

Abstract
Macrophages, have established roles in tumor growth and metastasis, but information about their role in lung tumor promotion is limited. To assess the role of macrophages in lung tumorigenesis, we developed a method of minimally-invasive, long-term macrophage depletion by repetitive intratracheal instillation of liposomal clodronate. Compared to controls treated with repetitive doses of phosphate buffered saline (PBS) containing liposomes, long-term macrophage depletion resulted in a marked reduction in tumor number and size at 4 months following a single intraperitoneal injection of the carcinogen urethane. After urethane treatment, lung macrophages developed increased M1 macrophage marker expression during the first 2–3 weeks, followed by increased M2 marker expression by week 6. Using a strategy to reduce alveolar macrophages during tumor initiation and early promotion stages (week 1–2) or during late promotion and progression stages (week 4–16), we found significantly fewer and smaller lung tumors in both groups compared to controls. Late stage macrophage depletion reduced VEGF expression and impaired vascular growth in tumors. In contrast, early stage depletion of alveolar macrophages impaired urethane-induced nuclear factor κB (NF-κB) activation in the lungs and reduced the development of premalignant atypical adenomatous hyperplasia (AAH) lesions at 6 weeks after urethane injection. Together, these studies elucidate an important role for macrophages in lung tumor promotion and indicate that these cells have distinct roles during different stages of lung carcinogenesis.
INTRODUCTION
Lung cancer is the leading cause of cancer death in the United States and worldwide(1). Although genetic mutations underlie malignant transformation, the presence of mutations by themselves is not sufficient for tumor formation, and additional alterations are necessary for the development of cancer(2,3). Among the multiple factors initiating and supporting tumor growth, inflammation plays one of the most important roles. In normal conditions, inflammation generated by the innate immune response is essential for host defense, clearance of damaged or transformed cells, and maintenance of tissue homeostasis. However, in the presence of tumors, inflammation may contribute to growth and invasion through elaboration of enzymes that induce tissue damage, release of reactive oxygen and nitrogen species that result in additional mutations, and production of angiogenic factors and cytokines that support survival of malignant cells(4).
Among inflammatory cell types in the lungs, macrophages are the most abundant and perhaps the most pleiotropic. Depending on microenvironmental cues, these cells can stimulate inflammatory responses by secretion of pro-inflammatory cytokines or suppress immune responses by releasing high levels of anti-inflammatory cytokines like interleukin (IL)-10 and transforming growth factor (TGF) β(4–7). Based on the phenotypic spectrum of these cells, Mantovani and colleagues classified macrophages into two groups: “classically activated” proinflammatory M1 and “alternatively activated” anti-inflammatory M2. Most studies describing the M1/M2 macrophage paradigm in tumors have been performed during progression stages of established tumors or using metastasis models. Evidence regarding the role of macrophages during early tumorigenesis is lacking. In addition, it is unclear whether carcinogen treatment affects the M1/M2 phenotype of lung macrophages during early stages of tumor formation.
In the current study, we investigated the impact of macrophages during different stages of urethane-induced lung carcinogenesis. Using a method of minimally invasive long-term alveolar macrophage depletion, we found that elimination of macrophages with repetitive doses of liposomal clodronate significantly reduced lung carcinogenesis. Depletion of macrophages during late promotion and progression stages, when there was an increase in cells polarized towards the M2 phenotype, reduced tumor angiogenesis and growth. Interestingly, depletion of macrophages during initiation and early promotion stages, when M1 polarized cells were increased, also resulted in attenuation of tumorigenesis. Early macrophage depletion reduced urethane-induced NF-κB activity and development of premalignant atypical adenomatous hyperplasia (AAH) lesions. Together, these data demonstrate separate but important roles for macrophages in early and late stages of urethane-induced lung tumor formation.
MATERIALS AND METHODS
Animal experiments
All animal care and experimental procedures were approved and conducted according to guidelines issued by the Vanderbilt University Institutional Animal Care and Use Committee. Sex, weight and age matched 8–10 week old wild type and NF-κB reporter (NGL) mice(8) on a FVB background were used. Tumors were induced by a single intraperitoneal (IP) injection of 1 g/kg urethane (ethyl carbamate, Sigma-Aldrich). Mice were euthanized at time points up to 4 months after injection of carcinogen. At the time of sacrifice, lungs were lavaged, perfused and fixed in ice cold Bouin’s fixative solution (Sigma-Aldrich) for 24 hours. After fixation, lungs were used for surface tumor number and diameter measurements and embedded in paraffin. Tumors on the lung surface were enumerated by at least two experienced readers, blinded to sample identifiers under a dissecting microscope; tumor counts were averaged and statistically analyzed. Tumor diameters were measured using Fisherbrand Traceable digital calipers (Fisher Scientific).
Depletion of macrophages with clodronate
Clodronate (dichloromethylene diphosphonic acid, Sigma-Aldrich) or sterile phosphate buffered saline (PBS)-containing liposomes were prepared as previously described(9). Liposomes were instilled intratracheally weekly for 4 months starting the day of urethane injection (4 month); on Days 0 and 7 after the injection of carcinogen for the depletion of macrophages during initiation and early promotion stages (early stage), and weekly starting at 4 weeks post-urethane for late promotion and progression stage macrophage depletion experiment (late stage). For liposome instillation, mice were anesthetized and intubated using a 1 ml syringe with a 6-mm-long, 22-gauge over-the-needle catheter (Abbocath-T, Venisystems) to inject intratracheally (IT) 100 µl of liposomal clodronate or vehicle control (PBS) liposomes.
Bronchoalveolar lavage (BAL)
BAL with total and differential cell counting was performed as previously described (10).
Bioluminescent Imaging
For in vivo imaging, NF-κB reporter (NGL) mice(8) were anesthetized, injected retro-orbitally with 1 mg of D-luciferin (Biosynth AG) in 100 µl isotonic saline and imaged using an IVIS cooled charged coupled device (Xenogen Corporation) as described previously(8,11). Data were collected and analyzed using Living Image v.2.50 (Xenogen Corporation) and IgorPro (Wavemetrics) software.
Tissue luciferase assay
Lungs were removed en bloc and homogenized in 1 ml of lysis buffer (Promega). After pulse centrifugation, luciferase activity was measured in a Monolight 3010 Luminometer (Analytical Luminescence Laboratory) after adding 100 µl of freshly reconstituted luciferase assay buffer to 20 µl of lung tissue homogenate. Results were expressed as relative light units (RLU) normalized for protein content, which was measured by Bradford assay (Bio-Rad).
Histology and immunohistochemistry
Fixed lung samples were embedded in paraffin, sectioned (5 µm), stained with H&E, and analyzed by a pathologist blinded to the experimental groups for evaluation of tumor and atypical adenomatous hyperplasia (AAH) lesions in three separate sections cut at predetermined depths. GFP immunostaining was performed as previously described(12). For arginase-1 localization, lung sections were immunostained with diaminobenzidene-peroxidase detection reagents using rabbit anti-arginase-1 antibody (clone H-52; Santa Cruz Biotechnology, Santa Cruz, CA). To evaluate tumor-infiltrating vessel density, lung sections were immunostained with rat anti-mouse CD34 antibodies (clone MEC14.7; BioLegend). Image analysis of digital photomicrographs was performed using Image-Pro Plus software (Media Cybernetics, Bethesda, MD).
Isolation of lung cells and flow cytometry
To obtain single cell suspensions, perfused lungs were digested in RPMI medium containing collagenase XI (0.7 mg/ml; Sigma-Aldrich) and type IV bovine pancreatic DNase (30 µg/ml; Sigma-Aldrich). After treatment with RBC Lysis Buffer (BioLegend), single cell suspensions were stained with antibodies: CD45-Alexa Fluor 700 (30-F11, E-Bioscience), and CD68-FITC (FA-11), CD206-PE (MR5D3), CD204-Alexa Fluor 647 (2F8), from AbD Serotec, CD86 PE-Cy7 (GL1), CD11b (M1/70) and Gr1 (RB6-8C5) from BD Bioscience. Flow cytometry was performed using BD LSR II flow cytometer (BD Bioscience) and data were analyzed with FlowJo software (TreeStar). For isolation of CD11b positive cells, lungs were treated as described above followed by magnetic separation using anti-CD11b microbeads (Miltenyi biotec).
Real-time PCR
RNA from whole lung tissue or lung CD11b-positive cells was isolated using the RNeasy Mini kit (Qiagen, Valencia, CA). DNase-treated samples were subjected to Real-Time PCR using SYBR Green PCR Master Mix (Applied Biosystems). PCR primers used were: IL-12p35 For: 5’-TGGACCTGCCAGGTGTCTTAG-3’, Rev: 5’-CAATGTGCTGGTTTGGTCCC-3’; CCL3 For: 5’-TGCCCTTGCTGTTCTTCTCT- 3’, Rev: 5’–GATGAATTGGCGTGGAATCT-3’; IL-1β For: 5’-TTGACGGACCCCAAAAGATG-3’; Rev: 5’-CAGGACAGCCCAGGTCAAA-3’; Mannose receptor: For 5’-CAAGGAAGGTTGGCATTTGT-3’, Rev: 5’-CCTTTCAGTCCTTTGCAAGC-3’; Ym1 For: 5’-GGGCATACCTTTATCCTGAG-3’, Rev: 5’-CCACTGAAGTCATCCATGTC-3’; IL-10 For: 5’-ACCTGCTCCACTGCCTTGCT-3’, Rev: 5’-GGTTGCCAAGCCTTATCGGA-3’; TNFa For: 5’-AAGCCTGTAGCCCACGTCGTA-3’, Rev 5’-GGCACCACTAGTTGGTTGTCTTTG-3’; iNOS For: 5’-CACCTTGGAGTTCACCCAGT-3’, Rev: 5’-ACCACTCGTACTTGGGATGC-3’; IL-6 For: 5’-TCCTCTGGTCTTCTGGAGTA-3’, Rev: 5’-CTTAGCCACTCCTTCTGTGA-3’; GAPDH For: 5’-TGAGGACCAGGTTGTCTCCT-3’, Rev: 5’-CCCTGTTGCTGTAGCCGTAT-3’. Relative mRNA expression in each sample was normalized to GAPDH and presented using the comparative Ct method (2ΔCt).
VEGF measurement
Concentration of VEGF was assayed in cell-free BAL supernatants using ELISA (R&D Systems, Minneapolis, MN), according to the manufacturer’s instructions.
Statistical analysis
For studies comparing differences between two groups, we used unpaired Student's t-tests. For differences between more than two groups we used one-way ANOVA with an appropriate post test. Comparisons of chest photon counts in PBS or clodronate liposome-treated NGL mice were performed using two-way ANOVA with a Bonferroni post test. Values are presented as mean ± SEM. P < 0.05 was considered statistically significant.
RESULTS
Depletion of alveolar macrophages reduces lung tumorigenesis
To assess the role of macrophages in lung tumorigenesis, we developed a method of minimally invasive, long-term alveolar macrophage depletion using repetitive intratracheal (IT) instillation of clodronate encapsulated liposomes, which cause selective apoptosis of macrophages(11,13–20). Consistent with prior results(15), a single IT injection of clodronate caused a 95% reduction in macrophages obtained by BAL on day 3 with partial recovery of this cell population by 7 days (Figure S1A). Analysis of macrophage morphology revealed two different cell populations: intact and remnant cells. Remnant macrophages were identified as cells with a condensed, pyknotic nucleus that lack a clear cytoplasm or limiting cell membrane, indicative of dead (or dying) cells (Figure S1C). While clodronate injection reduced total macrophages by 95% on day 3, only 15% of the remaining macrophages in BAL were intact, suggesting a very low number of functional macrophages. This percentage of intact macrophages increased to 53% and 79% on days 5 and 7 respectively (Figure S1B). The number and morphology of BAL macrophages in mice treated with vehicle (PBS) containing liposomes remained unchanged.
Based on these results, we used weekly IT injections of liposomal clodronate to obtain long-term reduction in macrophage numbers during the course of urethane-induced lung tumorigenesis. In these experiments, mice received a single IP injection of urethane and were treated weekly with liposomal clodronate for four months starting the day of urethane injection. Urethane-injected mice treated with PBS-containing “empty” liposomes and mice treated with urethane alone were used as control groups. At the time of harvest (4 months after urethane and 6 days after the final clodronate dose), mice treated with clodronate exhibited a significant reduction in total BAL cells (Figure 1A). Total and intact macrophages were reduced by clodronate treatment, while the relatively small number of neutrophils and lymphocytes in BAL were similar between clodronate and PBS liposome treated animals (Figure S2A and B). Depletion of macrophages during the four months of lung carcinogenesis resulted in a marked reduction in the number of lung surface tumors in comparison to the control groups (Figure 1B). In addition, the mean diameter of surface tumors was significantly reduced after macrophage elimination (Figure 1C).
Figure 1.

Depletion of alveolar macrophages attenuates urethane-induced lung tumorigenesis. A) Number of total cells in BAL from mice injected with urethane only (Ureth) or treated with weekly intratracheal (IT) injections of empty (PBS) liposomes or clodronate (Clod) liposomes for 4 month after urethane. B) Number of lung surface tumors and C) tumor diameter in mouse lungs. D) Histological assessment of the number of tumors per H&E stained lung section from tumor bearing mice treated with weekly IT injections of PBS liposomes or clodronate liposomes for 4 months. Data are presented as mean ± SEM of 6 mice for the urethane only group, 6 mice for the PBS group, and 10 mice for the clodronate group, *= p<0.05 compared to the other groups.
To confirm the finding of fewer lung tumors in macrophage depleted mice, we performed histological examination of lungs sectioned at three predetermined depths. Microscopic evaluation of tumor appearance revealed both adenomas and AAH lesions (Figure S2C). Similar to surface tumors, the number of adenomas per lung section from urethane-injected mice was 3.6-fold lower after macrophage depletion (Figure 1D). At the same time, we found no differences in AAH lesions between clodronate or PBS liposome treated animals (4.39±0.50 vs 4.33±0.33 AAH lesions per H&E stained lung section respectively). Our results indicate that urethane-induced lung tumorigenesis is highly dependent on the presence of alveolar macrophages.
Induction of lung tumorigenesis by urethane changes the phenotype of lung macrophages
Because macrophage polarization has been implicated in modulation of tumor progression, we characterized the phenotype of lung macrophages following urethane treatment. We measured expression of CD204 (scavenger receptor) and CD206 (mannose receptor) as markers of the M2 phenotype in the population of CD45+CD68+ lung macrophages by flow cytometry (Figure 2A). As shown in Figure 2B, the percentage of macrophages positive for CD204 and CD206 was significantly decreased during the first two weeks after urethane injection, followed by an increase at 6 weeks. To confirm the shift in polarization of lung macrophages, we isolated CD11b-positive cells from lungs during weeks 1–6 after urethane injection and assessed mRNA expression for IL-12, CCL3, IL-1β as markers of M1 macrophages and mannose receptor, Ym1, and IL-10 as indicators of cells with M2 characteristics(7). We found upregulation of M1 markers on CD11b+ cells at week 2–3 after urethane, including increased IL-12 at weeks 2–3 and increased CCL3 and IL-1β at week 3 (Figure 2C–E). Expression of M2 markers mannose receptor and Ym1 were increased by week 6 post-urethane, but IL-10 was not changed at any time point (Figure 2F–H). To examine the distribution of M2 polarized macrophages in the lungs, we immunostained lung sections from mice harvested at different time intervals after urethane exposure using anti-arginase-1 antibodies. We chose arginase-1 as the M2 marker for these studies due to the availability of an antibody with good sensitivity for immunohistochemistry. We detected very few positively-stained macrophages in lungs from naïve mice (Figure 2J); however, we observed a large increase in arginase-1 positive cells at 6 weeks after carcinogen exposure (Figure 2K). Since arginase-1 can be expressed by myeloid-derived suppressor cells (MDSCs) as well as macrophages(21), we investigated whether an increase in increase in MDSCs (defined as CD11b+Gr1+ cells) could account for the increase in arginase-1 positive cells identified at 6 weeks after urethane injection. We did not detect a significant increase in MDSCs in the lungs at this time point (5.97±1.50% of total CD45+ leukocytes in urethane treated mice vs. 4.85±0.79% in naïve mice), supporting the conclusion that increased arginase-1 staining was related to increased numbers of M2 polarized macrophages. Together, these studies indicate a shift in macrophage phenotype following carcinogen treatment. Depending on the measurement used, there appears to be a shift towards M1 macrophage marker expression during the first 2–3 weeks after urethane treatment, followed by a shift towards increased M2 polarization by 6 weeks.
Figure 2.
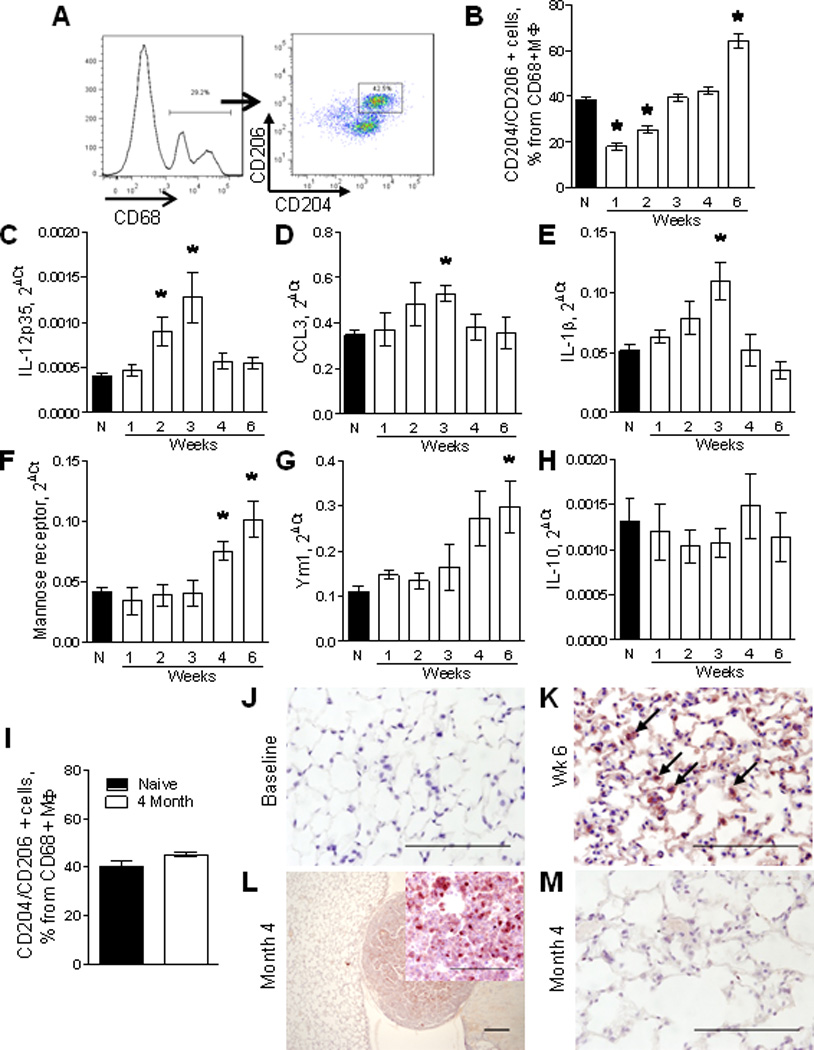
Characterization of macrophage phenotype in lungs following urethane treatment. A) Representative FACS plots demonstrating identification of M2 polarized macrophages in whole lungs of a naïve (untreated) mouse using anti-CD204 (scavenger receptor) and anti-CD206 (mannose receptor) antibodies. B) Analysis of the percentage of CD204 and CD206 double-positive cells within CD45+CD68+ macrophages in lungs of naïve mice (N) and mice at week 1–6 after urethane treatment. C–H) mRNA expression for M1 markers IL-12p35, CCL3, IL-1β and M2 markers mannose receptor, Ym1, IL-10 in total CD11b-positive cells isolated from lungs of naïve mice and 1–6 weeks after a single injection of urethane. n=4 mice per time point, *= p<0.05 compared to naïve mice. I) Percentage of CD204 and CD206 double-positive cells within lung CD45+CD68+ macrophages at 4 months after urethane injection. n=4 mice per group. J–M) Representative photomicrographs of lung sections stained with anti-arginase-1 antibodies at different time points after injection of urethane. Bars: 100µM.
At the time of lung tumor assessment (4 months after urethane injection), the percentage of lung macrophages expressing both CD204 and CD206 was not significantly increased in comparison to naïve mice (Figure 2I). Interestingly, macrophages positive for arginase-1 were found only within the tumor and peri-tumor areas, but not in normal-appearing alveoli (Figure 2L,M). These data suggest that in the presence of fully formed tumors, M2 polarized macrophages are limited to areas of tumor formation.
Elimination of alveolar macrophages during “early” and “late” stages of urethane-induced tumorigenesis reduces lung tumor formation
Based on information regarding timing of macrophage polarization described in the previous section, we sought to determine the role of macrophages in tumor formation during early (M1 predominant) and late (M2 predominant) time periods. Therefore, we treated mice with clodronate liposomes during the first two weeks post-urethane for “early stage” macrophage depletion or starting at four weeks post-urethane for “late stage” macrophage depletion (Figure 3A). Animals were sacrificed at four months post urethane. As expected, elimination of macrophages during the “late stage” significantly reduced the numbers of macrophages in BAL at four months (Figure 3B,C). These changes were accompanied by a decrease in number and diameter of lung surface tumors (Figure 3D,E). Histopathological examination of lungs sections confirmed the reduced numbers of tumors after depletion of macrophages during the “late stage” (Figure 3F), with no difference in the number of AAH lesions compared to the PBS liposome group (Figure 3G). These findings were similar to those obtained from experiments in which macrophages were depleted during the entire four months. However, to our surprise, we found that elimination of macrophages during the “early stage” had similar effects on BAL cellularity, the number and size of surface tumors, and histological assessment of lung tumors as compared to “late stage” macrophage depletion (Figure 3B–G). While the numbers and sizes of tumors after “early stage” macrophage depletion were slightly greater than in animals having depletion of cells during “late stage” tumorigenesis or during the entire 4 month experiment, the differences were not statistically significant. Together, our findings demonstrate that macrophages are required for urethane-induced lung carcinogenesis during both “early” and “late” stages and elimination of macrophages during either period markedly attenuates tumor formation.
Figure 3.

Depletion of lung macrophages during “early” and “late” phases of lung tumorigenesis reduces tumor number and size. A) Schematic representation of “early” and “late” stage macrophage depletion experiment. B) The number of total BAL cells and C) alveolar macrophages in BAL of urethane-injected mice treated with clodronate or PBS liposomes. D) The number of lung surface tumors, E) surface tumor diameter, F) tumor number per lung section, and G) AAH lesions per H&E stained lung section. Data are presented as mean ± SEM of 12 mice for the PBS group (early and late PBS control groups are combined) and 11 mice for each clodronate group, *= p<0.05 compared to PBS control group.
Depletion of lung macrophages during later stages of lung carcinogenesis reduces vascular growth in tumors
Because macrophages represent a primary source of pro-angiogenic proteins in the tumor microenvironment(22–24), we measured the concentration of VEGF in BAL from control and macrophage depleted mice. The level of VEGF in BAL of urethane-injected mice was reduced by depletion of macrophages during “late” carcinogenesis and by depletion of macrophages throughout the experiment (Figure 4A). We also assessed tumor angiogenesis by immunostaining for an endothelial cell marker of new blood vessels, CD34(25). Although the number of blood vessels infiltrating tumors was not significantly different in macrophage depletion groups compared to controls, the total vessel area in tumors was significantly reduced in both macrophage depletion groups compared to the PBS liposome-treated control group (Figure 4B–D). These data suggest that in our model macrophages are not required for the appearance of new tumor-infiltrating blood vessels, but rather are critical for supporting the growth of new and existing vessels.
Figure 4.
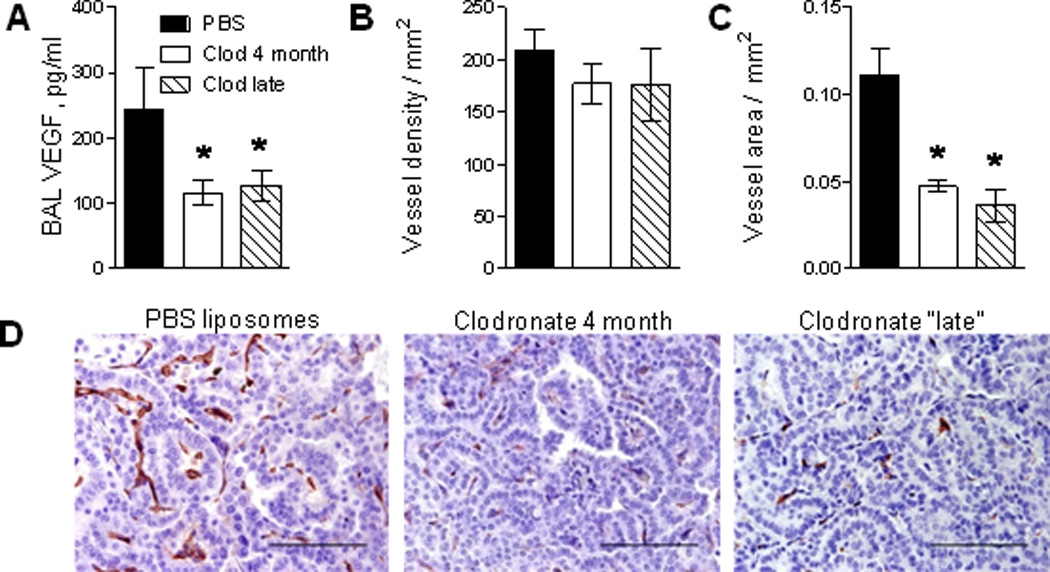
Depletion of alveolar macrophages reduces tumor angiogenesis. A) VEGF concentration in BAL from mice treated with weekly IT clodronate beginning at 4 weeks post-urethane (late stage) or throughout the 4 months period compared to control mice treated with PBS liposomes. B) Density of blood vessels in tumors and C) blood vessel area in tumors. The blood vessel density (or area) was assessed as the number (or area) of CD34+ endothelial cells per square millimeter of tumor. C) Representative photomicrographs of lung sections from tumor bearing mice immunostained for CD34. Bars: 100µM. N=10–12 mice per group, *= p<0.05 compared to PBS liposome controls.
Depletion of alveolar macrophages during early carcinogenesis blocks NF-κB activation and reduces AAH lesions
In a previous study, we demonstrated an important role for the NF-κB signaling pathway in urethane-induced lung carcinogenesis(10). In that study, we found that NF-κB was transiently activated in epithelial cells and macrophages of tumor-prone mouse strains. Here, we utilized NF-κB reporter mice (called NGL, FVB strain background) in which a synthetic NF-κB dependent promoter drives the expression of a GFP-luciferase fusion protein(8). NGL mice were injected with urethane and IT clodronate or PBS liposomes on day 0 followed by weekly treatments for four months. Repetitive noninvasive bioluminescent imaging of luciferase activity was performed immediately prior to each treatment with liposomes. Measurement of photon emission from the chest of mice treated with PBS liposomes revealed increased NF-κB-dependent luciferase activation during early stages of carcinogenesis with maximal values on day 14 (Figure 5A, B). Depletion of alveolar macrophages with clodronate substantially attenuated the increased NF-κB activity observed between days 7–21. Analysis of luciferase activity in lung homogenates confirmed a significant attenuation of NF-κB activity after clodronate treatment (Figure 5C). To confirm that these differences reflected lung specific NF-κB activation, we treated NGL mice with clodronate or PBS liposomes on days 0 and 7 after injection of urethane and harvested lungs on day 14. In this experiment, treatment with clodronate significantly reduced total BAL cells and alveolar macrophages (Figure S3A,B). On lung sections from these mice, immunohistochemistry for GFP showed a marked reduction in NF-κB-dependent GFP expression in the entire tissue section, particularly in airway epithelium (Figure 5D). The finding of attenuated NF-κB activity in airway epithelial cells after clodronate injection supports the conclusion that elimination of macrophages impairs pro-inflammatory signaling between macrophages and epithelial cells, thus reducing urethane-induced NF-κB activity in the entire lung compartment.
Figure 5.

Depletion of macrophages with liposomal clodronate attenuates activity of NF-κB and alters inflammatory mediator expression in the lungs after urethane. A) Time course of NF-κB-dependent lung bioluminescence in NF-κB reporter mice (NGL) treated weekly with PBS or clodronate liposomes. n=6 for PBS liposome and 10 for clodronate groups, *=p<0.05 compared to PBS liposome group at the same time point. B) Representative images for NF-κB-dependent lung bioluminescence, C) NF-κB-dependent luciferase activity (Relative light units, RLU) in lung homogenates, and D) representative photomicrographs of lung sections immunostained for GFP (Bars: 100µM., arrows point to GFP+ cells) at day 14 after urethane injection in mice treated with PBS or clodronate liposomes, n=5 per group. E–I) Expression of mRNA for IL-12p35, iNOS, IL-6, TNFα, and IL-10 in lung tissue at day 11 after urethane injection in mice treated with PBS or clodronate liposomes (n=5 per group, *=p<0.05). Animals were given PBS or clodronate liposomes on days 0 and 7 after injection of urethane.
To investigate the impact of macrophage depletion on inflammatory mediator production in the lungs, we measured mRNA expression of several inflammatory mediators in the lungs after treatment with clodronate or PBS liposomes on days 0 and 7. At day 11 post-urethane, IL-12 and iNOS expression were reduced after clodronate treatment and IL-6 showed a trend towards reduced expression (Figure 5E–G). In contrast, expression of TNFα and IL-10 was unchanged compared to the PBS liposome treated control group (Figure 5H,I). Although the precise mechanisms by which macrophages promote early stage tumorigenesis are uncertain, inflammatory mediator production may contribute to a pro-tumorigenic microenvironment.
We then asked whether “early” macrophage depletion and impaired NF-κB activation would result in a reduction of AAH lesions at 6 weeks post-urethane treatment, a time point at which AAH lesions become detectable in this model(26). In these studies, we depleted macrophages with clodronate on days 0 and 7 after urethane injection (week 1 and 2, “early stage”) and on week 4 and 5 (“late stage”) and harvested mice at week 6 (Figure 6A). As shown in Figure 6B, the number of AAH lesions was markedly reduced in mice following “early stage” macrophage depletion compared to mice with “late stage” macrophage depletion and mice treated with PBS liposomes. Thus, our data indicate that early macrophage depletion impairs NF-κB activation and early AAH formation, likely resulting in fewer tumors at 4 months.
Figure 6.

Depletion of alveolar macrophages during “early stage” lung carcinogenesis reduces formation of atypical adenomatous hyperplasia (AAH) lesions. A) Schematic representation of macrophage depletion experiment. B) Mean number of AAH lesions for each mouse counted per H&E stained lung section (3 per mouse) from mice treated with PBS liposomes or clodronate at day 0 and 7 (week 1,2) or week 4 and 5 and harvested at week 6 after injection of urethane (n=5 mice per group, *=p<0.05 compared to PBS liposome group). C) Total BAL cells and D) differential cell counts at week 6 from untreated mice (Naïve), mice treated with clodronate liposomes alone on day 0 and 7 and urethane-treated mice injected with clodronate or PBS liposomes on day 0 and 7, n=4–5 per group, *=p<0.05 compared to WT naive group.
In a separate experiment, BAL was performed on untreated control mice, mice treated with clodronate alone on day 0 and 7, and urethane-treated mice injected with clodronate or PBS liposomes on day 0 and 7. At 6 weeks after initiation of treatment, depletion of macrophages during the “early stage” after urethane injection resulted in similar numbers of total BAL cells compared to untreated (naïve) mice and mice treated with clodronate alone (Figure 6C). However, mice treated with PBS liposomes in the “early stage” after urethane injections had increased BAL macrophages compared to the other groups (Figure 6D). These results suggest that the long term reduction in lung macrophage numbers observed in mice subjected to the “early stage” macrophage depletion protocol following urethane (compared to urethane-treated controls) was not due to prolonged effects of liposomal clodronate. Rather, we suspect that the prolonged reduction in BAL macrophages following “early stage” macrophage depletion resulted from attenuation of the initial inflammatory response and subsequent differences in tumor multiplicity.
DISCUSSION
These studies demonstrate that macrophages are essential for promotion of carcinogen-induced lung tumors during both “early” (tumor initiation and early promotion) and “late” (late promotion and progression) stages of tumorigenesis. Although depletion of alveolar macrophages during either early or later stages reduces tumor size and multiplicity, the mechanisms involved appear to be different. Later stage macrophage depletion decreases VEGF concentration in BAL and reduces vascular growth in lung tumors. In contrast, early stage macrophage depletion, prior to the onset of M2 polarization, reduces urethane-induced NF-κB activation in the lungs, alters inflammatory mediator expression, and impairs formation of pre-malignant AAH lesions. Together, these studies indicate that macrophages support development and maintenance of a pro-tumorigenic microenvironment in the lungs in a variety of ways. As a result, a sustained presence of macrophages throughout the course of tumorigenesis is required for maximal tumor formation.
We and others previously demonstrated that macrophages can support tumor growth in a variety of animal models, including lung cancer, melanoma, breast cancer, and ovarian cancer models(11,13,14,16,17,19,20). In addition to animal model data, there is a strong correlation between macrophage density in tumors, microvessel counts, and relapse-free survival in humans with lung cancer(27). Importantly, prior studies have been performed using established cancer cell lines and metastasis models, or were carried out during progression stages when tumors were already established. Little is known regarding the role of macrophages in lung tumor promotion, but the prevailing opinion has been that during tumor initiation and early promotion macrophages primarily function to kill and remove malignant clones through their cytotoxic and phagocytic activities. In contrast, our studies indicate that macrophages have important pro-tumorigenic functions within the first few weeks after urethane injection. Early stage macrophage depletion by clodronate injection on day 0 and day 7 post-urethane almost completely eliminated AAH lesions in the lung at 6 weeks after urethane treatment, indicating a critical role for macrophages in early tumor promotion. Our findings suggest that macrophages promote tumorigenesis by enhancing survival and/or proliferation of mutated cells in the first few weeks after urethane treatment.
In these studies, we found that elimination of macrophages blocks NF-κB activation in the lungs of urethane-treated mice, which occurs transiently during the first 2–3 weeks after urethane treatment(10). We previously reported that urethane induces tumors only in mouse strains where NF-κB is activated and that inhibition of NF-κB in airway epithelial cells reduces lung tumorigenesis(10). These findings were recently corroborated and expanded by other reports demonstrating a requirement for NF-κB signaling in tobacco-smoke and oncogenic Kras-induced lung carcinogenesis(28–30). In addition, mice with impaired NF-κB activation in myeloid cells showed reduced tumor size and multiplicity in a lung tumor model in which mutant Kras expressing mice were exposed to tobacco smoke(28). It has been suggested that chronic inhibition of NF-κB signaling in tumor associated macrophages switches them to an activated phenotype that is cytotoxic to tumor cells(31). Together, available data indicate that the NF-κB pathway is an important regulator of lung tumorigenesis in mouse models and may play cell specific roles in epithelial and myeloid cells. Our studies contribute to this emerging picture by showing that cross-talk between macrophages and epithelial cells is required for NF-κB activation in lung epithelium after urethane exposure. As we have previously shown, NF-κB signaling impacts survival of epithelial cells after urethane treatment through expression of anti-apoptotic mediators like Bcl-2(10). Therefore, we speculate that early macrophage depletion impacts tumorigenesis by depriving epithelial cells of crucial survival and proliferation signals via NF-κB pathway signaling.
Classically activated M1 macrophages have been reported to play important roles in host defense and tumor resistance(7,22,32). In lungs, M1 polarization of macrophages has been associated with prolonged survival time in patients with non-small cell lung cancer(33–35). Despite reported anti-tumor roles for M1 polarized macrophages, persistent activation of these cells may lead to tissue damage and thereby support malignant transformation(5,16,36). Pro-inflammatory cytokines produced by M1 macrophages such as IL-6 and TNFα have been reported to protect initiated epithelial cells from apoptosis and promote the proliferation of these cells(37–40). Other inflammatory cells recruited by macrophages, particularly neutrophils, can impact malignant transformation of epithelial cells through release of reactive oxygen and nitrogen species(5,16,41,42). Our studies support the concept that the overall impact of macrophages is pro-tumorigenic throughout the entire course of carcinogen-induced tumor formation.
During the course of lung tumor development, lung macrophages shift their polarization to the M2 phenotype, particularly in and around tumors. Redente and colleagues demonstrated a shift in polarization of lung macrophages from M1 towards M2 at 3 weeks after urethane exposure in A/J mice(43). In FVB mice, we found an increase in M1 polarized macrophages during the first 2–3 weeks after urethane followed by an increase in M2 cells by 6 weeks post-urethane. Therefore, reduction of macrophages during later stages of tumor development has a disproportional effect on cells with M2 characteristics. In contrast to M1 polarized macrophages, alternatively activated (M2) polarized macrophages have been shown to support tumor growth(4,7,22,44). Mice lacking IFNγ exhibit larger lung tumors due to M2 macrophage polarization, whereas IL-4R knockout mice have smaller tumors(44). Since M2 polarized macrophages are known to secrete pro-angiogenic cytokines, these cells can support tumor progression through increased vascularization of neoplastic lesions. Based on this concept, M2 polarization of macrophages has been suggested as an important prognostic factor for many tumors, including lung cancer(4,7,22,32,34,35). Although the level of VEGF was lower in BAL from macrophage depleted mice in our studies compared to controls, the density of microvessels within the tumors was not decreased. However, elimination of alveolar macrophages significantly reduced the size of vessels in lung tumors. These data suggest that in the urethane model alveolar macrophages are dispensable for formation of new blood vessels in tumors but are required for the growth of new and existing vessels. Thus, an important pro-tumorigenic aspect of lung macrophages appears to be modulation of vascular growth.
Macrophages are very plastic cells and depending on the microenviromental cues may exhibit diverse functions(45). We identified macrophages phenotype as M1 or M2 based on FACS analysis, mRNA expression for a panel of markers, and immunohistochemistry for arginase-1. Although we found phenotypic differences in macrophages over time in response to urethane injection, a diverse population of macrophages was present in the lungs at each time point studied. In addition, partial macrophage phenotypes that do not fit neatly within the M1/M2 paradigm and cells that express low levels of both M1 and M2 markers appear to be common. So while it is tempting to try to explain the role and function of cell types based on simple constructs, global interventions involving macrophages or other complex cell types impact cells with a spectrum of functions and characteristics. Therefore, these interventions are likely to result in complex changes to the local microenvironment.
Accumulating evidence supports a strong association between airway inflammation and lung cancer, particularly in patients with chronic obstructive pulmonary disease (COPD)(46). Retrospective studies have suggested that treatment with anti-inflammatory drugs such as corticosteroids reduce the incidence of lung cancer, supporting an important role for inflammation in lung tumorigenesis(47). Macrophages are prominently increased in the airways of COPD patients(27); therefore, these cells may be involved in mediating increased lung cancer risk in these individuals. Since our studies suggest that macrophages may be more uniformly pro-tumorigenic than previously appreciated, macrophages and their products may be important targets for future chemoprevention and treatment studies to reduce the incidence of lung cancer and improve the clinical outcome and quality of life for patients.
Supplementary Material
Acknowledgments
This work was supported by Department of Veterans Affairs (T.S.B), Vanderbilt Cancer Center Support Grant 2010 (T.S.B), National Institutes of Health Grants 5T32HL094296-03 (R.Z.) and CA113734 (F.E.Y).
Reference List
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J. Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Coussens LM, Werb Z. Inflammatory cells and cancer: think different! J. Exp. Med. 2001;193:F23–F26. doi: 10.1084/jem.193.6.f23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 4.Gordon S. Alternative activation of macrophages. Nat. Rev. Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 5.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin WW, Karin M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J. Clin. Invest. 2007;117:1175–1183. doi: 10.1172/JCI31537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23:549–555. doi: 10.1016/s1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- 8.Everhart MB, Han W, Sherrill TP, Arutiunov M, Polosukhin VV, Burke JR, Sadikot RT, Christman JW, Yull FE, Blackwell TS. Duration and intensity of NF-kappaB activity determine the severity of endotoxin-induced acute lung injury. J. Immunol. 2006;176:4995–5005. doi: 10.4049/jimmunol.176.8.4995. [DOI] [PubMed] [Google Scholar]
- 9.Everhart MB, Han W, Parman KS, Polosukhin VV, Zeng H, Sadikot RT, Li B, Yull FE, Christman JW, Blackwell TS. Intratracheal administration of liposomal clodronate accelerates alveolar macrophage reconstitution following fetal liver transplantation. J. Leukoc. Biol. 2005;77:173–180. doi: 10.1189/jlb.1203647. [DOI] [PubMed] [Google Scholar]
- 10.Stathopoulos GT, Sherrill TP, Cheng DS, Scoggins RM, Han W, Polosukhin VV, Connelly L, Yull FE, Fingleton B, Blackwell TS. Epithelial NF-kappaB activation promotes urethane-induced lung carcinogenesis. Proc. Natl. Acad. Sci. U. S. A. 2007;104:18514–18519. doi: 10.1073/pnas.0705316104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stathopoulos GT, Sherrill TP, Han W, Sadikot RT, Yull FE, Blackwell TS, Fingleton B. Host nuclear factor-kappaB activation potentiates lung cancer metastasis. Mol. Cancer Res. 2008;6:364–371. doi: 10.1158/1541-7786.MCR-07-0309. [DOI] [PubMed] [Google Scholar]
- 12.Connelly L, Robinson-Benion C, Chont M, Saint-Jean L, Li H, Polosukhin VV, Blackwell TS, Yull FE. A transgenic model reveals important roles for the NF-kappa B alternative pathway (p100/p52) in mammary development and links to tumorigenesis. J. Biol. Chem. 2007;282:10028–10035. doi: 10.1074/jbc.M611300200. [DOI] [PubMed] [Google Scholar]
- 13.Gazzaniga S, Bravo AI, Guglielmotti A, van Rooijen N, Maschi F, Vecchi A, Mantovani A, Mordoh J, Wainstok R. Targeting tumor-associated macrophages and inhibition of MCP-1 reduce angiogenesis and tumor growth in a human melanoma xenograft. J. Invest Dermatol. 2007;127:2031–2041. doi: 10.1038/sj.jid.5700827. [DOI] [PubMed] [Google Scholar]
- 14.Halin S, Rudolfsson SH, van Rooijen N, Bergh A. Extratumoral macrophages promote tumor and vascular growth in an orthotopic rat prostate tumor model. Neoplasia. 2009;11:177–186. doi: 10.1593/neo.81338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koay MA, Gao X, Washington MK, Parman KS, Sadikot RT, Blackwell TS, Christman JW. Macrophages are necessary for maximal nuclear factor-kappa B activation in response to endotoxin. Am. J. Respir. Cell Mol. Biol. 2002;26:572–578. doi: 10.1165/ajrcmb.26.5.4748. [DOI] [PubMed] [Google Scholar]
- 16.Luo YP, Zhou H, Krueger J, Kaplan C, Liao D, Markowitz D, Liu C, Chen T, Chuang TH, Xiang R, Reisfeld RA. The role of proto-oncogene Fra-1 in remodeling the tumor microenvironment in support of breast tumor cell invasion and progression. Oncogene. 2010;29:662–673. doi: 10.1038/onc.2009.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meng Y, Beckett MA, Liang H, Mauceri HJ, van Rooijen N, Cohen KS, Weichselbaum RR. Blockade of tumor necrosis factor alpha signaling in tumor-associated macrophages as a radiosensitizing strategy. Cancer Res. 2010;70:1534–1543. doi: 10.1158/0008-5472.CAN-09-2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mircescu MM, Lipuma L, van Rooijen N, Pamer EG, Hohl TM. Essential role for neutrophils but not alveolar macrophages at early time points following Aspergillus fumigatus infection. J. Infect. Dis. 2009;200:647–656. doi: 10.1086/600380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robinson-Smith TM, Isaacsohn I, Mercer CA, Zhou M, van Rooijen N, Husseinzadeh N, McFarland-Mancini MM, Drew AF. Macrophages mediate inflammation-enhanced metastasis of ovarian tumors in mice. Cancer Res. 2007;67:5708–5716. doi: 10.1158/0008-5472.CAN-06-4375. [DOI] [PubMed] [Google Scholar]
- 20.Zeisberger SM, Odermatt B, Marty C, Zehnder-Fjallman AH, Ballmer-Hofer K, Schwendener RA. Clodronate-liposome-mediated depletion of tumour-associated macrophages: a new and highly effective antiangiogenic therapy approach. Br. J. Cancer. 2006;95:272–281. doi: 10.1038/sj.bjc.6603240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Condamine T, Gabrilovich DI. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. 2011;32:19–25. doi: 10.1016/j.it.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Allavena P, Sica A, Solinas G, Porta C, Mantovani A. The inflammatory micro-environment in tumor progression: the role of tumor-associated macrophages. Crit Rev. Oncol. Hematol. 2008;66:1–9. doi: 10.1016/j.critrevonc.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 23.Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124:263–266. doi: 10.1016/j.cell.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 24.Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer. 2004;4:71–78. doi: 10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- 25.Carothers AM, Davids JS, Damas BC, Bertagnolli MM. Persistent cyclooxygenase-2 inhibition downregulates NF-{kappa}B, resulting in chronic intestinal inflammation in the min/+ mouse model of colon tumorigenesis. Cancer Res. 2010;70:4433–4442. doi: 10.1158/0008-5472.CAN-09-4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O'Donnell EP, Zerbe LK, Dwyer-Nield LD, Kisley LR, Malkinson AM. Quantitative analysis of early chemically-induced pulmonary lesions in mice of varying susceptibilities to lung tumorigenesis. Cancer Lett. 2006;241:197–202. doi: 10.1016/j.canlet.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 27.Chen JJ, Lin YC, Yao PL, Yuan A, Chen HY, Shun CT, Tsai MF, Chen CH, Yang PC. Tumor-associated macrophages: the double-edged sword in cancer progression. J. Clin. Oncol. 2005;23:953–964. doi: 10.1200/JCO.2005.12.172. [DOI] [PubMed] [Google Scholar]
- 28.Takahashi H, Ogata H, Nishigaki R, Broide DH, Karin M. Tobacco smoke promotes lung tumorigenesis by triggering IKKbeta- and JNK1-dependent inflammation. Cancer Cell. 2010;17:89–97. doi: 10.1016/j.ccr.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Basseres DS, Ebbs A, Levantini E, Baldwin AS. Requirement of the NF-kappaB subunit p65/RelA for K-Ras-induced lung tumorigenesis. Cancer Res. 2010;70:3537–3546. doi: 10.1158/0008-5472.CAN-09-4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meylan E, Dooley AL, Feldser DM, Shen L, Turk E, Ouyang C, Jacks T. Requirement for NF-kappaB signalling in a mouse model of lung adenocarcinoma. Nature. 2009;462:104–107. doi: 10.1038/nature08462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hagemann T, Lawrence T, McNeish I, Charles KA, Kulbe H, Thompson RG, Robinson SC, Balkwill FR. "Re-educating" tumor-associated macrophages by targeting NF-kappaB. J. Exp. Med. 2008;205:1261–1268. doi: 10.1084/jem.20080108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mantovani A, Sica A. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr. Opin. Immunol. 2010;22:231–237. doi: 10.1016/j.coi.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 33.Taniguchi H, Shimada Y, Sawachi K, Hirota K, Inagawa H, Kohchi C, Soma G, Makino K, Terada H. Lipopolysaccharide-activated alveolar macrophages having cytotoxicity toward lung tumor cells through cell-to-cell binding-dependent mechanism. Anticancer Res. 2010;30:3159–3165. [PubMed] [Google Scholar]
- 34.Ma J, Liu L, Che G, Yu N, Dai F, You Z. The M1 form of tumor-associated macrophages in non-small cell lung cancer is positively associated with survival time. BMC. Cancer. 2010;10:112. doi: 10.1186/1471-2407-10-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ohri CM, Shikotra A, Green RH, Waller DA, Bradding P. Macrophages within NSCLC tumour islets are predominantly of a cytotoxic M1 phenotype associated with extended survival. Eur. Respir. J. 2009;33:118–126. doi: 10.1183/09031936.00065708. [DOI] [PubMed] [Google Scholar]
- 36.Erez N, Truitt M, Olson P, Arron ST, Hanahan D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-kappaB-Dependent Manner. Cancer Cell. 2010;17:135–147. doi: 10.1016/j.ccr.2009.12.041. [DOI] [PubMed] [Google Scholar]
- 37.Balkwill F. Tumour necrosis factor and cancer. Nat. Rev. Cancer. 2009;9:361–371. doi: 10.1038/nrc2628. [DOI] [PubMed] [Google Scholar]
- 38.Moore RJ, Owens DM, Stamp G, Arnott C, Burke F, East N, Holdsworth H, Turner L, Rollins B, Pasparakis M, Kollias G, Balkwill F. Mice deficient in tumor necrosis factor-alpha are resistant to skin carcinogenesis. Nat. Med. 1999;5:828–831. doi: 10.1038/10552. [DOI] [PubMed] [Google Scholar]
- 39.Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, Karin M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317:121–124. doi: 10.1126/science.1140485. [DOI] [PubMed] [Google Scholar]
- 40.Popivanova BK, Kitamura K, Wu Y, Kondo T, Kagaya T, Kaneko S, Oshima M, Fujii C, Mukaida N. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J. Clin. Invest. 2008;118:560–570. doi: 10.1172/JCI32453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Biswas SK, Sica A, Lewis CE. Plasticity of macrophage function during tumor progression: regulation by distinct molecular mechanisms. J. Immunol. 2008;180:2011–2017. doi: 10.4049/jimmunol.180.4.2011. [DOI] [PubMed] [Google Scholar]
- 42.Pang B, Zhou X, Yu H, Dong M, Taghizadeh K, Wishnok JS, Tannenbaum SR, Dedon PC. Lipid peroxidation dominates the chemistry of DNA adduct formation in a mouse model of inflammation. Carcinogenesis. 2007;28:1807–1813. doi: 10.1093/carcin/bgm037. [DOI] [PubMed] [Google Scholar]
- 43.Redente EF, Orlicky DJ, Bouchard RJ, Malkinson AM. Tumor signaling to the bone marrow changes the phenotype of monocytes and pulmonary macrophages during urethane-induced primary lung tumorigenesis in A/J mice. Am. J. Pathol. 2007;170:693–708. doi: 10.2353/ajpath.2007.060566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Redente EF, Dwyer-Nield LD, Barrett BS, Riches DW, Malkinson AM. Lung tumor growth is stimulated in IFN-gamma−/− mice and inhibited in IL-4Ralpha−/− mice. Anticancer Res. 2009;29:5095–5101. [PMC free article] [PubMed] [Google Scholar]
- 45.Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat. Immunol. 2010;11:889–896. doi: 10.1038/ni.1937. [DOI] [PubMed] [Google Scholar]
- 46.Punturieri A, Szabo E, Croxton TL, Shapiro SD, Dubinett SM. Lung cancer and chronic obstructive pulmonary disease: needs and opportunities for integrated research. J. Natl. Cancer Inst. 2009;101:554–559. doi: 10.1093/jnci/djp023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kiri VA, Fabbri LM, Davis KJ, Soriano JB. Inhaled corticosteroids and risk of lung cancer among COPD patients who quit smoking. Respir. Med. 2009;103:85–90. doi: 10.1016/j.rmed.2008.07.024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.