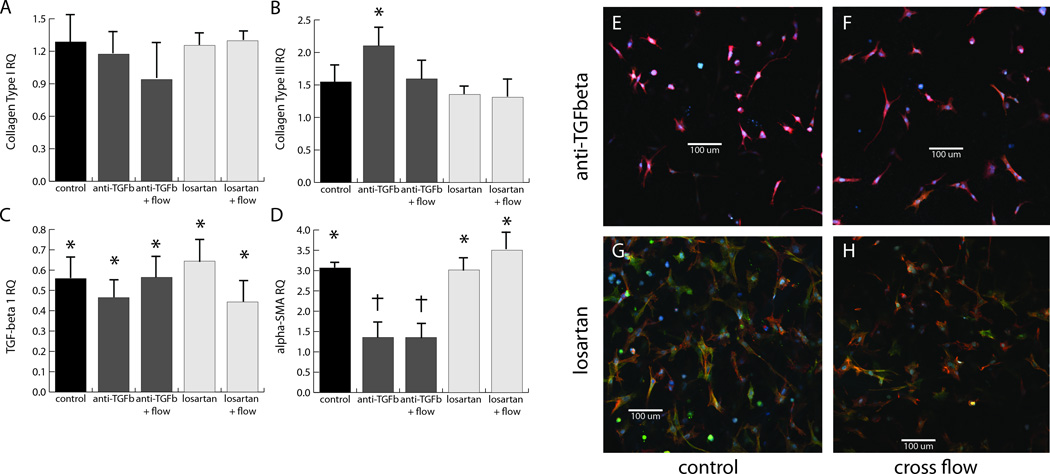
Effects of inhibition of AT1R and TGF-β on message levels of collagen type I (A), collagen type III (B), TGF-β1 (C), and α-SMA (D), using a pan-specific TGF-β blocking antibody (dark grey bars) and the AT1R antagonist, losartan (light grey bars). Immunofluorescence images of cardiac fibroblasts treated with anti-TGF-β (E, F) and losartan (G, H) at both the 48 h control (E, G) and cross flow conditions (F, H). Blue corresponds to DAPI, red to phalloidin, and green to anti-α-SMA. All images are 600 × 600 µm.
TGF-β signaling is known to be enhanced by activation of angiotensin II receptors, and AT1R in particular has been linked to flow-induced mechanotransduction [27–34]. We therefore examined the role of AT1R in the cross flow response by blocking this receptor using losartan. The antagonist had a similar effect to blocking of TGF-β on the message levels of collagens type I and III and TGF-β1 (Fig. 5a–c), providing further evidence for a relationship between AT1R-activated signaling pathways and the production of TGF-β1 by the cell for autocrine/paracrine signaling. In contrast to the TGF-β blocking antibody, the addition of losartan did not affect the baseline increase in α-SMA message levels (Fig. 5d). However, the presence of losartan in the perfusing media did block the flow-induced effect on message levels (Fig. 5d). These results suggest that cross flow may act through AT1R, which when activated elicits TGF-β1 expression and thereby initiates the myofibroblast transition. Immunofluorescence was also used to validate the observed trends in α-SMA message levels, and to gain insight into the effect of these blocking agents on cell morphology. Cells treated with the TGF-β blocking antibody exhibited similar morphology and α-SMA expression to the initial controls (Fig. 5e, f). Losartan-treated cells appeared similar to the 48 h controls (Fig. 5g, h). This is evident for both the control conditions (Fig. 5e, g) and for the cells exposed to cross flow (Fig. 5f, h).
Losartan may affect the TGF-β receptor and a previous study demonstrated its ability to block angiotensin II type 2 receptors [39]. Therefore, since losartan is capable of blocking both AT and TGF-β pathways, demonstration of the role of AT1R-mediated signaling in the cellular response required more specific targeting of the receptor. Lentiviral transduction was used to insert a shRNA against AT1R into cardiac fibroblasts, and results are shown in Figure 6. AT1R shRNA knocked down AT1R levels to around 55% of the levels present in cells infected with a scrambled shRNA (Fig. 6a). Cells treated with shRNA were then exposed to the cross flow regimen. Figures 6b–e show message levels for samples exposed to shRNA (AT1R and scrambled) at the 48 h control and the cross flow condition. In general, cells infected with scrambled shRNA responded similarly to non-infected cells, with the exception of a statistically significant increase in collagen type I and III expression for the 48 h time point (Fig. 6b–c). Moreover, cross flow did not significantly increase collagen type I or III expression relative to initial controls (Fig. 6c). Scrambled shRNA produced the same patterns of TGF-β1 and α-SMA expression as non-infected cells (Fig. 6d–e). In general, the incorporation of the AT1R shRNA produced similar effects to losartan treatment. However, the combination of AT1R shRNA and cross flow caused a significant increase in collagen type I compared to initial control levels (Fig. 6b). Moreover, AT1R shRNA prevented a significant increase in collagen type III expression in the 48 h control samples (Fig. 6c). Nonetheless, AT1R shRNA prevented the rise in TGF-β1 message levels to the initial control levels with the addition of cross-flow (Fig. 6d). Similarly, AT1R shRNA attenuated the rise in α-SMA expression in response to cross flow, while still allowing for the baseline increase in this message level at the 48 h time point. Taken together, these results suggest that AT1R directly participates in the transduction of the mechanical stress exerted by cross flow.
Figure 6. Knockdown of AT1R using shRNA.

The effects of shRNA knockdown of AT1R on flow-mediated phenotype shifts in cardiac fibroblasts. A) Immunoblot verifying knockdown of AT1R. B–E) Message levels of key phenotype markers in response to treatment with AT1R shRNA and scrambled shRNA, as well as cross flow and 48 h controls. Relative quantities were normalized to initial controls treated with scrambled shRNA. AT1R shRNA had no significant effect on initial controls infected with scrambled shRNA.
The effect of cyclic strain on smad2 phosphorylation
Because our results suggested that cross flow acts through AT1R to facilitate the myofibroblast transition, experiments were conducted to determine if cyclic strain also works through a TGF-β-related pathway in order to clarify the degree and type of crosstalk between these stimuli. In particular, we examined the phosphorylation of smad2, a downstream effector of TGF-β, in cells exposed to cyclic strain, as shown in Figure 7. Immunoblotting (Fig. 7a–b) showed a band at 60 kDa, which represents smad2 levels (the 52 kDa band in the phospho-smad blot represents cross-reactivity of the antibody with smad3). The amount of protein varied between blots (Fig. 7c) due to the difficulty in isolating cellular proteins from the protein-based hydrogels, however silver staining showed that the relative levels of smad2 were not significantly different across all samples. Comparison of the ratio of phosphorylated to total smad2 (Fig. 7d) showed that relative phosphorylation of smad2 increased in control samples as time progressed, which mirrored the observed increases in collagen type III and α-SMA message levels. Interestingly, the application of cyclic strain attenuated the phosphorylation to initial control levels, which also matched the effect of strain on α-SMA message levels. To verify the results of immunoblotting, the levels of smad2 phosphorylation were also examined using immunohistochemistry, as shown in Figure 7e. Previous studies have shown that once phosphorylated, smad2/3 is shepherded into the nucleus by smad4 to mediate transcription (Fig. 7g). Examination of the ratio of fluorescence intensity between the nucleus and cytoplasm (Fig. 7f) showed that the 48 and 96 h controls and cross flow condition had significantly higher levels of phosphorylated smad2 compared to initial controls and cyclically strained samples, which aligns with the immunoblotting results. Overall, these data suggest that the observed changes phosphorylated smad2 were due to transduction of the mechanical strain stimulus.
Figure 7. The effect of strain and flow on smad phosphorylation.

Western blot of cardiac fibroblasts exposed to cyclic strain using smad2/3 (A) and phospho-smad2 (B) antibodies. Representative lanes from silver staining (C) indicated uneven protein loading caused by separation of cellular protein from the collagen hydrogel. The ratio of phosphor-smad2 to total smad2 is quantified in (D). Immunohistochemistry of smad2/3 and its localization to the nucleus during phosphorylation. E) shows images of cardiac fibroblasts stained for smad2/3 and F) shows quantification of nuclear localization. G) illustrates how smad2/3 is shuttled into the nucleus to affect transcription.
Discussion
There is increasing evidence that mechanical stimulation is a key regulator of the fibroblast to myofibroblast transition in the myocardium. Here, we demonstrated that cardiac fibroblasts distinguish between fluid-induced and strain-induced mechanical stresses, though there is crosstalk between the mechanotransduction mechanisms. The cellular response to both types of mechanical stress involves the TGF-β and AT1R signaling pathways, and is shown schematically in Figure 8. Direct activation of AT1R by fluid-induced shear stimulates the production of TGF-β1, which in turn acts in an autocrine and paracrine manner to create a positive feedback loop promoting the myofibroblast phenotype. Blocking of AT1R using losartan or shRNA negates this effect. At the same time, cyclic strain attenuates the phosphorylation of smad2, a downstream effector of TGF-β signaling, and therefore prevents the myofibroblast transition. These mechanisms indicate that increased interstitial perfusion would instigate the fibrotic response, while also suggesting a protective effect of cyclic strain in preventing cardiac fibrosis. This finding may have a direct impact on both the understanding and treatment of fibrosis in the myocardium, since our study suggests AT1R is a potential target to block the mechanical stress-induced transition to the myofibroblast phenotype. These results also provide insight into recent clinical findings that AT1R inhibitors used to treat hypertension can also improve the function of fibrotic hearts, and reduce the occurrence of heart failure [40].
Figure 8. Summary of proposed mechanism.
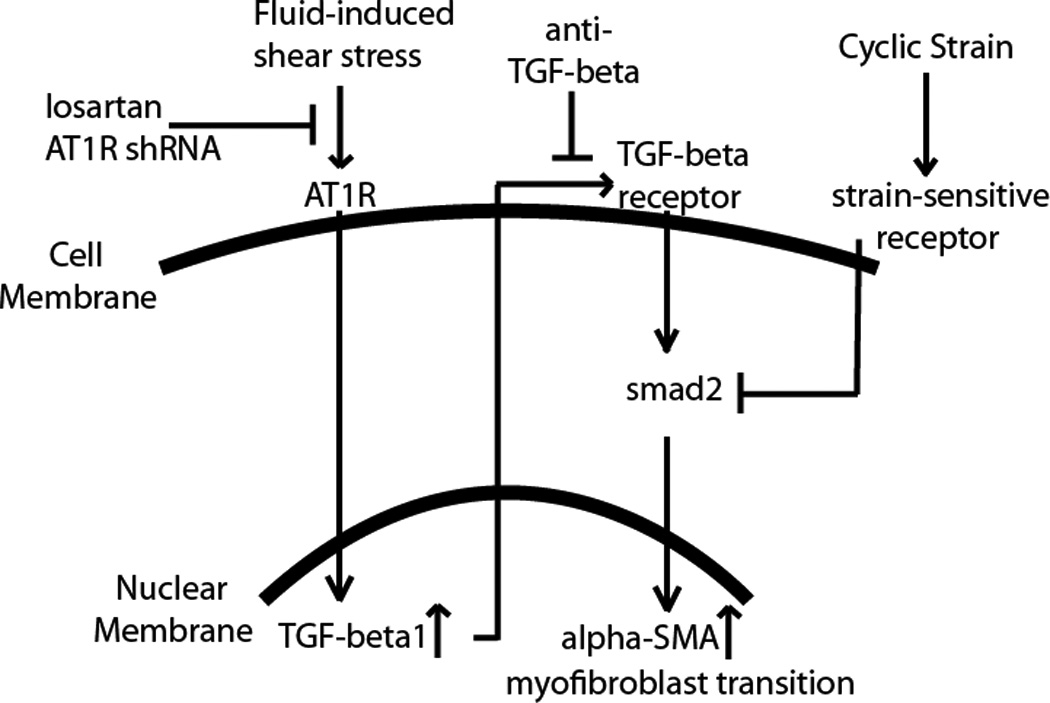
Summary of the crosstalk between AT1R activation and TGF-β signaling in cardiac fibroblasts undergoing fluid- and strain-induced mechanical stimulation.
Changes in α-SMA message levels provided the clearest demonstration of the divergent signal transduction of cross flow and cyclic strain. Cross flow alone caused a nearly nine-fold increase in message, while cyclic strain caused a decrease to the initial control level. However, the combination of cyclic strain and cross flow resulted in α-SMA message levels not significantly different from the 48 h control, suggesting that the two modes of mechanical stimulation act through connected signaling pathways. Inhibition experiments indicated that inactivation of AT1R attenuated the cross flow-induced effect, but only blocking TGF-β negated both the flow-induced α-SMA message increase and the baseline rise between the initial and 48 h controls. This result not only identifies AT1R as a transduction receptor for cross flow, but also suggests that TGF-β1 is a master regulator of the mechanical stress-induced myofibroblast transition. Further evidence for the role of TGF-β1 as a regulator of mechanotransduction was provided by experiments in which fibroblasts were exposed to hypoxic conditions. The low oxygen environment stimulated the myofibroblast transition, as evidenced by increased collagen type III message levels, but did so without the increase in TGF-β1 expression observed in cells exposed to cross flow. Finally, immunoblotting for phosphorylated smad2 indicated that cyclic strain blocked TGF-β signaling by interfering with smad2 phosphorylation, though the receptor responsible for strain transduction was not elucidated. Overall, these data provide a link between the transduction of flow and strain, and show that these mechanical stimuli can have competing effects.
Computational studies of the mechanics of hydrogels tested in our flow-strain bioreactor have shown that the deformation of the hydrogel and interstitial fluid flow are also strongly coupled in this system [41], as they are in vivo [20]. Due to the high porosity of the 3D protein hydrogel, cyclic strain can cause substantial interstitial fluid flow within the matrix, even in the absence of cross flow. In contrast, cross flow itself induces negligible strain-induced stress within the hydrogel. In the present study only the cross flow condition caused a marked increase in α-SMA, although the computational model predicted a considerable amount of interstitial flow for the all three mechanical stimulation conditions examined (flow, strain, and combined flow/strain). Therefore, application of mechanical strain clearly attenuated the rise in α-SMA, even though it also induced interstitial flow. Taken together, these results suggest that interstitial fluid flow is a potent initiator of signaling through AT1R, but that cyclic deformation of the extracellular matrix is able to override or diminish these effects.
The findings of the present study serve to illuminate the fibrotic response in cardiac tissue. Since a fibrotic scar is characterized by excessive extracellular matrix deposition, it is stiffer than surrounding myocardium. And because a cardiac scar is also characterized by an absence of healthy, contracting cardiomyocytes, there is considerably less strain in the scar than in surrounding regions. However, the scar region is still perfused by capillary networks [2] and interstitial fluid may be increased by edema. Our study provides evidence that this combination of mechanical factors may play a role in causing the transition of healthy fibroblasts to the myofibroblast phenotype, thereby propagating the fibrotic response. Our findings also provide a potential mechanism for the effects of losartan in treating fibrotic hearts, since blocking of flow-mediated phenotype shifts in cardiac fibroblasts may be of particular benefit. The in vitro model we used is necessarily a simplified version of the complex cellular milieu of the myocardium. However it is a promising system for isolating the effects of mechanical stresses in 3D systems, and therefore is of potential importance to clinical trials focusing on the anti-fibrotic potential of losartan and its conjugates [42].
Highlights.
interstitial flow promotes myofibroblast transition; cyclic strain inhibits transition
effect of flow negated by blocking AT1R using losartan and shRNA
anti-TGF-β blocks flow-induced activation of cardiac fibroblasts
smad2 phosphorylation reduced in response to cyclic strain
results illuminate mechanisms for sensing flow/strain by cardiac fibroblasts
Supplementary Material
Acknowledgements
This work was supported in part by the Microfluidics in Biomedical Sciences Training Program at the University of Michigan, sponsored by the National Institute of Biomedical Imaging and Bioengineering [to P.G.]. Additional support was provided by a pilot grant from the Center for Cardiovascular Sciences at the University of Michigan [to M.V.W., M.W.R, and J.P.S.].
Abbreviations
- AT1R
angiotensin 2 type 1 receptor
- α-SMA
alpha-smooth muscle actin
- TGF-β
transforming growth factor beta
- 3D
three dimensional
- MMP1
matrix metalloproteinase 1
- FBS
fetal bovine serum
- HEPES
(4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid)
- PDMS
polydimethylsiloxane
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Swynghedauw B. Molecular mechanisms of myocardial remodeling. Physiol. Rev. 1999;79:215–262. doi: 10.1152/physrev.1999.79.1.215. [DOI] [PubMed] [Google Scholar]
- 2.Boudoulas KD, Hatzopoulos AK. Cardiac repair and regeneration: the Rubik's cube of cell therapy for heart disease. Dis. Model. Mech. 2009;2(7–8):344–358. doi: 10.1242/dmm.000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baudino T, Carver W, Giles W, Borg T. Cardiac fibroblasts: friend or foe? Am. J. Physiol. Heart Circ. Physiol. 2006;291:H1015–H1026. doi: 10.1152/ajpheart.00023.2006. [DOI] [PubMed] [Google Scholar]
- 4.Galie P, Stegemann JP. Reduced serum content and increased matrix stiffness promote the cardiac myofibroblast transition in 3D collagen matrices. Cardiovascular Pathology. 2011 doi: 10.1016/j.carpath.2010.10.001. [EPub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dobaczewski M, Bujak M, Li N, Gonzalez-Quesada C, Mendoza LH, Wang XF, Frangogiannis NG. Smad3 signaling critically regulates fibroblast phenotype and function in healing myocardial infarction. Circ. Res. 2010;107:418–428. doi: 10.1161/CIRCRESAHA.109.216101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang J, Seth A, McCulloch CAG. Force regulates smooth muscle actin in cardiac fibroblasts. Am. J. Physiol. Heart Circ. Physiol. 2000;279:H2776–H2785. doi: 10.1152/ajpheart.2000.279.6.H2776. [DOI] [PubMed] [Google Scholar]
- 7.Swaney JS, Roth DM, Olson ER, Naugle JE, Meszaros JG, Insel PA. Inhibition of cardiac myofibroblast formation and collagen synthesis by activation and overexpression of adenylyl cyclase. P.N.A.S. 2004;10(2):437–442. doi: 10.1073/pnas.0408704102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baxter SC, Morales MO, Goldsmith EC. Adaptive changes in cardiac fibroblast morphology and collagen organization as a result of mechanical environment. Cell Biochem. Biophys. 2008;51:33–44. doi: 10.1007/s12013-008-9013-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Poobalarahi F, Baicu CF, Bradshaw AD. Cardiac myofibroblasts differentiated in 3D culture exhibit distinct changes in collagen I production, processing, and matrix deposition. Am. J. Physiol. Heart Circ. Physiol. 2006;291:H2924–H2932. doi: 10.1152/ajpheart.00153.2006. [DOI] [PubMed] [Google Scholar]
- 10.Ng CP, Hinz B, Swartz MA. Interstitial fluid flow induces myofibroblast differentiation and collagen alignment in vitro. J. Cell Science. 2005;118:4731–4739. doi: 10.1242/jcs.02605. [DOI] [PubMed] [Google Scholar]
- 11.Asazuma-Nakamura Y, Dai P, Yoshinori H, Jiang Y, Hamaoka K, Takamatsu T. Cx43 contributes to TGF-β signaling to regulate differentiation of cardiac fibroblasts into myofibroblasts. Experimental Cell Research. 2009;315(7):1190–1199. doi: 10.1016/j.yexcr.2008.12.021. [DOI] [PubMed] [Google Scholar]
- 12.Li P, Wang D, Lucas J, Oparil S, Xing D, Cao X, Novak L, Renfrow MB, Chen YF. Atrial natriuretic peptide inhibits transforming growth factor induced smad signaling and myofibroblast transformation in mouse cardiac fibroblasts. Circ. Res. 2008;102:185–192. doi: 10.1161/CIRCRESAHA.107.157677. [DOI] [PubMed] [Google Scholar]
- 13.Leask A. Potential Therapeutic targets for cardiac fibrosis: TGF, angiotensin, endothelin, CCN2, and PDGF: partners in fibroblast activation. Circ. Res. 2010;106:1675–1680. doi: 10.1161/CIRCRESAHA.110.217737. [DOI] [PubMed] [Google Scholar]
- 14.Walker GA, Masters KS, Shah DN, Anseth KS, Leinwand LA. Valvular myofibroblast activation by transforming growth factor- β: implications for pathological extracellular matrix remodeling in heart valve disease. Circ. Res. 2004;95:253–260. doi: 10.1161/01.RES.0000136520.07995.aa. [DOI] [PubMed] [Google Scholar]
- 15.Cucoranu I, Clempus R, Dikalova A, Phelan PJ, Ariyan S, Dikalov S, Sorescu D. NAD(P)H oxidase 4 mediates transforming growth factor-β1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ. Res. 2005;97:900–907. doi: 10.1161/01.RES.0000187457.24338.3D. [DOI] [PubMed] [Google Scholar]
- 16.Zou Y, Akazawa H, Qin Y, Sano M, Takano H, Minamino T, Makita N, Iwanaga K, Zhu W, Kudoh S, Toko H, Tamura K, Kihara M, Nagai T, Fukamizu A, Umemura S, Iiri T, Fujita T, Komuro I. Mechanical stress activates anngiotensin II type 1 receptor without involvement of angiotensin II, Nature. Cell Biology. 2004;6(6):499–506. doi: 10.1038/ncb1137. [DOI] [PubMed] [Google Scholar]
- 17.Yasuda N, Miura S, Akazawa H, Tanaka T, Qin Y, Kiya Y, Imaizumi S, Fujino M, Ito K, Zou Y, Fukuhara S, Kunimoto S, Koichi F, Sato T, Ge J, Mochizuki N, Nakaya H, Saku K, Kumoro I. Conformational switch of angiotensin II type I receptor underlying mechanical stress-induced activation. EMBO reports. 2008;9(2):179–186. doi: 10.1038/sj.embor.7401157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yasuda N, Akazawa H, Qin Y, Zou Y, Komuro I. A novel mechanism of mechanical stress-induced angiotensin II type I receptor activation without the involvement of angiotensin II. Arch. Pharmacol. 2008;377:393–399. doi: 10.1007/s00210-007-0215-1. [DOI] [PubMed] [Google Scholar]
- 19.Ramkhelawon J, Vilar J, Rivas D, Mees B, de Crom R, Tedgui A. Shear stress regulates angiotensin II type I receptor expression in endothelial cells. Circ. Res. 2009;105:869–875. doi: 10.1161/CIRCRESAHA.109.204040. [DOI] [PubMed] [Google Scholar]
- 20.Ashikaga H, Coppola BA, Yamazaki KG, Villarreal FJ, Omens JH, Covell JW. Changes in regional myocardial volume during the cardiac cycle: implications for transmural blood flow and cardiac structure. A.J.P. Heart Circ. Phys. 2008;295(2):H610–H618. doi: 10.1152/ajpheart.00107.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laine GA, Allen SJ. Left ventricular myocardial edema. Lymph flow, interstitial fibrosis, and cardiac function. Circ. Res. 1991;68:1713–1721. doi: 10.1161/01.res.68.6.1713. [DOI] [PubMed] [Google Scholar]
- 22.Butt RP, Bishop JE. Mechanical load enhances the stimulatory effect of serum growth factors on cardiac fibroblast procollagen synthesis. J. Mol. Cell Cardiology. 1997;29(4):1141. doi: 10.1006/jmcc.1996.0347. [DOI] [PubMed] [Google Scholar]
- 23.Lew AM, Glogauer M, McCulloch CAG. Specific inhibition of skeletal alpha-actin gene transcription by applied mechanical forces through integrins and actin. Biochem. J. 1999;341:647–653. [PMC free article] [PubMed] [Google Scholar]
- 24.Costa APD, Nadruz W, Franchini KG. FAK mediates the activation of cardiac fibroblasts induced by mechanical stress through regulation of the mTOR complex. Cardiovasc. Res. 2010;86:421–431. doi: 10.1093/cvr/cvp416. [DOI] [PubMed] [Google Scholar]
- 25.Wang J, Su M, Fan J, Seth A, McCulloch CAG. Transcriptional regulation of contractile gene by mechanical forces applied through integrins in osteoblasts. J.B.C. 2002;277(25):22889–22895. doi: 10.1074/jbc.M203130200. [DOI] [PubMed] [Google Scholar]
- 26.MacKenna DA, Dolfi F, Vuori K, Ruoslahti E. Extracellular signal- regulated kinase and c-Jun NH2-terminal kinase activation by mechanical stretch is integrin-dependent and matrix-specific in rat cardiac fibroblasts. J. Clin. Invest. 1998;101:301–310. doi: 10.1172/JCI1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang S, Tarbell JM. Effect of Fluid Flow on Smooth Muscle Cells in a 3-Dimensional Collagen Gel Model. Arterioscler. Thromb. Vasc. Biol. 2000;20:2220–2225. doi: 10.1161/01.atv.20.10.2220. [DOI] [PubMed] [Google Scholar]
- 28.Bonvin C, Overney J, Shieh AC, Dixon JB, Swartz MA. A multichamber fluidic device for 3D cultures under interstitial flow with live imaging: Development, characterization, and applications. Biotech and Bioeng. 2010;105(5):982–991. doi: 10.1002/bit.22608. [DOI] [PubMed] [Google Scholar]
- 29.Radisic M, Yang L, Boublik J, Cohen RJ, Langer R, Freed LE, Vunjak-Novakovic G. Medium perfusion enables engineering of compact and contractile cardiac tissue. Am. J. Physiol. Heart Circ. Physiol. 2004;286:H507–H516. doi: 10.1152/ajpheart.00171.2003. [DOI] [PubMed] [Google Scholar]
- 30.Shi ZD, Tarbell JM. Fluid flow mechanotransduction in vascular smooth muscle cells and fibroblasts. Annals of Biomedical Engineering. 2011;39(6):1608–1619. doi: 10.1007/s10439-011-0309-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ng CP, Swartz MA. Fibroblast alignment under interstitial fluid flow using a novel 3-D tissue culture model. Am. J. Physiol. Heart Circ. Physiol. 2003;284:H1771–H1777. doi: 10.1152/ajpheart.01008.2002. [DOI] [PubMed] [Google Scholar]
- 32.Shi ZD, Wang H, Tarbell JM. Heparan sulfate proteoglycans mediate interstitial flow mechanotransduction regulating MMP-13 expression and cell motility via FAK-ERK in 3D collagen. PLoS One. 2011;6:e15956. doi: 10.1371/journal.pone.0015956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shi ZD, Ji XY, Qazi H, Tarbell JM. Interstitial flow promotes vascular fibroblast, myofibroblast, and smooth muscle cell motility in 3-D collagen I via upregulation of MMP-1. Am. J. Physiol. Heart Circ. Physiol. 2009;297:H1225–H1234. doi: 10.1152/ajpheart.00369.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shi ZD, Abraham G, Tarbell JM. Shear Stress Modulation of Smooth Muscle Cell Marker Genes in 2-D and 3-D Depends on Mechanotransduction by Heparan Sulfate Proteoglycans and ERK1/2. P.L.o.S. O.N.E. 2010;5(8):e12196. doi: 10.1371/journal.pone.0012196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zlochiver S, Munoz V, Vikstrom K, Taffet S, Berenfeld O, Jalife J. Electrotonic myofibroblast-to-myocyte coupling increases propensity to reentrant arrhythmias in two dimensional cardiac monolayers. Biophysical Journal. 2008;95:4469–4480. doi: 10.1529/biophysj.108.136473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Galie PA, Stegemann JP. Simultaneous Application of Interstitial Flow and Cyclic Mechanical Strain to a Three-Dimensional Cell-Seeded Hydrogel. Tissue Eng. Part C. 2011;17(5):527–536. doi: 10.1089/ten.tec.2010.0547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Westfall MV, Rust EM, Metzger JM. Slow skeletal troponin I gene transfer, expression, and myofilament incorporation enhances adult cardiac myocyte contractile function. P.N.A.S. 1997;94(10):5444–5449. doi: 10.1073/pnas.94.10.5444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ihaka R, Gentleman RR. A language for data analysis and graphics. J. Comp. Graph. Stat. 1996;5(3):299–314. [Google Scholar]
- 39.Habashi JP, Doyle JJ, Holm TM, Aziz H, Schoenhoff F, Bedja D, Chen Y, Modiri AN, Judge DP, Dietz HC. Angiotensin II Type 2 Receptor Signaling Attenuates Aortic Aneurysm in Mice Through ERK Antagonism. Science. 2011;332(6027):361–365. doi: 10.1126/science.1192152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Konstam MA, Neaton JD, Dickstein K, Drexler H, Komajda M, Martinez FA, Riegger GAJ, Malbecq W, Smith RD, Guptha S, Poole-Wilson PA. Effects of high-dose versus low-dose losartan on clinical outcomes in patients with heart failure (HEAAL study): a randomised, double-blind trial. Lancet. 2009;374(9704):1840–1848. doi: 10.1016/S0140-6736(09)61913-9. [DOI] [PubMed] [Google Scholar]
- 41.Galie PA, Spilker RL, Stegemann JP. A linear, biphasic model incorporating a Brinkman term to describe the mechanics of cell-seeded collagen hydrogels. Annals of Biomedical Engineering. 2011 doi: 10.1007/s10439-011-0371-9. [EPub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.National Institutes of Health. ClinicalTrials.gov [Internet] Bethesda (MD): National Library of Medicine (US); 2000. [cited 2011 Aug 26]. Massachusetts General Hospital. Effect of Losartan in Patients With Nonobstructive Hypertrophic Cardiomyopathy. Available from: http://clinicaltrials.gov/show/NCT01150461. NCT ID: NCT01150461. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.