

Abstract
A detailed EPR and computational study of a key paramagnetic form of xanthine oxidase (XO) has been performed which serves as a basis for developing a valence bond description of C-H activation and transition state stabilization along the reaction coordinate with aldehyde substrates. EPR spectra of aldehyde Inhibited XO have been analyzed in order to provide information regarding the relationship between the g-, 95,97Mo hyperfine (AMo), and the 13C hyperfine (AC) tensors. The analysis of the EPR spectra have allowed for greater insight into the electronic origin of key delocalizations within the Mo-Oeq-C fragment, and how these contribute to C-H bond activation/cleavage and transition state (TS) stabilization. A natural bond orbital analysis of the enzyme reaction coordinate with aldehyde substrates shows that both Mo=S π→C-H σ* (ΔE= 24.3 kcal/mol) and C-H σ → Mo=S π* (ΔE = 20.0 kcal/mol) back donation are important in activating the substrate for C-H bond for cleavage. Additional contributions to C-H activation derive from Oeq lp→C-H σ* (lp = lone pair; ΔE = 8.2 kcal/mol), and S lp→C-H σ* (ΔE = 13.2 kcal/mol) stabilizing interactions. The Oeq donor ligand that derives from water is part of the Mo-Oeq-C fragment probed in the EPR spectra of XO Inhibited, and the observation of Oeq lp→C-H σ* back donation indicates a key role for the Oeq in activating the substrate C-H bond for cleavage. We also show that the Oeq donor plays an even more important role in transition state (TS) stabilization. We find that Oeq→(Mo + C) charge transfer dominantly contributes to stabilization of the TS (ΔE = 89.5 kcal/mol) and the Mo-Oeq-C delocalization pathway reduces strong electronic repulsions that contribute to the classical TS energy barrier. The Mo-Oeq-C delocalization at the TS allows for the TS to be described in valence bond terms as a resonance hybrid of the reactant (R) and product (P) valence bond wavefunctions.
Keywords: Hydroxylation, molybdenum enzymes, xanthine oxidase, aldehyde oxidase hydride transfer, electron paramagnetic resonance, EPR spectroscopy, natural bond orbital
INTRODUCTION
Xanthine oxidoreductase (XO) is a molybdenum hydroxylase and the prototypical member of the XO pyranopterin molybdenum enzyme family.1,2 XO catalyzes the oxidative hydroxylation of a wide variety of substrates, including purines and other aromatic heterocycles, aldehydes, and formamide by formal oxygen atom insertion into the substrate C-H bond. A key difference between the molybdenum hydroxylases and monooxygenase enzymes is that the inserted oxygen atom derives from metal-activated water instead of dioxygen, and reducing equivalents are generated rather than consumed in the reductive half reaction.3 Both XO and the structurally related aldehyde oxidase (AO) have been implicated in pro-drug activation and drug metabolism.4–9 Although little is known regarding the pathophysiological relevance of AO,10 the enzyme catalyzes the reduction of sulfa drugs, activates anticancer prodrugs,11 and has recently been shown to metabolize famciclovir to the potent antiviral penciclovir,12,13 which has been found to be effective against such viral infections as herpes simplex (types 1 and 2), Epstein-Barr, varicella zoster, and hepatitis B.14 The structure of the oxidized XO active site (XOox) is 5-coordinate square pyramidal (Figure 1A) with an apical oxo ligand, and equatorial sulfido, hydroxyl and pyranopterin-dithiolene sulfur ligands.2,15–18 It is now generally believed that substrate hydroxylation is initiated by nucleophilic attack of metal-activated water (i.e. hydroxide) on the substrate carbon atom that is to be hydroxylated, and this results in a tetrahedral intermediate (IM, Figure 1B) or transition state (TS, Figure 1C).1,2,15,19,20 This mechanistic hypothesis is supported by spectroscopic and structural studies on enzyme-product complexes that confirm a Mo-Oeq-Cproduct linkage.15,21 A large kinetic isotope effect (KIE) is observed upon deuteration of the C8-H position of xanthine, and this is consistent with at least partial Csubstrate-H bond-breaking occurring at the TS.22 This Csubstrate-H bond-breaking step has been described as a transfer of hydride from the substrate to the sulfido (S2−) ligand at the active site, resulting in a two-electron reduction of Mo(VI) and protonation of the terminal sulfido (Figure 1D). The transfer of a hydride is of keen interest with respect to how large electronic repulsions and charge buildup associated with the sulfido-hydride interaction are compensated along the reaction coordinate. Here we present a detailed EPR and computational study of a key paramagnetic form of xanthine oxidase (XO) that serves as a basis for understanding important active site – substrate interactions along the reaction coordinate from IM to TS..
Figure 1.

Proposed reaction mechanism for XO. (A) oxidized active site, (B) tetrahedral intermediate (IM) resulting from nucleophilic attack of metal activated water (i.e. HO−) on aldehyde carbonyl carbon and proton transfer to the general base E1261, (C) putative transition state (TS) showing hydrogen migration between substrate carbon and the terminal sulfido ligand, and (D) the reduced Mo(IV) site following product release and binding of H2O.
This paramagnetic Mo(V) form of XO can be generated using aldehyde substrates to generate the aldehyde Inhibited enzyme form. Aldehyde Inhibited XO has been shown to exhibit 17O, 33S, 13C, and 1,2H hyperfine coupling with the Mo(V) spin=½ center,23–26 and Bray and coworkers suggested two structures for Inhibited that possess tetrahedral carbon centers (Figure 2). Recent 1,2H ENDOR studies on formaldehyde Inhibited XO have provided evidence for structure B in Figure 2.27 XO Inhibited possesses structural features that are common to the putative tetrahedral intermediate (IM) and other non-planar substrate carbon geometries along the reaction coordinate that we, and others,19,20,28,29 have calculated for XO with aldehyde and heterocyclic aromatic organic (i.e. purine) substrates. These include the presence of Mo-S-R(H) ligation in place of a terminal sulfido (Mo=S), an Mo-O-R linkage, and a tetrahedral carbon center that derives from the aldehyde substrate. Thus, XO Inhibited can be thought to represent a rudimentary paramagnetic analogue of enzyme structures found between IM and the TS (Figure 1).
Figure 2.

Structures originally proposed by Bray for XO aldehyde Inhibited that possess a tetrahedral carbon center. Recent ENDOR studies provide evidence for structure B.27
The relationship between the g-, 95,97Mo hyperfine (AMo), and the 13C hyperfine (AC) tensors in aldehyde Inhibited XO has been used to understand the electronic origin of key delocalizations within the Mo-Oeq-C fragment. These experimental data have been used to interpret the results of detailed bonding calculations for XO at geometries between IM and TS. Our combined spectroscopic and bonding study has provided new insight into the nature of the hydride transfer process in XO family enzymes. We have used a natural bond orbital (NBO) formalism to show how specific donor-acceptor interactions contribute to C-H bond activation/cleavage and TS stabilization. The results highlight the importance of Mo-Oeq-C, C-H, and Mo-Ssulfido bond covalency in mediating efficient electronic communication between substrate and the Mo center, and have allowed us to develop a valence bond description of C-H activation and transition state stabilization along the reaction coordinate with aldehyde substrates.
EXPERIMENTAL
Enzyme Preparation for Spectroscopic Studies
B. taurus XO was either purchased from Sigma Aldrich or was isolated and purified from unpasteurized cow’s milk according to the method of Massey and coworkers.30,31 Both commercial and isolated XO samples had A280/A450 ratio of about 11.7 in bicine/NaOH buffer, pH 8.2 and had an activity to flavin ratio (AFR) of 200 (fully functional enzyme reported by Massey et al, had an AFR of 210 and an A280/A450 ratio of about 5.3 to 5.8).32 Enzyme concentrations were determined using the extinction coefficient at 450 nm (37,800 M−1cm−1). The Inhibited enzyme form was generated using the reducing substrates formaldehyde, acetaldehyde, 2-pyridinecarboxaldehyde and 3-pyridinecarboxaldehyde (Sigma Aldrich). XO was dialyzed against 50 mM-Bicine/NaOH buffer at pH 8.2. XO samples (0.07 to 0.1 mM) were placed in quartz EPR tubes and a large excess of the substrate (0.03 to 0.06 M) was then added. This mixture was then bubbled gently with oxygen with frequent shaking to ensure aerobiosis. These reaction mixtures were then incubated for times that ranged from minutes to hours depending on the nature of the substrate. Samples were then frozen in liquid N2 for EPR measurements after appropriate incubation times.
EPR spectroscopy
EPR spectra of XO were collected at X-band (9.4 GHz) using a Bruker EMX spectrometer with associated Bruker magnet control electronics and microwave bridges. A microwave power of 20 dB was used for all experiments. Spectra were collected at 40 and 100 K using an Oxford Instruments liquid helium flow cryostat. Simulations of the EPR spectra were performed using the MATLAB toolbox EasySpin,33 with further analyses performed using in-house written scripts for the program Visual Molecular Dynamics.34
Electronic Absorption Spectroscopy
Electronic absorption spectra were collected using a Hitachi U-3501 UV–Vis–NIR dual-beam spectrometer capable of scanning a wavelength region between 185 and 3200 nm. XO enzyme samples (~5 μM) were incubated with aldehyde substrates (1mM) in 50mM Bicine/NaOH buffer solution at pH 8.2. The electronic absorption spectra were measured in a 1-cm pathlength, 100 μL, black-masked, quartz cuvette (Starna Cells, Inc.) equipped with a Teflon stopper. All electronic absorption spectra were collected at room temperature.
Computational Methods
Spin-unrestricted gas-phase geometry optimizations calculations for two “inhibited” structures were performed at the density functional level of theory using the ORCA,35 ADF,36 and Gaussian 03W37 software packages. All Gaussian 03W calculations employed the B3LYP hybrid exchange-correlation functional.38 A 6-31G(d, p) basis set, a split valence basis set with added polarization functions, was used for all atoms except Mo, where the LANL2DZ basis set, which includes an effective core potential, was used. The tetrahedral computational models for the aldehyde Inhibited structures were those originally proposed by Bray (1: [(dt)MoO(S-C(R)(H)-O)] and 2: [(dt)MoO(S-C(R)(OH)-O)], where dt = 1,2-dimethyl-ene-1,2-dithiolate and R = H, methyl, 2-pyridine, or 3-pyridine) (Figure 2). Input files were prepared using GaussView and ADFInput, as appropriate. EPR parameters were calculated at the B3LYP/TZVP/ZORA38–41 level using ORCA 2.7.0.35,42,43. EPR calculations used a decontracted basis set and increased radial integration accuracy for Mo (specialgridintacc set to 7). ADF calculations used a triple-zeta basis set (TZP in the ADF basis set notation) and the PBE44 GGA density functional. Relativistic corrections were incorporated self-consistently in the ADF and ORCA calculations with the ZORA scalar relativistic Hamiltonian.45,46 ADF geometry calculations (geometry optimizations, transition state searches, and intrinsic reaction coordinate) used frozen core basis sets (C: 1s, O: 1s, S: 1s2s2p, Mo: 1s2s2p3s4p) and default integration accuracies. Optimized geometries run as single-point calculations used no frozen cores and increased integration accuracy (integration key set to 6).
Reaction path geometries were located as follows (using ADF2010): first, a transition state guess was located with a linear transit run and then converged with a TS search. The transition state was verified by the observation of a single large (−572 cm−1) negative eigenvalue in the frequency calculation; this corresponded to the C-H stretching mode. The TS geometry and Hessian were then used as an input for an intrinsic reaction coordinate (IRC) calculation. Selected IRC path points were then run as single-point jobs, and the natural bond orbital (NBO) analysis was performed using the adfnbo package.
RESULTS AND DISCUSSION
Nature of the Ground State Wavefunction for the Aldehyde Inhibited form of Xanthine Oxidase
Low-temperature (100 K) X-band (~9.4 GHz) EPR spectra for aldehyde Inhibited bovine XO using formaldehyde and 3-pyridinecarboxaldehyde are shown in Figure 3 (black). The data for 3-pyridinecarboxaldehyde Inhibited XO displays well-resolved 95,97Mo hyperfine splitting. The formaldehyde Inhibited EPR spectrum shows evidence for 1H coupling that results in increased spectral line broadening. Additionally, the formaldehyde Inhibited EPR spectrum displays 95,97Mo (I=5/2) hyperfine splitting and well-resolved 13C (I=1/2) hyperfine splitting when using H13COH. The rhombic nature of the g-tensor anisotropy necessitates the use of an orthorhombic or lower-symmetry spin-Hamiltonian in simulations of the EPR spectra (Equation 1).
Figure 3.

Aldehyde Inhibited X-band (9.41 GHz) EPR spectra for XO (0.07mM) in 50 mM Bicine/NaOH buffer, pH 8.2. The aldehyde substrates are 3-pyridinecarboxaldehyde (a), HCOH (b), and H13COH (c). Experimental spectra are in black and spectral simulations are in red. Spin-Hamiltonian parameters are given in Table 1.
| Equation 1 |
Here, g is the g-tensor, β is the Bohr magneton, B is the applied magnetic field, An are the nuclei-specific hyperfine coupling tensors (n = 95,97Mo, 1H, 13C), S̑ is the electron spin operator, and Ȋ is the nuclear spin operator. The 95,97Mo and 13C hyperfine tensors, AMo and AC, are comprised of an isotropic Fermi contact term, , a spin dipolar term, , and an orbital dipolar term, , the latter of which is typically small.47 Although is proportional to the spin density at the nucleus of interest, the nature of the anisotropic term results from the spatial distribution of the spin density around the nucleus. Therefore, the anisotropy in contains important information regarding the delocalized nature of the singly-occupied molecular orbital (SOMO) wavefunction and electronic communication between the Mo center and a tetrahedral carbon center.47
Spectral simulations of the Inhibited EPR spectra are presented in Figure 3 (red), and the relevant spin-Hamiltonian parameters given in Table 1. Formaldehyde Inhibited XO displays a strongly coupled proton48,49 that is not evident in the 3-pyridinecarboxaldehyde spectra, and this indicates that the aldehydic R group is oriented in the “up” or M≡O direction (Figure 2). A previous EPR study of Inhibited utilized a 95,97Mo isotope perturbation to obtain the hyperfine parameters and .48 The anisotropy in the dipolar term, , is unusual, and indicative of a Mo(z2) singly occupied molecular orbital (SOMO) wave function.50 In order to better understand the nature of the Inhibited SOMO we have determined the relative orientation of the g- and AMo-tensors. We note that the structure of Inhibited as presented in Figure 2 possesses a pseudo mirror plane (y-z) that bisects the dithiolene sulfur donors and contains the Mo≡O unit. Detailed EPR studies on Tp*MoOX2 (X = F, Cl, Br) oxomolybdenum complexes that possess Cs (mirror) symmetry show that an increase in Mo-ligand covalency results in progressively larger rotations of the g-tensor relative to AMo in the mirror plane.51 Furthermore, Cs Tp*MoO(bdt) (bdt = benzene-2,3-dithiolate), also displays large rotations (~45°) of the g-tensor relative to AMo in the mirror plane of this molecule.52,53 We note that in Cs symmetry, one component of the g-tensor and AMo-tensor are collinear (x-direction).50 All of our EPR simulations for Inhibited were obtained with Euler rotations (i.e. β) that primarily rotate the g-tensor in the molecular y-z plane relative to AMo. This results in principle components of that indicate a normal Mo(x2-y2) ground state for Inhibited with the Mo(x2-y2) redox orbital oriented orthogonal to the Mo≡O bond. The nature of the AC tensor is consistent with unpaired spin being delocalized from the Mo center to the C spy hybrid atomic orbital that derives from the carbonyl carbon of the aldehyde substrate. The orientation of relative to the molecular frame places along the lobes of the Mo(x2-y2) orbital that bisect the dithiolene Sdt donors and point in the general direction of the tetrahedral C center (Figure 4). The calculated angle between and , the largest component of , is 160° and this defines the relative orientation of the Mo(x2-y2) and C(spy) orbitals with respect to each other. The deviation from 180° is indicative of an asymmetry within the Mo(-O-C-S-) four membered chelate ring. Calculations reveal non-zero Mo-C, Mo-Oeq, and Oeq-C overlaps in the singly occupied Mo(x2-y2) wavefunction. These non-zero overlaps provide a potentially direct pathway for Mo→C spin delocalization27 as well as a covalent delocalization pathway mediated by the Mo-Oeq-C linkage. However, despite the fact that the Mo(x2-y2) and C(spy) orbitals are aligned for a potential net psuedo- σ bonding interaction, the calculated Mo-C Mayer bond order54 in Inhibited is zero.
Table I.
EPR Parameters for XO Inhibited (bovine)a
| Axis | Average | Euler Angles (zyz) | |||||
|---|---|---|---|---|---|---|---|
| Parameters | z | y | x | <A> | α | β | γ |
| Formaldehyde | |||||||
| g | 2.0002 (1.9972) | 1.9856 (1.9802) | 1.9610 (1.9525) | 1.9823 (1.9766) | |||
| A(95,97Mo; ×10−4 cm−1) | 59.50 (54.14) | 23.00 (18.45) | 22.50 (17.97) | 35.00 (30.19) | 86.75° | 138.59° | 8.28° |
| A(13C; ×10−4 cm−1) | 12.33 (11.34) | 16.67 (11.69) | 16.00 (14.72) | 15.00 (12.58) | 90.00° | 69.15° | 56.58° |
| A(1H; ×10−4 cm−1) | 3.33 (3.65) | 3.33 (3.97) | 5.00 (5.84) | 3.89 (4.49) | 9.51° | 102.89° | −81.87° |
| 2-Pyridinecarboxaldehyde | |||||||
| g | 2.0025 (1.9997) | 1.9878 (1.9812) | 1.9614 (1.9559) | 1.9839 (1.9789) | |||
| A(95,97Mo; ×10−4 cm−1) | 57.00 (53.99) | 22.33 (18.52) | 21.67 (17.80) | 33.67 (29.65) | 69.00° | 137.00° | 17.62° |
| 3-Pyridinecarboxaldehyde | |||||||
| g | 1.9995 (2.0001) | 1.9824 (1.9815) | 1.9578 (1.9570) | 1.9799 (1.9795) | |||
| A(95,97Mo; ×10−4 cm−1) | 56.33 (53.59) | 20.67 (18.10) | 20.67 (17.26) | 32.33 (29.65) | 72.00° | 140.00° | 12.00° |
Parameters in parentheses are the calculated spin-Hamiltonian Parameters. Euler rotations are defined as a rotation of the gi axes relative to the A axis frame. Using a right hand coordinate frame, the rotation α is about the Az-axis, the rotation β is about the new y-axis, and the rotation γ is about the new z-axis. A counterclockwise rotation is positive. EPR parameters were calculated at the B3LYP/TZVP/ZORA40 level using ORCA 2.7.0.35,42,43.
Figure 4.

Principal component orientations of the g, AMo,, and AC tensors for XO Inhibited superimposed on the DFT calculated β-LUMO wavefunction. The largest component of AMo (AMomax) is oriented 4.75° off of the Mo≡O bond. The largest component of AC (ACmax) forms a 160° angle with AMomid (see text).
Spin Population Analysis and Metal-Ligand Covalency
The good agreement between the calculated and experimental 95,97Mo hyperfine, 13C hyperfine, and g-tensors, as well as the relationship between their experimentally determined Euler rotational matrices (Table 1, Figure 4), allow us to explore the electronic origin of these parameters in Inhibited in terms of the calculated spin density and atomic spin populations. We recently used this procedure to calibrate the experimentally determined N hyperfine interaction (hfi) with calculated N atomic spin populations on the acceptor fragment in a series of Donor-Acceptor biradical systems.55 An excellent correlation was obtained between the calculated N spin populations and the experimentally determined hfi, and this was evaluated in the context of detailed bonding calculations that provided insight into the electronic origins of covalent spin delocalization and spin polarization contributions to the observed N hfi. Specifically, this work showed that the nature of the spin density distribution can serve as a probe of Donor→Acceptor, and by inference, ligand-to-metal charge transfer (LMCT).
Inspection of the calculated spin density (Figure 5) and atomic spin populations in Inhibited show covalent delocalization of positive spin from the d1 Mo(V) center onto the Oeq and C centers of the four membered chelate ring. The observation of net positive spin populations on O and C contrasts with the negative net spin population on the equatorial chelate ring S donor (Seq). The observation of a net negative spin population on Seq is unusual, and derives from a spin polarization mechanism that results from configurational mixing (CI) of Seq→Mo ligand-to-metal charge transfer (LMCT) states that are formed from promotion of an α (spin-up) electron localized in a doubly occupied molecular orbital to an unoccupied virtual orbital localized on Mo. Seq→Mo LMCT transitions will only contribute to positive spin populations on Seq if the acceptor orbital in these Seq→Mo LMCT transitions is the half-occupied Mo(x2-y2) acceptor orbital. This idea is supported by our calculated LMCT oscillator strengths for Inhibited, which indicate the absence of intense (i.e. ε > 400 M−1cm−1) low-energy (E < 22,000 cm−1) Seq→ Mo(x2-y2) LMCT transitions. The observation of a negative spin population on Seq is also consistent with the small degree of Mo(x2-y2)-Seq covalency calculated for Inhibited, where the spin-bearing α-HOMO (HOMO = highest occupied molecular orbital) possesses <2% Seq(p) character. The Ooxo-Mo-Seq-C dihedral angle in Inhibited is calculated to be ~107.0° and this geometry precludes dominant Mo(x2-y2)-Seq(p) π-type orbital interactions since the in-plane (x-y plane) Seq(p) orbital capable of forming a π-bond with Mo is involved in strong σ-bonding with the chelate ring C.56,57 Such low Mo(x2-y2)-Sthiolate covalency in an oxomolybdenum thiolate is rare, but has recently been realized in Tp*MoO(mba) (mba = mercaptobenzyl alcohol), which possesses a 95.5° Ooxo-Mo-Sthiolate-C dihedral angle.58 Detailed sulfur K-edge X-ray absorption spectroscopic (XAS) studies have provided an experimental estimate of ~0% S thiolate character admixed into the Mo redox orbital of Tp*MoO(mba).58 Thus, the low Mo(x2-y2)-Seq covalency found in Inhibited is a direct consequence of the Ooxo-Mo-Seq-C dihedral angle, which precludes any appreciable Mo(x2-y2)-Seq(p) π-type covalency. In marked contrast to the < 2% Seq(p) character admixed into the Inhibited spin-bearing α-HOMO, the Oeq donor contributes ~10% character to this orbital. Therefore, the Mo(x2-y2)-Oeq bond covalency in Inhibited, coupled with the structural similarities between XO Inhibited and XO along the IM→TS reaction coordinate point to a potentially important role for Oeq in 1) C-H bond activation and 2) facilitating electronic communication between the Mo and C centers along the reaction coordinate via a direct Mo(x2-y2)-Oeq-C pathway.
Figure 5.
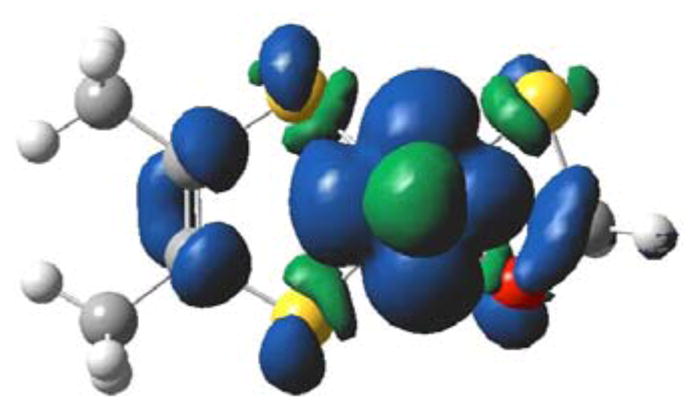
Calculated spin density distribution for 1. The orientation is down the z-axis of Figure 4. Positive spin density (blue) is delocalized from the Mo center onto Oeq and the tetrahedral C. Negative spin density (green) is found on Seq. Calculated atomic spin populations: Mo = +99.5%; Seq = −0.57%; C = +2.03%; Oeq = +1.91%.
The Role of Mo-S, Mo-Oeq-C, and C-H Donor-Acceptor Interactions in C-H Bond Activation and TS Stabilization
The calculated spin density and atomic spin populations for Inhibited reveal a covalent spin delocalization pathway that involves the Mo ion, Oeq, and the tetrahedral C center. The observation of this delocalization, coupled with the structural similarity between Inhibited and active site structures that evolve along the IM→TS reaction coordinate, has spurred our interest in electronic structure contributions to the XO reaction coordinate with aldehyde substrates. The IM→TS activation energy, ΔE†, calculated with acetaldehyde as substrate is found to be 12.6 kcal/mol and the IM→TS reaction profile as a function of acetaldehyde C-H distance is presented in Figure 6. The results for the IM→TS reaction profile are in general agreement with those obtained using detailed QM/MM approaches.19 A key question that has remained unanswered regarding electronic structure contributions to reactivity in XO relates to how the enzyme facilitates activation and scission of the substrate C-H bond along the reaction coordinate. In order to address this issue, we have employed a natural bond orbital (NBO) analysis that provides insight into electronic structure contributions to C-H activation and key charge transfer stabilizations that lead to a reduction in repulsive interactions along the reaction coordinate and, ultimately, to lowering the activation energy for enzymatic aldehyde oxidation.
Figure 6.

Calculated energy profile along the reaction coordinate from IM to TS as a function of the substrate (acetaldehyde) C-H distance. Note that the charge of the transferred H is positive and does not change along this coordinate, in agreement with prior work.19
NBOs conveniently represent localized lone-pair and bonding regions59 and provide a convenient way to develop a valence bond, or Lewis structure, description of important bonding interactions along the IM→TS reaction coordinate in XO catalyzed oxidations. In order to reach the TS and ultimately form product (P), the C-H σ- and Mo=S π-bonds must be broken concomitantly with the formation of an S-H σ-bond and a reduction of the Mo(VI) state to Mo(IV). The calculated Wiberg bond order60 changes for the Mo=S, C-H, and S-H bonds along the IM→TS reaction coordinate are presented in Figure 7 and clearly reflect these key bond making and bond breaking steps. Reducing the aldehyde C-H bond order and activating this bond for cleavage can involve two electron occupation of the C-H σ* antibonding orbital (Figure 8A) or a reduction of electron density in the C-H σ bonding orbital (Figure 8B). Therefore, it is of interest to evaluate the relative contributions of the complementary charge transfer interactions in Figure 8 to C-H activation and eventual TS stabilization. The calculated C-H σ-bonding and Mo=S π-bonding NBOs for XO at the IM geometry are presented in Figure 9, and occupation numbers are given in Table II. The C-H bonding interaction is formed from C(2p)-H(1s) orbital overlap. Due to the fact that the H 1s orbital possess no radial nodes, the resulting heteronuclear diatomic type NBO takes on the appearance of an atomic s-p hybrid orbital. Both the C-H σ-bonding and C-H σ-antibonding NBOs possess large amplitudes on the C-H hydrogen atom with a larger radial extension observed for the C-H σ-antibonding orbital. The Mo=S π-bonding NBO is very covalent, with approximately 60% S and 40% Mo character, while the corresponding Mo=S π* antibonding NBO possesses 40% S and 60% Mo character. The nature of the Mo=S π* antibonding NBO is in excellent agreement with the results of S K-edge X-ray absorption spectroscopic (XAS) studies on the Mo(VI) model compound Tpi-PrMoOS(OPh) which showed 30–41% sulfido character in the oxidized LUMO (Mo=S π*) wavefunction.61 The Mo 4d – S 3p interaction that comprises the Mo=S bonding scheme results in high degree of covalency due to the large radial expansion of the Mo 4d and S 3p atomic orbitals and the small energy difference between these orbitals. This results in a calculated Mo=S π - Mo=S π* energy gap at the IM geometry of only 21,350 cm−1 (2.7 eV). Thus, the Mo=S bonding scheme represents a unique electronic structure poised to activate substrate C-H bonds through C-H σ → Mo=S π* and Mo=S π → C-H σ* donations, with both reducing the C-H bond order and activating this bond for cleavage.
Figure 7.

Wiberg bond orders60 calculated along the reaction coordinate of Figure 6. Blue: Mo=S; Black: C-H; Red: S-H.
Figure 8.
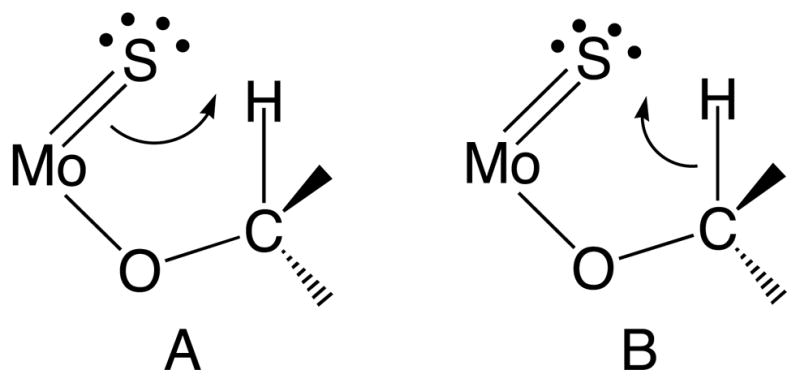
Lewis structures that describe Mo=S π → C-H σ* charge transfer resulting in S-H bond formation (A) and C-H σ → Mo=S π* charge transfer resulting in S-H bond formation (B).
Figure 9.
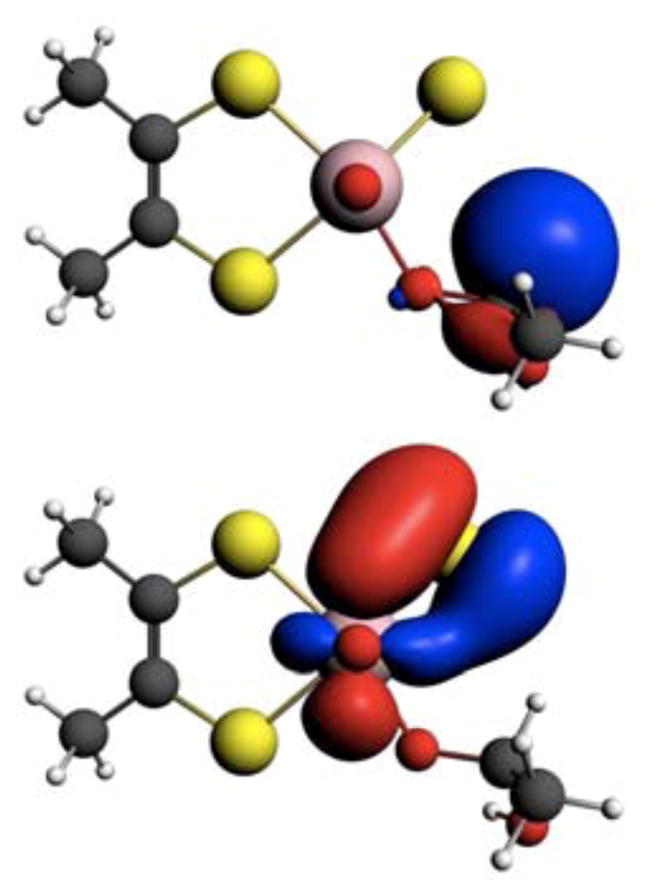
NBOs for the C-H σ-bond (Top) and the Mo=S π-bond (bottom) at the IM geometry.
Table II.
NBO Occupation Numbers for Points IM to 3
| Point | C-H σ | C-H σ* | Mo-S π | Mo-S π* | Oeq lp | S lp |
|---|---|---|---|---|---|---|
| IM | 1.97e | 0.06e | 1.92e | 0.23e | 1.66e | 1.99e |
| 1 | 1.80 e | 0.18e | 1.89e | 0.42e | 1.67e | 1.97e |
| 2 | 1.73e | 0.24e | 1.87e | 0.51e | 1.67e | 1.97e |
| 3 | 1.63e | 0.32e | 1.84e | 0.60e | 1.65e | 1.96e |
Figure 10 displays key bonding (b) and lone-pair (lp) donor NBOs (left on each donor-acceptor orbital diagram), and antibonding (ab*) acceptor NBOs (right on each donor-acceptor orbital diagram) for point 2 along the reaction coordinate of Figure 6. Here, the substrate hydrogen being transferred to the terminal sulfido ligand is approximately midway between the C-H distance of IM and the C-H distance in the TS. The stabilization energy (ΔE) resulting from these bonding interactions can be approximated using second-order perturbation theory via59
Figure 10.

NBOs for point 2 showing CT energy stabilizations and charge transferred (Q) between donor and acceptor NBO’s.
| Equation 2 |
where 〈b(lp)|F̂|ab*〉 is the Fock matrix element between the donor (b or lp) and acceptor (ab*) NBOs, and Eab*–Eb(lp) is the energy difference between a corresponding pair of donor and acceptor NBOs. The charge transferred, Qb(lp)→ab*, between donor and acceptor NBOs is defined as59
| Equation 3 |
The dominant donor→acceptor interactions which contribute to C-H bond activation derive from the Mo=S π→C-H σ* interaction (ΔE= 24.3 kcal/mol) with a comparable contribution from C-H σ → Mo=S π* (ΔE= 20.0 kcal/mol). Additional appreciable contributions originate from Oeq lp→C-H σ* (ΔE= 8.2 kcal/mol), and S lp→C-H σ* (ΔE= 13.2 kcal/mol) stabilizing interactions. Furthermore, the charge transfers derived from Equation 2 show that, at position 2 along the reaction coordinate, 0.11e has been back donated into the Mo=S π* antibond from the C-H σ bond. A markedly larger 0.19e has been transferred into the C-H σ* antibond from the Mo-S π bond and the S and Oeq lp orbitals due to a combination of Mo=S π→C-H σ*, Oeq lp→C-H σ* and S lp→C-H σ* charge transfer interactions. Values for the calculated NBO occupation numbers, energy stabilizations, and the Fock matrix elements at points IM, 1, 2, and 3 are provided in Tables 2–4.
Table IV.
Fock Matrix Elements (Fij) and Energy Gaps (ΔE) for Points IM to 3
| Point | C-H σ → Mo-S π* | Mo-S π → C-H σ* | Oeq lp → C-H σ* | S lp → C-H σ* | ||||
|---|---|---|---|---|---|---|---|---|
| Fij (eV) | ΔE(eV) | Fij (eV) | ΔE(eV) | Fij (eV) | ΔE(eV) | Fij (eV) | ΔE(eV) | |
| IM | 0.14 | 10.07 | 0.49 | 15.78 | 1.31 | 15.51 | - | - |
| 1 | 1.50 | 8.16 | 2.12 | 11.97 | 1.36 | 15.51 | 2.37 | 24.22 |
| 2 | 1.91 | 7.35 | 2.37 | 10.07 | 1.55 | 12.79 | 2.67 | 22.04 |
| 3 | 2.40 | 6.53 | 2.56 | 8.44 | 1.71 | 10.61 | 2.88 | 19.86 |
A covalency induced “electronic buffer effect” has been observed in oxomolybdenum-dithiolene model compounds.62,63 This has been postulated to take advantage of the redox non-innocent behavior of the coordinated dithiolene to modulate64 the reduction potential of the Mo site during the course of catalysis via Mo-dithiolene forward and back donation. In the Mo hydroxylases, we find that similar charge transfer stabilizations occur along the reaction coordinate that effectively buffer the active site against the large formal charge changes that accompany C-H bond cleavage. Here, the key processes are the complimentary forward donation from the C-H σ bond to Mo=S π*, and back donation into the C-H σ* from the Mo=S π bond. This directly results from the highly covalent Mo=S bonding interaction61 (Figure 9) which markedly reduces charge buildup on the transferred H and minimizes repulsive interactions along the C---H---S reaction coordinate.
With respect to the S-H bond forming and C-H bond breaking steps that were anticipated from Figure 8, the calculated C-H σ bond order is reduced to 0.29 at the TS, while the corresponding S-H σ bond order has increased to 0.64 (Figure 7). Additionally, the Mo=S bond order has been reduced from 1.65 at IM to 1.06 at the TS due to a weakening of the Mo=S π bond. A weak Mo=S π bonding interaction is still present at the TS, and this results from (Mo − C) →S-H σ* back donation as depicted in Figure 11. Following these large changes in bond order, the key question now shifts from how the C-H bond is activated to describing the nature of TS stabilization in XO. We note that the use of these interaction energies may not be quantitatively accurate for the description of transition states that are highly delocalized65 since these NBO stabilization energies derive from a perturbative treatment. However, these energies can be used in a qualitative way to provide an intuitive description of the key donor-acceptor contributions to TS stabilization.
Figure 11.

CT energy stabilizations between donor and acceptor NBO’s at the XO/XDH TS.
In contrast to earlier points (see 1–3 in Figure 6) along the reaction coordinate, the donor-acceptor interactions that dominantly contribute to the stabilization of point 4 and the TS are different. Important donor and acceptor NBOs and their respective donor-acceptor interactions at the TS are presented in Figure 11. The donor (Mo − C; 70%Mo/30%C) and acceptor (Mo + C; 30%Mo/70%C) NBOs are best described as linear combinations of product (Mo lone-pair) and reactant (C lone-pair) orbitals. Here it is observed that the dominant donor→acceptor interaction that contributes to TS stabilization derives from the Oeq→(Mo + C) charge transfer (ΔE= 89.5 kcal/mol). The nature of the Oeq→(Mo + C) charge transfer interaction is important since it results in a nascent product C=O π bonding interaction and it contributes to Mo reduction. The effects of this delocalization are clearly observed in the doubly occupied natural localized molecular obital (NLMO) of Figure 12. We note that 95% of this NLMO derives from the Oeq→(Mo + C) charge transfer interaction observed in Figure 11. Other stabilizing donor-acceptor interactions include S-H σ → (Mo + C) (ΔE= 51.5 kcal/mol) and the aforementioned (Mo − C) →S-H σ* (ΔE= 40.2 kcal/mol) charge transfer that contributes to Mo=S π bonding.
Figure 12.
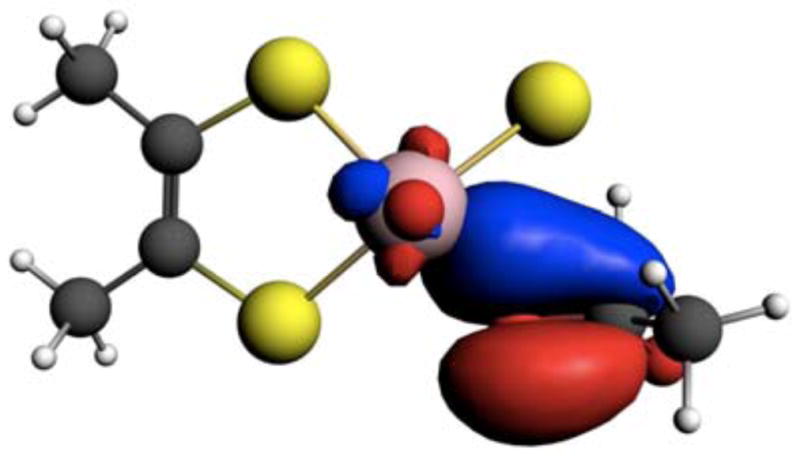
NLMO that derives from dominant Oeq→(Mo + C) charge transfer showing nacent C=O π-bonding leading to product formation.
Remarkably, the Oeq→(Mo + C) charge transfer results in the same type of Mo-Oeq-C delocalization that was described earlier in our analysis of the EPR derived hyperfine interactions and the calculated positive spin density delocalization in aldehyde Inhibited XO. That such similar Mo-Oeq-C delocalizations could be so important in stabilizing the TS in XO and promoting a positive spin population on the tetrahedral C center in Inhibited supports our earlier statement that XO Inhibited may be thought of as a rudimentary paramagnetic analogue of the TS. In addition to contributing to Mo reduction and product C=O π bond formation, the Oeq→(Mo + C) charge transfer strongly contributes to lowering the energy of the TS. This results from the fact that this charge transfer interaction effectively reduces the strong electronic repulsions present at the TS that contribute to the classical energy barrier; a concept that is central to transition state theory.59,66.
CONCLUSION
The Csubstrate-H bond-breaking step in XO has been previously described as a transfer of hydride from the substrate to the sulfido (S2−) ligand at the active site, resulting in a two-electron reduction of Mo(VI) and protonation of the terminal sulfido. Our interest in XO mediated hydride transfer has been focused on how the enzyme reduces the large electronic repulsions and charge buildup associated with C-H bond scission along the reaction coordinate. We have used EPR spectroscopy to study an XO Inhibited enzyme form and the results have been used to support detailed bonding calculations along the XO reaction coordinate. Our analysis of aldehyde Inhibited EPR spectra has yielded the relative orientations of the g-tensor, and the 95,97Mo and 13C hyperfine tensors. The anisotropy in the 95,97Mo hyperfine tensor is now interpreted in terms of proper Euler rotations that support a Mo(x2-y2) ground state for this species. The calculated spin density and atomic spin populations for Inhibited have been used to highlight a key Mo-Oeq-C delocalization pathway that contributes to appreciable spin delocalization onto the tetrahedral carbon center that derives from the aldehyde substrate. A natural bond orbital analysis of the XO reaction coordinate with acetaldehyde as reducing substrate shows that the dominant donor→acceptor interactions that facilitate weakening and concomitant activation of the substrate C-H bond derive from Mo=S π → C-H σ* and C-H σ → Mo=S π* back donations. These competing donor-acceptor interactions allow the substrate C-H hydrogen to be transferred to the terminal sulfido with only a slight positive charge, and this charge does not change along the reaction coordinate. An additional contribution originating from Oeq lp→C-H σ* charge transfer is also present. The Oeq donor plays an even more important role in TS stabilization. We find that the Oeq→(Mo + C) charge transfer process dominantly contributes to TS stabilization. Thus, the Mo-Oeq-C delocalization pathway observed in aldehyde Inhibited reduces strong electronic repulsions that contribute to the classical TS energy barrier in the enzyme. In summary, this work has increased our understanding of substrate C-H bond activation and the nature of through-bond donor-acceptor electronic couplings that allow for efficient electronic communication between substrate and the Mo center along the XO reaction coordinate.
Table III.
Donor→Acceptor Energy Stabilizations for Points IM to 3 (Energies in kcal/mol)
| Point | C-H σ → Mo-S π* | Mo-S π → C-H σ* | Oeq lp → C-H σ* | S lp → C-H σ* |
|---|---|---|---|---|
| IM | 0.08 | 0.73 | 4.6 | < 0.05 |
| 1 | 11.2 | 17.0 | 5.1 | 9.9 |
| 2 | 20.0 | 24.3 | 8.2 | 13.2 |
| 3 | 36.2 | 33.4 | 12.6 | 16.5 |
Acknowledgments
M.L.K. acknowledges the National Institutes of Health grant GM 057378 for financial assistance.
References
- 1.Hille R. Arch Biochem Biophys. 2005;433:107. doi: 10.1016/j.abb.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 2.Kirk ML, Knottenbelt S, Habtegabre A. In: Computational Inorganic and Bioinorganic Chemistry. Solomon EI, Scott RA, King BR, editors. Wiley; 2009. p. 614. [Google Scholar]
- 3.Hille R. Chem Rev. 1996;96:2757. doi: 10.1021/cr950061t. [DOI] [PubMed] [Google Scholar]
- 4.Purifoy D, Beauchamp L, Demiranda P, Ertl P, Lacey S, Roberts G, Rahim S, Darby G, Kretinsky T, Powell K. J Med Vir. 1993;S1:139. doi: 10.1002/jmv.1890410527. [DOI] [PubMed] [Google Scholar]
- 5.Demiranda P, Burnette T. Drug Metab Dispos. 1994;22:55. [PubMed] [Google Scholar]
- 6.Fowles S, Pratt S, LaRoche J, Prince W. Eur J Clin Pharmacol. 1994;46:355. doi: 10.1007/BF00194405. [DOI] [PubMed] [Google Scholar]
- 7.Krasny H, Beauchamp L, Krenitsky T, Demiranda P. Drug Metab Dispos. 1995;23:1242. [PubMed] [Google Scholar]
- 8.Obach RS. Drug Metab Dispos. 2004;32:89. doi: 10.1124/dmd.32.1.89. [DOI] [PubMed] [Google Scholar]
- 9.Smith MA, Marinaki AM, Arenas M, Shobowale-Bakre M, Lewis CM, Ansari A, Duley J, Sanderson JD. Alimentary Pharmacology & Therapeutics. 2009;30:375. doi: 10.1111/j.1365-2036.2009.04057.x. [DOI] [PubMed] [Google Scholar]
- 10.Garattini E, Mendel R, Romao MJ, Wright R, Terao M. Biochem J. 2003;372:15. doi: 10.1042/BJ20030121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rooseboom M, Commandeur JNM, Vermeulen NPE. Pharm Rev. 2004;56:53. doi: 10.1124/pr.56.1.3. [DOI] [PubMed] [Google Scholar]
- 12.Hodge R. Antiviral Chemistry and Chemotherapy. 1993;4:67. [Google Scholar]
- 13.Rashidi M, Smith J, Clarke S, Beedham C. Drug Metab Dispos. 1997;25:805. [PubMed] [Google Scholar]
- 14.Hille R. J Biol Inorg Chem. 1996;1:397. [Google Scholar]
- 15.Pauff JM, Cao H, Hille R. J Biol Chem. 2009;284:8751. doi: 10.1074/jbc.M804517200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pauff JM, Zhang JJ, Bell CE, Hille R. J Biol Chem. 2008;283:4818. doi: 10.1074/jbc.M707918200. [DOI] [PubMed] [Google Scholar]
- 17.Asai R, Nishino T, Matsumura T, Okamoto K, Igarashi K, Pai EF. J Biochem. 2007;141:525. doi: 10.1093/jb/mvm054. [DOI] [PubMed] [Google Scholar]
- 18.Enroth C, Eger B, Okamoto K, Nishino T, Nishino T, Pai E. Proc Nat Acad Sci USA. 2000;97:10723. doi: 10.1073/pnas.97.20.10723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Metz S, Thiel W. J Am Chem Soc. 2009;131:14885. doi: 10.1021/ja9045394. [DOI] [PubMed] [Google Scholar]
- 20.Zhang XH, Wu YD. Inorg Chem. 2005;44:1466. doi: 10.1021/ic048730l. [DOI] [PubMed] [Google Scholar]
- 21.Hemann C, Ilich P, Stockert AL, Choi EY, Hille R. J Phys Chem B. 2005;109:3023. doi: 10.1021/jp046636k. [DOI] [PubMed] [Google Scholar]
- 22.D’Ardenne S, Edmondson D. Biochemistry. 1990;29:9046. doi: 10.1021/bi00490a023. [DOI] [PubMed] [Google Scholar]
- 23.Howes B, Bennett B, Bray R, Richards R, Lowe D. J Am Chem Soc. 1994;116:11624. [Google Scholar]
- 24.Howes B, Pinhal N, Turner N, Bray R, Anger G, Ehrenberg A, Raynor J, Lowe D. Biochemistry. 1990;29:6120. doi: 10.1021/bi00478a002. [DOI] [PubMed] [Google Scholar]
- 25.Morpeth F, George G, Bray R. Biochem J. 1984;220:235. doi: 10.1042/bj2200235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Malthouse J, George G, Lowe D, Bray R. Biochem J. 1981;199:629. doi: 10.1042/bj1990629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shanmugam M, Zhang B, McNaughton RL, Kinney RA, Hille R, Hoffman BM. J Am Chem Soc. 2010;132:14015. doi: 10.1021/ja106432h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alfaro JF, Joswig-Jones CA, Ouyang W, Nichols J, Crouch GJ, Jones JP. Drug Metab Dispos. 2009;37:2393. doi: 10.1124/dmd.109.029520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bayse CA. Dalton Trans. 2009:2306. doi: 10.1039/b821878a. [DOI] [PubMed] [Google Scholar]
- 30.Komai H, Massey V, Palmer G. J Biol Chem. 1969;244:1692. [PubMed] [Google Scholar]
- 31.Hille R, Kim JH, Hemann C. Biochemistry. 1993;32:3973. doi: 10.1021/bi00066a018. [DOI] [PubMed] [Google Scholar]
- 32.Massey V, Komai H, Palmer G, Elion GB. J Biol Chem. 1970;245:2837. [PubMed] [Google Scholar]
- 33.Stoll S, Schweiger A. J Magn Reson. 2006;178:42. doi: 10.1016/j.jmr.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 34.Humphrey W, Dalke A, Schulten K. J Mol Graph. 1996;14:33. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 35.Neese F. ORCA, an ab initio, density functional, and semi-empirical program package. University of Bonn; Germany: [Google Scholar]
- 36.ADF2009.01. SCM, Theoretical Chemistry. Vrije Universiteit; Amsterdam, The Netherlands: http://www.scm.com. [Google Scholar]
- 37.Gaussian 03. R. C. G., Inc; Pittsburgh, PA: 2003. [Google Scholar]
- 38.Becke A. J Chem Phys. 1993;98:5648. [Google Scholar]
- 39.van Lenthe E, Baerends EJ, Snijders JG. J Chem Phys. 1994;101:9783. [Google Scholar]
- 40.Weigend F, Ahlrichs R. Phys Chem Chem Phys. 2005;7:3297. doi: 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- 41.van Lenthe E, Baerends EJ, Snijders JG. J Chem Phys. 1993;99:4597. [Google Scholar]
- 42.Neese F. J Chem Phys. 2001;115:11080. [Google Scholar]
- 43.Neese F. J Chem Phys. 2003;118:3939. [Google Scholar]
- 44.Perdew JP, Burke K, Ernzerhof M. Phys Rev Lett. 1996;77:3865. doi: 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- 45.Vanlenthe E, Baerends EJ, Snijders JG. J Chem Phys. 1993;99:4597. [Google Scholar]
- 46.Vanlenthe E, Baerends EJ, Snijders JG. J Chem Phys. 1994;101:9783. [Google Scholar]
- 47.Solomon EI. In: Comm Inorg Chem. Sutin N, editor. Vol. 3 Gordon and Breach; New York: 1984. [Google Scholar]
- 48.Pick FM, McGartol Ma, Bray RC. Eur J Biochem. 1971;18:65. doi: 10.1111/j.1432-1033.1971.tb01215.x. [DOI] [PubMed] [Google Scholar]
- 49.Barber MJ, Bray RC, Lowe DJ, Coughlan MP. Biochem J. 1976;153:297. doi: 10.1042/bj1530297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mabbs FE, Collison D. Electron Paramagnetic Resonance of d Transition Metal Compounds. Elsevier; Amsterdam: 1992. [Google Scholar]
- 51.Nipales NS, Westmoreland TD. Inorg Chem. 1997;36:756. [Google Scholar]
- 52.Drew SC, Hill JP, Lane I, Hanson GR, Gable RW, Young CG. Inorg Chem. 2007;46:2373. doi: 10.1021/ic060585j. [DOI] [PubMed] [Google Scholar]
- 53.Drew SC, Young CG, Hanson GR. Inorg Chem. 2007;46:2388. doi: 10.1021/ic060586b. [DOI] [PubMed] [Google Scholar]
- 54.Bridgeman AJ, Cavigliasso G, Ireland LR, Rothery J. Dalton Trans. 2001:2095. [Google Scholar]
- 55.Kirk ML, Shultz DA, Habel-Rodriguez D, Schmidt RD, Sullivan U. J Phys Chem B. 2010;114:14712. doi: 10.1021/jp102955j. [DOI] [PubMed] [Google Scholar]
- 56.McNaughton RL, Helton ME, Cosper MM, Enemark JH, Kirk ML. Inorg Chem. 2004;43:1625. doi: 10.1021/ic034206n. [DOI] [PubMed] [Google Scholar]
- 57.McNaughton RL, Tipton AA, Conry RR, Kirk ML. Abstracts of Papers of the American Chemical Society. 2000;219:U782. [Google Scholar]
- 58.Peariso K, Helton ME, Duesler EN, Shadle SE, Kirk ML. Inorg Chem. 2007;46:1259. doi: 10.1021/ic061150z. [DOI] [PubMed] [Google Scholar]
- 59.Reed AE, Curtiss LA, Weinhold F. Chem Rev. 1988;88:899. [Google Scholar]
- 60.Wiberg KB. Tetrahedron. 1968;24:1083. [Google Scholar]
- 61.Doonan CJ, Rubie ND, Peariso K, Harris HH, Knottenbelt SZ, George GN, Young CG, Kirk ML. J Am Chem Soc. 2008;130:55. doi: 10.1021/ja068512m. [DOI] [PubMed] [Google Scholar]
- 62.Westcott BL, Gruhn NE, Enemark JH. J Am Chem Soc. 1998;120:3382. [Google Scholar]
- 63.Inscore FE, Knottenbelt SZ, Rubie ND, Joshi HK, Kirk ML, Enemark JH. Inorg Chem. 2006;45:967. doi: 10.1021/ic0506815. [DOI] [PubMed] [Google Scholar]
- 64.Kirk ML, McNaughton RL, Helton ME. Progress in Inorganic Chemistry: Synthesis, Properties, and Applications. 2004;52:111. [Google Scholar]
- 65.Alabugin IV, Manoharan M. J Phys Chem A. 2002;107:3363. [Google Scholar]
- 66.Pross A, Shaik SS. Acc Chem Res. 1983;16:363. [Google Scholar]