

Abstract
Dysfunctional neural control of airway smooth muscle (ASM) is involved in inflammatory diseases, such as asthma. However, neurogenesis in the lung is poorly understood. This study uses mouse models to investigate developmental mechanisms of ASM innervation, a process that is highly coordinated with ASM formation during lung branching morphogenesis. We show that brain-derived neurotrophic factor (BDNF) is an essential ASM-derived signal for innervation. Although BDNF mRNA expression is temporally dissociated with ASM formation and innervation, BDNF protein is coordinately produced through post-transcriptional suppression by miR-206. Using a combination of chemical and genetic approaches to modulate sonic hedgehog (Shh) signaling, a pathway essential for lung branching and ASM formation, we show that Shh signaling blocks miR-206 expression, which in turn increases BDNF protein expression. Together, our work uncovers a functional cascade that involves Shh, miR-206 and BDNF to coordinate ASM formation and innervation.
Introduction
The respiratory tract, which consists of the trachea, bronchi, and lung, is innervated by two types of neurons that are distinguished by the location of their cell bodies. Intrinsic neurons have cell bodies clustered along the smooth muscle stripe in the trachea and major bronchi (Myers et al., 1990; Undem et al., 1990; Weichselbaum and Sparrow, 1999; Tollet et al., 2001; Langsdorf et al., 2011). These neurons have short axons that innervate nearby targets (Weigand and Myers, 2010). In contrast, cell bodies of extrinsic neurons are located in jugular/nodose ganglia, dorsal root ganglia, and sympathetic ganglia, while their axons extend into the lung to innervate airway smooth muscle (ASM) and neuroendocrine cells (Adriaensen and Scheuermann, 1993; Canning, 2006).
Neural innervation of the respiratory tract plays a key role in physiological reflexes, such as coughing, and regulates smooth muscle tone (Canning, 2006; Kc and Martin, 2010). In addition, neural inputs to neuroendocrine cells may be involved in regeneration of airway epithelium after injury (Reynolds et al., 2000). Furthermore, changes in innervation are associated with the development of airway diseases (Undem et al., 2000; Kerzel et al., 2003; Joos, 2001).
Our previous studies in mouse demonstrate that the glial cell line-derived neurotrophic factor (GDNF) family is essential for intrinsic neurogenesis (Langsdorf et al., 2011). Intrinsic neurons not only respond to members of the GDNF family in culture, they also require GDNF family for survival, as the number of intrinsic neurons is diminished by genetic disruption of GDNF family signaling (Tollet et al., 2002; Langsdorf et al., 2011). In contrast, ASM innervation in the distal lung remains unaffected in GDNF family mutants, indicating that the lung is primarily innervated by extrinsic neurons that require a different neurogenic signal.
Sonic hedgehog (Shh) signaling is required at the tip of lung bud for branching and in the stalk to induce mesenchyme specification into ASM (Cardoso and Lü, 2006; Morrisey and Hogan, 2010). Shh is expressed in the lung epithelium and signals to the neighboring mesenchyme by binding to its receptor Patched. Upon binding, Shh triggers a cascade of signaling events that involves activation of Smoothened, transcription factor Glis, and expression of target genes, such as Gli1 and Patched-1.
The early smooth muscle marker, α-smooth muscle actin (SMA), appears in the major bronchi but not in the rest of lung buds at embryonic day (E)12.5, when axons are identified within the major bronchi but do not extend into the lung (Tollet et al., 2001; Langsdorf et al., 2011). At E13.5, axons extend into the distal lung and are found in approximation with developing ASM. Axons continue to grow and elaborate into a complex neural network during later embryonic stages (Tollet et al., 2001). The temporal and spatial coupling between axon outgrowth and ASM formation suggest that an ASM-derived neurotrophic factor coordinates these two processes.
Here, we investigate molecular mechanisms that coordinate ASM innervation and formation. We identified brain-derived neurotrophic factor (BDNF) as an essential ASM-derived trophic factor for innervation and uncover a functional interaction between Shh signaling and BDNF expression via miR-206.
Materials and Methods
Mice.
Dermo1-Cre line (stock number: 008712), Rosa(SmoM2) line, which carries the constitutive active SmoM2 allele in the ROSA26 locus (stock number: 004339), BDNF+/− line (stock number: 002266), and Ptc1lacZ/+ line (stock number: 003081) were purchased from The Jackson Laboratories. The SMA-HrGFP transgenic line, in which a green fluorescent protein (hrGFP) is expressed under control of the rat α-SMA promoter, was generated at Boston University School of Medicine, Boston, MA (Ghosh et al., 2011). The miR-206−/− line was kindly provided by Dr. Eric Olson at University of Texas Southwestern Medical Center, Dallas, TX (Williams et al., 2009). For timed pregnancy, male and female mice were mated overnight and the morning of finding the vaginal plug was considered as E0.5. Embryos of either sex were used in our experiments. All animal studies were approved by the Institutional Animal Care and Use Committee at Boston University Medical Campus, Boston, MA.
Histology and immunohistochemistry.
The lungs were fixed in 4% paraformaldehyde/PBS at 4°C overnight. After cryoprotection in 30% sucrose/PBS, 10 μm sections were collected. Sections were blocked for 1 h in antibody dilution buffer (3% BSA, 10% goat serum, 2% horse serum, and 0.1% Triton X-100 in PBS) and primary antibody applied for 2 h at room temperature or overnight at 4°C. Antibodies against nerve growth factor (NGF) family members were purchased from Santa Cruz Biotechnology, including rabbit anti-NGF (1:100, sc-548), BDNF (1:100, catalog no. sc-546), NT3 (1:50, catalog no. sc-547), and NT4 (1:50, catalog no. sc-545). Other primary antibodies include biotinylated TuJ1 antibody against neuron-specific class III β-tubulin (1:200, BAM1195, R&D Systems) and mouse anti-smooth muscle actin (1:500, MS-113, Thermo Scientific). Antigen-antibody complexes were detected either by fluorescence or by chromogenic substrates. Fluorescently labeled whole lungs were imaged using a Zeiss Axiovert 100M LSM 510 equipped with an argon and two HeNe lasers. Compressed, Z-stack and titled confocal images were presented. To quantify innervation density, adjacent lung sections were immunolabeled with the TuJ1 and SMA antibodies. Immunoreactive areas within a section were measured using NIH ImageJ, and the ratio between the TuJ1+ area and the SMA+ area was calculated as innervation density. X-gal staining to detect β-galactosidase expression in Ptc1lacZ/+ embryos was performed as described previously (Langsdorf et al., 2011).
Lung organ culture.
Lungs were dissected from E11.5 mouse embryos and maintained at air-medium interface on Transwell inserts (Langsdorf et al., 2011). Embryonic lung organ cultures were treated with cyclopamine (0.5 μm, Calbiochem, catalog no. 239804) and SAG (Smoothened Agonist, 1 μg/ml, Calbiochem, catalog no. 566660) for 48 h before protein and gene expression analysis.
Constructs and luciferase assay.
The 1.5 kb BDNF 3′-UTR was cloned by reverse transcription (RT)-PCR and then inserted downstream of firefly luciferase gene in psiCHECK-2 vector (Promega) using XhoI/NotI cuts. miR-206 and a mutant miR-206 with a deleted seed sequence (miR-206mut) were cloned into a lentiviral vector pLVTHM using SpeI restriction enzyme cut site. 293T cells were transfected with the psiCHECK-2_BDNF 3′-UTR and miR-206 using FuGENE 6 transfection reagent (Roche), and luciferase activity was measured by dual luciferase assay following manufacturer's protocol (Promega). miR-206 was cloned by PCR using following primers: forward primer: 5′-GGACTAGTGGTCCTTGATCTCAGACTGAA-3′; reverse primer: 5′-GGACTAGTAGCCATATGAGCAGAGGAA-3′.
miRNA array.
Affymetrix GeneChip miRNA array was used for miRNA profiling in control and cyclopamine-treated lung organ cultures. Array and result analyses were performed by Boston University Microarray Facility with an on-site biostatistician. The Affymetrix miRNA QC tool was used for background subtraction, calculation of detection p value, and array normalization. Briefly, background signals generated by anti-genomic probes with similar GC content are used for background subtraction for those miRNA probes with the same GC content. Detection p value for each specific probe was calculated based on a Wilcoxon Rank-Sum test of the miRNA probe set signals compared to the distribution of signals from GC content-matched anti-genomic probes. To address the variation of each chip (each experiment), quintile normalization approach will be used to normalize probe intensities across chips. Experiments were performed in triplicate. A two-sample t test was performed to identify upregulated miRNA expression in cyclopamine-treated cultures (p < 0.05 was considered significant).
In situ hybridization. In situ hybridization to detect BDNF mRNA and miR-206 expression was performed on 15 μm frozen sections using digoxigenin-labeled antisense RNA probe and LNA (locked nucleic acid) probe (Exiqon), respectively. The detailed protocol was described previously (Ai et al., 2007; Obernosterer et al., 2007). To generate the antisense probe for BDNF mRNA, a 126 bp DNA fragment was cloned into pGEM-T vector (Promega) after RT-PCR using the following primers: BDNF_forward: 5′-CCACTGCCGGGGATCCGAGA-3′; BDNF_reverse: 5′TTTCATGGGCGCCGCCTTCA-3′.
Quantitative RT-PCR.
Lungs were collected for RNA isolation using RNeasy Mini Kit (Qiagen). To quantify BDNF mRNA levels, total RNA (1 μg) was reverse transcribed using SuperScript III First-Strand Synthesis SuperMix (Invitrogen). Quantitative PCR was performed using Power SYBR Green PCR Master Mix (Applied Biosystems) and StepOnePlus Real-Time PCR System. The relative level of BDNF mRNA expression was calculated by normalizing to β-actin mRNA using ΔCt (cycle threshold difference). Primers used in PCR are as follows: BDNF_forward: 5′-CCACTGCCGGGGATCCGAGA-3′; BDNF_reverse: 5′-TTTCATGGGCGCCGCCTTCA-3′; β-actin_forward: 5′-GCAGCTCCTTCGTTGCCGGT-3′; β-actin_reverse: 5′-TACAGCCCGGGGAGCATCGT-3′.
To quantify miR-206 levels, 10 ng of RNA was subjected to reverse transcription using the TaqMan Micro-RNA Reverse Transcription Kit (Applied Biosystems). TaqMan MicroRNA assays (Applied Biosystems) were used for quantitative RT-PCR. Relative level of miR-206 expression was calculated by normalizing to U6 using ΔCt. Each experiment was performed in triplicate.
Western blot analysis.
Embryonic lungs were homogenized in RIPA buffer containing proteinase inhibitor (Roche Diagnostics) using PowerGen 125 (Fisher Scientific). Cell lysates were collected and mixed with an equal volume of 2× Laemmli buffer. Proteins in the cell lysate (20 μg) were separated by NuPAGE Bis-Tris gels (12%, Invitrogen) before transference onto Hybond-ECL nitrocellulose membrane (GE Healthcare). Membranes were blocked with 5% BSA for an hour and incubated with primary antibodies (rabbit anti-BDNF (1:500, H-117, catalog no. sc-20981), rabbit anti-GAPDH (1:100,000, Ab8245, Abcam) in 2.5% BSA overnight at 4°C. After washing, the membrane was incubated in the secondary antibody followed by detection with ECL reagents.
Statistical analysis.
The Student's t test was used for all analyses.
Results
BDNF is required for ASM innervation
The NGF family plays essential roles in neural innervation of target tissues (Tucker et al., 2001; Ginty and Segal, 2002). To identify neurotrophic factors that are required for ASM innervation, we characterized the expression of the NGF family in the embryonic lung. We found that BDNF was expressed selectively by ASM and vascular smooth muscle cells at E14.5, while NGF and neurotrophin 3 (NT3) were not detected (Fig. 1A). Neurotrophin 4 (NT4), which binds to the same high affinity TrkB receptor as BDNF, is also expressed by ASM but is restricted to proximal airways (Fig. 1A).
Figure 1.

BDNF is essential for the innervation of ASM. A, Immunohistochemistry to analyze the expression of the NGF family members in the E14.5 lung. Arrows point to ASM that is immunolabeled with the BDNF or NT4 antibody. Asterisk marks a blood vessel. Notably, the vascular smooth muscle (marked by an arrow) also expresses BDNF, while endothelial cells are not labeled by the BDNF antibody. B, Whole-mount TuJ1 immunolabeling of axons within the left lobe of wild-type and BDNF−/− embryos at E14.5. Inserts show enlarged images of TuJ1 immunolabeling of the distal lung. C, Immunolabeling of TuJ1+ axons and SMA+ ASM in wild-type and BDNF−/− lungs at E16.5. Arrows point to TuJ1-immunoreactive axons that are in the proximity of the ASM. D, Quantification of ASM innervation density (TuJ1+ area/SMA+ area) at E16.5. Data represent mean and SEM of quantification from 15 nonoverlapping sections, 5 from each timed embryos; *p = 0.023. E, BDNF deficiency has no effect on intrinsic neurons. Intrinsic neurons in the main bronchi at E16.5 were immunolabeled with the TuJ1 antibody. Similar to wild-type controls, BDNF −/− embryos have clusters of the cell bodies of intrinsic neurons (pointed by arrowheads) in the major bronchi without significant difference in their abundance. Scale bars, 50 μm. All images represent results from three age-matched embryos.
To investigate the role of BDNF in ASM innervation, we compared neurogenesis in wild-type and BDNF−/− lungs by immunohistochemistry using a TuJ1 antibody that recognizes the pan neuronal β-tubulin marker. BDNF deficiency had no effect on lung branching morphogenesis or the expression of SMA by ASM (Fig. 1B,C). However, compared to wild-type lungs, BDNF−/− lungs exhibit a significant reduction in the outgrowth and branching of axons, as shown by TuJ1 immunolabeling of the whole left lung lobes of E14.5 embryos and mid-lung sections of E16.5 embryos (Fig. 1B,C). We quantified ASM innervation density by-calculating the ratio between TuJ1 immunoreactivity and SMA immunoreactivity in E16.5 lung sections. BDNF−/− lungs had ∼50% reduction in ASM innervation density compared to that of wild-type controls (Fig. 1C,D), demonstrating that BDNF is an essential target-derived neurotrophic factor for ASM innervation. Notably, residual innervation in BDNF−/− lungs is mostly detected in the proximal airway where NT4 is expressed (Fig. 1A,B), suggesting that NT4 plays a partially redundant role in ASM innervation (García-Suárez et al., 2009). In addition, the intrinsic neurons in the main bronchi were not affected in BDNF−/− embryos (Fig. 1E). This observation provides further evidence that distinct signals are required for intrinsic and extrinsic neurogenesis in the respiratory tract, and that intrinsic neurons contribute little to ASM innervation in the distal lung.
BDNF expression is regulated post-transcriptionally
To determine whether BDNF expression is temporally associated with axon outgrowth, we analyzed BDNF protein level in the lung between E11.5 and E15.5 by Western blot analysis. BDNF protein is undetectable until E12.5, when SMA expression is initiating in the lung bud (Tollet et al., 2001; Langsdorf et al., 2011). At E13.5, BDNF protein is detected in the lung, and the level peaks around E14.5 (Fig. 2A,B). Therefore, BDNF protein expression is temporally coordinated during ASM formation and initiation of axon outgrowth into the lung.
Figure 2.

Temporal expression of BDNF mRNA and protein in the embryonic lung. A, Analysis of the temporal expression of BDNF in the developing lung between E11.5 and E15.5 by Western blot analysis. B, Relative levels of BDNF protein were quantified by normalizing to the GAPDH internal control; **p = 0.007, *p = 0.03. C, Temporal quantification of BDNF mRNA levels by quantitative RT-PCR using β-actin mRNA as internal control; *p = 0.04. D, In situ hybridization to detect BDNF mRNA expression in E12.5 and E13.5 lungs. Lung epithelium is outlined by dotted lines. The sense probe was used as a control for background signal; n.s., Not significant. Data represent mean and SEM of three independent experiments.
The dynamics of BDNF mRNA expression differ from those of the protein in the developing lung. Quantitative RT-PCR assays showed that BDNF mRNA is detectable as early as E11.5. The relative level of BDNF mRNA is reduced at E14.5 and E15.5 compared to earlier embryonic stages (Fig. 2C). We confirmed BDNF mRNA expression in the lung mesenchyme at E12.5 and in ASM at E13.5 by in situ hybridization (Fig. 2D). The dissociation between the time courses of BDNF protein expression and mRNA expression suggests that a post-transcriptional mechanism represses BDNF protein production before the formation of ASM and innervation.
Shh signaling promotes BDNF expression via a post-transcriptional mechanism
Shh signaling is required for ASM development (Litingtung et al., 1998; Pepicelli et al., 1998). We assayed Shh signaling activity in the embryonic lung by X-gal staining of a reporter Ptc1lacZ/+ line (Goodrich et al., 1997). Shh signaling is active in the lung mesenchyme at E12.5, before SMA expression, and persists in ASM up to E16.5 (Fig. 3A) (Bellusci et al., 1997). Based on our finding that BDNF protein expression is temporally associated with ASM formation (Fig. 2A), we speculated that Shh signaling may regulate BDNF expression. To test this hypothesis, we perturbed the Shh signaling activity by small molecule compounds and assessed the effect of these perturbations on BDNF expression in lung organ cultures. Lungs were dissected from E11.5 embryos and cultured in an air–media interface for 48 h before protein and total RNA were extracted for Western blot analysis and quantitative RT-PCR. Compared to the DMSO control, treatment with cyclopamine (0.5 μm), which blocks Shh signaling (Fig. 3B,C), diminished BDNF protein level by >7-fold (Fig. 3D). In contrast, treatment with the Shh signaling agonist SAG (1 μg/ml) (Fig. 3B,C) increased BDNF protein level by 4.8-fold compared to controls (Fig. 3D). However, BDNF mRNA levels were not significantly affected by these treatments (Fig. 3E), suggesting that Shh signaling post-transcriptionally induces changes in BDNF protein expression. This observation, together with the previous finding of temporally dissociated BDNF protein and mRNA expressions (Fig. 2), suggest that Shh signaling functions to release post-transcriptional inhibition of BDNF expression in ASM.
Figure 3.
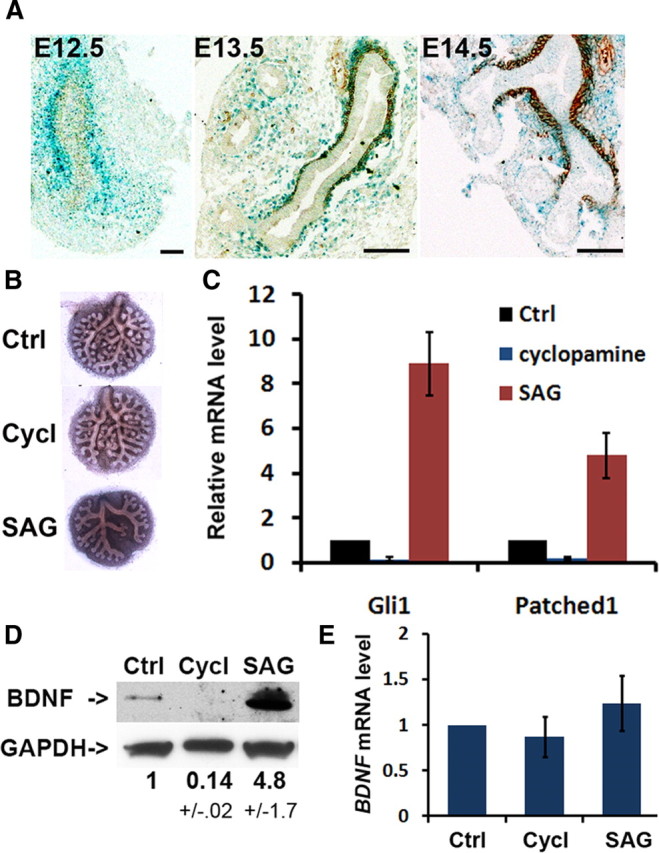
Shh signaling post-transcriptionally activates BDNF expression in lung organ cultures. A, Shh signaling in the lung mesenchyme and ASM in the embryonic lung assayed by X-gal staining of Ptc1LacZ/+ mouse embryos followed by immunolabeling with the SMA antibody to detect ASM. Shh signaling is activated in the progenitors of ASM before SMA expression at E12.5 and persists at E13.5 and E14.5 in the SMA+ ASM. B–E, E11.5 lung cultures were cultured in the presence of DMSO as control (Ctrl), 0.5 μm cyclopamine (Cycl, inhibitor), or 1 μg/ml SAG (agonist) for 48 h before morphological and gene expression analysis. Cyclopamine and SAG differentially affects lung branching morphogenesis in B and mRNA expression of Shh target genes in C. BDNF protein and mRNA expression in lung explants were analyzed by Western blot in D and quantitative RT-PCR in E, respectively. Data represent mean and SEM of results from three independent experiments.
Shh signaling inhibits miR-206 expression
To characterize downstream effectors of the Shh pathway that regulate gene expression at a post-transcriptional level, we performed miRNA profiling with control and cyclopamine-treated embryonic lung explants. miRNAs are small noncoding RNAs that suppress gene expression by inducing cleavage and/or translational repression of mRNAs after binding to mRNAs through their seed sequences (Mendell, 2005). In the context of our findings, we sought to identify miRNAs that are upregulated by cyclopamine treatment. Five miRNAs were identified in our screen (Table 1). Among these five, miR-206 was the only miRNA that is conserved across species and has BDNF as one of its predicted gene targets. Therefore, we selected miR-206 for further analysis. We confirmed that the miR-206 level is regulated by Shh signaling in lung explants by quantitative PCR (qPCR) (Fig. 4A). Consistent with the results of miRNA array, cyclopamine treatment increased miR-206 levels by ∼8-fold, while SAG reduced miR-206 expression by ∼4-fold in cultured lung explants (Fig. 4A).
Table 1.
miRNAs whose expression was induced by cyclopamine treatment in lung organ cultures
| miRNA | Fold of change |
|---|---|
| miR-125b-3p | 2.11 |
| miR-694 | 4.44 |
| miR-206 | 7.51 |
| miR-697 | 8.26 |
| miR-764-5p | 9.23 |
Embryonic lung organ cultures were treated with DMSO control or 0.5 μm cyclopamine for 48 h followed by RNA isolation and miRNA profiling. miRNAs with more than 2-fold increase in expression by cyclopamine and greater than p < 0.05 were considered significant.
Figure 4.

Shh signaling represses miR-206 expression. A, Quantification of miR-206 level in lung explants that were treated with DMSO control (Ctrl), cyclopamine (Cycl), or SAG by qPCR; **p < 0.01. B, In situ hybridization to detect miR-206 expression in the embryonic lungs at E12.5, E13.5, and E15.5. Images show miR-206 signal after 4 h of color development at 37°C. Inset in the E15.5 image shows miR-206 signal in the lung mesenchyme after additional overnight color development. Scale bar, 50 μm. C, Quantitative PCR to assay miR-206 expression in sorted GFP+ cells and control GFP− cells from lungs of SMA-hrGFP transgenic mice at E13.5 and E15.5. D, Quantitative PCR to assess the expression of Gli1 and Patched 1, two target genes of Shh signaling, and miR-206 in the lung of Dermo1-Cre;Rosa(SmoM2) embryos at E16.5. Littermates that were Dermo1-Cre− were used as controls; *p = 0.03, **p = 0.007. Data represent mean and SEM of results from three independent experiments.
To test whether Shh signaling regulates miR-206 expression in vivo, we first assessed whether miR-206 is expressed in the lung mesenchyme by in situ hybridization. The specificity of the miR-206 antisense probe was validated by detection of specific signals in tissues that are previously shown to express miR-206 (skeletal muscle, neural tube, and sensory ganglion) (data not shown) (Rao et al., 2006; Williams et al., 2009). Utilizing similarly stringent in situ conditions, we detected miR-206 in the lung mesenchyme between E12.5 and E15.5 (Fig. 4B). miR-206 was hardly detectable at E15.5 compared to E12.5 (Fig. 4B), suggesting that miR-206 expression is reduced at E15.5. miR-206 expression in the lung mesenchyme at E15.5 became evident after an extended period of detection (Fig. 4B, inset).
To further prove that miR-206 is preferentially expressed by lung mesenchyme, we performed a second assay using purified cells from the embryonic lung of a SMA-hrGFP transgenic mouse that selectively expresses a green fluorescent protein, hrGFP, under the control of a rat SMA promoter (Ghosh et al., 2011). Using this mouse, we sorted the GFP+ cells from the lungs of E13.5 and E15.5 embryos by flow cytometry. As an internal control, the CD31−;CD45−;GFP− population was also collected. Cells from the whole litter were pooled, and qPCR was performed with extracted total RNA. Consistent with our in situ hybridization results, miR-206 was found to be preferentially expressed in the lung mesenchyme compared to negative internal controls (Fig. 4C), thereby providing further evidence for an interaction between Shh signaling and miR-206 expression. Notably, the miR-206 level in SMA-GFP+ cells was ∼20-fold lower at E15.5 than at E12.5 (Fig. 4C), while BDNF protein expression had an opposite temporal change (Fig. 2A), consistent with a role of miR-206 in post-transcriptional silencing of BDNF gene expression.
To further demonstrate that Shh signaling blocks miR-206 expression in vivo, we analyzed changes in miR-206 levels in a Dermo1-Cre;Rosa(SmoM2) mouse line. SmoM2 contains a W539L point mutation and, upon Cre-mediated excision of a stop fragment in the Rosa(SmoM2) allele, Shh signaling is constitutively active in a ligand-independent manner (Mao et al., 2006). By crossing with a Dermo1-Cre line (Yin et al., 2008), Shh signaling is activated in the lung mesenchyme of Dermo1-Cre;Rosa(SmoM2)embryos. We analyzed changes in expression of Shh signaling target genes and miR-206 at E16.5 in mutant lungs, a time-point when endogenous Shh signaling activity is reduced in wild-type lungs compared to earlier stages (Bellusci et al., 1997). A large majority of Dermo1-Cre;Rosa(SmoM2) embryos die before E16.5. Among the few that survived, qPCR analysis showed that Gli1 and Patched1 mRNA levels were increased in the mutant lung (Fig. 4D), confirming hyperactive Shh signaling. miR-206 levels were significantly reduced by threefold in these mutant lungs compared to Rosa(SmoM2) controls (Fig. 4D), providing evidence that Shh signaling blocks miR-206 expression in vivo.
miR-206 directly represses BDNF expression in the ASM to affect innervation
A direct role for miR-206 in suppressing BDNF gene expression was tested by luciferase assay. We cloned a ∼1.5 kb BDNF 3′-UTR fragment that includes all three predicted miR-206 binding sites (positions 212–218, 406–412, and 1305–1311; www.targetscan.org) into the 3′-end of the firefly luciferase cDNA. Luciferase reporter assays were performed in 293T cells that were cotransfected with the luciferase construct and a lentiviral miR-206 expression vector. A mutant miR-206 that has the deleted 6 bp seed sequence (miR-206mut) and therefore can no longer bind to mRNA was used as a control. Compared to miR-206mut, miR-206 reduced the luciferase activity by fivefold, indicating that miR-206 is a direct post-transcriptional repressor of BDNF expression (Fig. 5A).
Figure 5.

miR-206 represses BDNF expression. A, Functional analysis of miR-206 binding sites at BDNF 3′-UTR by luciferase assay. 293T cells were transfected with the luciferase construct and a lentiviral vector to overexpress miR-206. miR-206 with deleted seed sequences (miR-206mut) was used as a control. Experiments were repeated three times in triplicate; **p = 0.004. B, Immunostaining of E16.5 lungs of wild-type and miR-206−/− embryos with the SMA antibody. C, Western blot analysis to assess changes in BDNF protein level in E14.5. GAPDH was the control for loading. D, Quantification of normalized BDNF levels at E13.5 and E14.5 in wild-type and miR-206−/− lungs; *p = 0.03, **p < 0.01. E, Quantitative RT-PCR to assay BDNF mRNA levels in wild-type littermates and miR-206−/− lungs at E14.5. Three pairs of wild-type and miR-206−/− littermates were used for each assay except in panel C, where six pairs of wild-type and miR-206−/− embryos at E13.5 were assayed.
To test the role of miR-206 in regulating BDNF expression in vivo, we compared BDNF mRNA and protein levels in the lungs of wild-type and miR-206−/− embryos. miR-206 deficiency had no effect on lung morphogenesis or ASM development (Williams et al., 2009) as based on histology and SMA immunolabeling (Fig. 5B). However, compared to wild-type lungs at E13.5 and E14.5, the level of BDNF protein was approximately doubled in miR-206−/− lungs (Fig. 5C,D), while BDNF mRNA level is not changed (Fig. 5E). Notably, the increase in BDNF protein levels in miR-206−/− lungs was higher at E13.5 than at E14.5, consistent with the expression dynamics and a role of miR-206 in post-transcriptional repression of BDNF expression.
We assessed the effect of elevated BDNF levels on ASM innervation in miR-206−/− embryos by whole-mount fluorescent TuJ1 staining followed by confocal imaging. We compiled Z-stack images of immunolabeled lung lobes to follow major axons and their branches. Compared to wild-type controls, miR-206−/− lungs not only show more axon branches, but axons also extend more peripherally into the distal lung lubes (Fig. 6A). In addition, we observed significantly increased ASM innervation density in E14.5 miR-206−/− lungs compared to WT lungs after quantification based on immune-labeled mid-lung sections with the SMA and TuJ1 antibodies (Fig. 6B,C).
Figure 6.
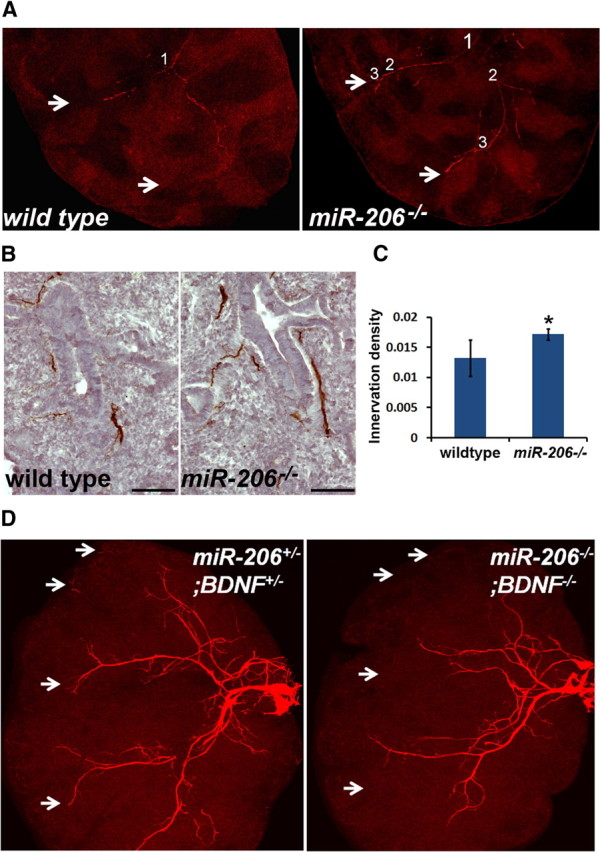
miR-206−/− embryos exhibit increased ASM innervation that is dependent on BDNF. A, Confocal images of TuJ1 immunolabeled right apical lung lobes from E13.5 wild-type and miR-206−/− embryos. Compiled Z-stack images showed branching of a major axon in the lower half of the lobe. Branching points are marked by numbers. White arrows mark the tips of axons in miR-206−/− lobes and corresponding locations in wild-type lobes. B, C, Quantification of innervation density in E14.5 wild-type and miR-206−/− lungs. TuJ1 immunolabeled lung sections were shown in B. Scale bars, 50 μm. Innervation density (TuJ1+ area/SMA+ area) was quantified based on adjacent lung sections that were immunolabeled with the TuJ1 and SMA antibodies. A total of 15 nonoverlapping sections from 3 embryos were quantified for each marker, 5 sections per embryo; *p = 0.017. D, Confocal images of TuJ1-immunolabeled axons in the right apical lung lobe of BDNF+/−;miR-206+/− littermate controls and BDNF−/−;miR-206−/− embryos at E14.5. White arrow points to the tips of extended axons in the control distal lung and corresponding location in the lung of BDNF−/−;miR-206−/− embryos. Three wild-type and miR-206−/− littermates were analyzed in each experiment.
miR-206 deficiency may affect ASM innervation by affecting the expression of BDNF or other target genes. To further establish a causal link between elevated BDNF levels and increased innervation in miR-206−/− lungs, we generated miR-206−/−;BDNF−/− double-mutant embryos and assessed the innervation defects in double mutant lungs. If miR-206 represses BDNF protein expression to affect ASM innervation, miR-206−/−;BDNF−/− embryos would exhibit similarly defective ASM innervation as BDNF−/− embryos. Otherwise, the double mutant embryos would have a different phenotype from that of BDNF−/− embryos. Whole-mount TuJ1 immunolabeling of the E14.5 right apical lobes showed that BDNF deficiency in the miR-206−/− background diminishes innervation in the distal lung, a phenotype reminiscent of that of BDNF−/− lungs (Fig. 6D, compared to Fig. 1B). In addition, we quantified TuJ1 immunoreactive areas using compressed Z-stack confocal images. miR-206−/−;BDNF−/− lungs showed a ∼33% reduction in TuJ1 immunoreactivity, as compared to double heterozygous littermate controls at E14.5. Notably, this reduction was identical to that of BDNF−/− lungs. Therefore, miR-206 directly represses BDNF expression to regulate ASM innervation.
Discussion
Our study investigates mechanisms that control ASM innervation during lung morphogenesis. We identified BDNF as an essential ASM-derived neurotrophic signal for extrinsic neurons to extend their axons into the distal lung. Further investigation reveals a functional interaction between Shh signaling and BDNF protein expression. This interaction is mediated by miR-206, a direct post-transcriptional inhibitor of BDNF gene expression. In this regard, we showed that Shh signaling represses miR-206 expression, thereby elevating BDNF protein expression levels. Our findings uncover a role for Shh signaling in BDNF-dependent ASM innervation. In our model (Fig. 7), Shh signaling not only induces the specification of ASM (Litingtung et al., 1998; Pepicelli et al., 1998; Cardoso and Lü, 2006; Morrisey and Hogan, 2010), it also represses an inhibitory miR-206 to activate BDNF protein expression, which in turn facilitates ASM innervation. The dual roles of Shh signaling in the lung mesenchyme provide a mechanism to coordinate ASM formation and innervation during airway branching (Fig. 7).
Figure 7.

Model of coordinated ASM formation and BDNF-dependent innervation by Shh regulation of miR-206. Shh signaling induces ASM formation and lung branching morphogenesis. In addition, Shh signaling releases miR-206-mediated suppression of BDNF expression in the ASM, thereby inducing axon outgrowth. The dual roles of Shh signaling coordinate ASM formation and innervation in the embryonic lung.
The cascade of Shh/miR-206/BDNF is not only supported by our functional studies, but is also consistent with the timing of Shh signaling activation and the dynamics of miR-206 and BDNF expression. Shh signaling is detectable as early as lung bud forms (Bellusci et al., 1997). In addition, Shh signaling is activated in the lung mesenchyme and in ASM up until E16.5, and its activity starts to decrease after E16.5 (Fig. 3A) (Bellusci et al., 1997). Between E12.5 and E15.5, when ASM forms and gets innervated, we show that miR-206 levels in GFP+ mesenchymal cells decrease. This is consistent with persistent activation of the inhibitory Shh signaling pathway during this developmental window. Simultaneously, we show that BDNF protein levels increase at this time. Based on our results that the BDNF gene is a direct target of miR-206, the increase in BDNF levels during ASM innervation is likely due to reduced expression of miR-206.
Mechanisms that underlie Shh regulation of miR-206 levels will require further investigation. One possibility is that Shh signaling regulates the transcription of miR-206. However, none of the transcriptional factors that are predicted to bind to the miR-206 promoter were found to be significantly affected by cyclopamine treatment in lung organ cultures (Rao et al., 2006). Therefore, it is unlikely that Shh signaling represses the transcription of miR-206. Alternatively, Shh signaling may regulate miR-206 levels via post-transcriptional mechanisms. Two recent studies show that activation of the TGFβ/BMP pathways triggers the binding of their downstream factors, Smads, to the miR-206 precursor, which in turn blocks processing of the miR-206 precursor into its mature form (Sato et al., 2009; Winbanks et al., 2011). These findings suggest the possibility that Shh signaling, which activates BMP signaling in the lung mesenchyme (Bellusci et al., 1996; Weaver et al., 2003), may indirectly block maturation of miR-206.
Results of our luciferase assays demonstrate that miR-206 directly binds to the BDNF 3′-UTR to block BDNF expression. However, a previous study proposed an indirect role of miR-206 in silencing BDNF expression (Kim et al., 2006). In that study, overexpressed miR-206 potently reduced BDNF protein levels in C2C12 myoblast cells but had little effect on the luciferase activity (Kim et al., 2006). To explain this discrepancy, we compared BDNF 3′-UTR sequences tested in both luciferase assays. Our study tested a 1.5 kb fragment that contains all three miR-206 binding sites, while a 1.0 kb fragment tested by the other group contained partial BDNF coding sequences and only one miR-206 binding site within the 3′-UTR closest to the stop codon (Kim et al., 2006). The difference in cloned BDNF 3′-UTR sequences likely explains discrepant results between the two luciferase assays. It also suggests that one miR-206 binding site in the BDNF 3′-UTR may be insufficient for miR-206 suppression. Interestingly, multiple BDNF mRNA variants are expressed due to different promoters, alternative splicing, and multiple poly(A) sites (Ohara et al., 1992; Timmusk et al., 1993). The possibility that BDNF mRNA variants with differences in miR-206 binding sites at 3′-UTR are differentially regulated by miR-206 adds further potential complexities to BDNF expression regulation and function.
Our current study further establishes that distinct neurogenic signals are required for intrinsic versus extrinsic neurogenesis in the developing respiratory tract. Unlike intrinsic neurons that are dependent on GDNF family signals for survival and differentiation (Langsdorf et al., 2011), extrinsic neurons require BDNF, and likely NT4, for axon outgrowth to innervate a large majority of ASM in the lung (García-Suárez et al., 2009).
The essential role of neurotrophins in ASM innervation during embryonic development may provide insights into pathogenic changes in innervation in airway diseases (de Vries et al., 2006; Rochlitzer et al., 2006). It has been reported that the levels of NGF family members, such as NGF, BDNF and NT4, are elevated in bronchial lavage fluid of patients with inflammatory airway diseases, such as asthma (Bonini et al., 1996; Tortorolo et al., 2005). It is possible that changes in neurotrophin expression leads to alterations in ASM innervation, thereby playing a role in airway hyper-reactivity. If true, blockade of neurotrophin signaling may provide a novel approach to treat asthma.
Footnotes
This study was supported by a Boston University Medical Campus (BUMC) startup fund (to X.A.) and grants from U.S. National Institutes of Health (to J.L. and A.F.). We thank the microarray facility at BUMC for analysis and assistance with statistics, Dr. Eric Olson at University of Texas Southwestern Medical Center, Dallas, TX for providing miR-206−/− mice, and Dr. Matt Layne, Boston University Medical School, for critical reading and commenting on this manuscript.
The authors declare no competing financial interests.
References
- Adriaensen D, Scheuermann DW. Neuroendocrine cells and nerves of the lung. Anat Rec. 1993;236:70–86. doi: 10.1002/ar.1092360111. [DOI] [PubMed] [Google Scholar]
- Ai X, Kitazawa T, Do AT, Kusche-Gullberg M, Labosky PA, Emerson CP., Jr SULF1 and SULF2 regulate heparan sulfate-mediated GDNF signaling for esophageal innervation. Development. 2007;134:3327–3338. doi: 10.1242/dev.007674. [DOI] [PubMed] [Google Scholar]
- Bellusci S, Henderson R, Winnier G, Oikawa T, Hogan BL. Evidence from normal expression and targeted misexpression that bone morphogenetic protein (Bmp-4) plays a role in mouse embryonic lung morphogenesis. Development. 1996;122:1693–1702. doi: 10.1242/dev.122.6.1693. [DOI] [PubMed] [Google Scholar]
- Bellusci S, Furuta Y, Rush MG, Henderson R, Winnier G, Hogan BL. Involvement of Sonic hedgehog (Shh) in mouse embryonic lung growth and morphogenesis. Development. 1997;124:53–63. doi: 10.1242/dev.124.1.53. [DOI] [PubMed] [Google Scholar]
- Bonini S, Lambiase A, Bonini S, Angelucci F, Magrini L, Manni L, Aloe L. Circulating nerve growth factor levels are increased in humans with allergic diseases and asthma. Proc Natl Acad Sci U S A. 1996;93:10955–10960. doi: 10.1073/pnas.93.20.10955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canning BJ. Reflex regulation of airway smooth muscle tone. J Appl Physiol. 2006;101:971–985. doi: 10.1152/japplphysiol.00313.2006. [DOI] [PubMed] [Google Scholar]
- Cardoso WV, Lü J. Regulation of early lung morphogenesis: questions, facts and controversies. Development. 2006;133:1611–1624. doi: 10.1242/dev.02310. [DOI] [PubMed] [Google Scholar]
- de Vries A, Engels F, Henricks PA, Leusink-Muis T, McGregor GP, Braun A, Groneberg DA, Dessing MC, Nijkamp FP, Fischer A. Airway hyper-responsiveness in allergic asthma in guinea-pigs is mediated by nerve growth factor via the induction of substance P: a potential role for trkA. Clin Exp Allergy. 2006;36:1192–1200. doi: 10.1111/j.1365-2222.2006.02549.x. [DOI] [PubMed] [Google Scholar]
- García-Suárez O, Pérez-Pinera P, Laurà R, Germana A, Esteban I, Cabo R, Silos-Santiago I, Cobo JL, Vega JA. TrkB is necessary for the normal development of the lung. Respir Physiol Neurobiol. 2009;167:281–291. doi: 10.1016/j.resp.2009.06.001. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Paez-Cortez JR, Boppidi K, Vasconcelos M, Roy M, Cardoso W, Ai X, Fine A. Activation dynamics and signaling properties of Notch3 in the developing pulmonary artery. J Biol Chem. 2011;286:22678–22687. doi: 10.1074/jbc.M111.241224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginty DD, Segal RA. Retrograde neurotrophin signaling: Trk-ing along the axon. Curr Opin Neurobiol. 2002;12:268–274. doi: 10.1016/s0959-4388(02)00326-4. [DOI] [PubMed] [Google Scholar]
- Goodrich LV, Milenković L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277:1109–1113. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- Joos GF. The role of neuroeffector mechanisms in the pathogenesis of asthma. Curr Allergy Asthma Rep. 2001;1:134–143. doi: 10.1007/s11882-001-0081-8. [DOI] [PubMed] [Google Scholar]
- Kc P, Martin RJ. Role of central neurotransmission and chemoreception on airway control. Respir Physiol Neurobiol. 2010;173:213–222. doi: 10.1016/j.resp.2010.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerzel S, Päth G, Nockher WA, Quarcoo D, Raap U, Groneberg DA, Dinh QT, Fischer A, Braun A, Renz H. Pan-neurotrophin receptor p75 contributes to neuronal hyperreactivity and airway inflammation in a murine model of experimental asthma. Am J Respir Cell Mol Biol. 2003;28:170–178. doi: 10.1165/rcmb.4811. [DOI] [PubMed] [Google Scholar]
- Kim HK, Lee YS, Sivaprasad U, Malhotra A, Dutta A. Muscle-specific microRNA miR-206 promotes muscle differentiation. J Cell Biol. 2006;174:677–687. doi: 10.1083/jcb.200603008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langsdorf A, Radzikinas K, Kroten A, Jain S, Ai X. Neural crest cell origin and signals for intrinsic neurogenesis in the mammalian respiratory tract. Am J Respir Cell Mol Biol. 2011;44:293–301. doi: 10.1165/rcmb.2009-0462OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litingtung Y, Lei L, Westphal H, Chiang C. Sonic hedgehog is essential to foregut development. Nat Genet. 1998;20:58–61. doi: 10.1038/1717. [DOI] [PubMed] [Google Scholar]
- Mao J, Ligon KL, Rakhlin EY, Thayer SP, Bronson RT, Rowitch D, McMahon AP. A novel somatic mouse model to survey tumorigenic potential applied to the Hedgehog pathway. Cancer Res. 2006;66:10171–10178. doi: 10.1158/0008-5472.CAN-06-0657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell JT. MicroRNAs: critical regulators of development, cellular physiology and malignancy. Cell Cycle. 2005;4:1179–1184. doi: 10.4161/cc.4.9.2032. [DOI] [PubMed] [Google Scholar]
- Morrisey EE, Hogan BL. Preparing for the first breath: genetic and cellular mechanisms in lung development. Dev Cell. 2010;18:8–23. doi: 10.1016/j.devcel.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers AC, Undem BJ, Weinreich D. Electrophysiological properties of neurons in guinea pig bronchial parasympathetic ganglia. Am J Physiol. 1990;259:L403–L409. doi: 10.1152/ajplung.1990.259.6.L403. [DOI] [PubMed] [Google Scholar]
- Obernosterer G, Martinez J, Alenius M. Locked nucleic acid-based in situ detection of microRNAs in mouse tissue sections. Nat Protoc. 2007;2:1508–1514. doi: 10.1038/nprot.2007.153. [DOI] [PubMed] [Google Scholar]
- Ohara O, Gahara Y, Teraoka H, Kitamura T. A rat brain-derived neurotrophic factor-encoding gene generates multiple transcripts through alternative use of 5′ exons and polyadenylation sites. Gene. 1992;121:383–386. doi: 10.1016/0378-1119(92)90148-i. [DOI] [PubMed] [Google Scholar]
- Pepicelli CV, Lewis PM, McMahon AP. Sonic hedgehog regulates branching morphogenesis in the mammalian lung. Curr Biol. 1998;8:1083–1086. doi: 10.1016/s0960-9822(98)70446-4. [DOI] [PubMed] [Google Scholar]
- Rao PK, Kumar RM, Farkhondeh M, Baskerville S, Lodish HF. Myogenic factors that regulate expression of muscle-specific microRNAs. Proc Natl Acad Sci U S A. 2006;103:8721–8726. doi: 10.1073/pnas.0602831103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds SD, Giangreco A, Power JH, Stripp BR. Neuroepithelial bodies of pulmonary airways serve as a reservoir of progenitor cells capable of epithelial regeneration. Am J Pathol. 2000;156:269–278. doi: 10.1016/S0002-9440(10)64727-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochlitzer S, Nassenstein C, Braun A. The contribution of neurotrophins to the pathogenesis of allergic asthma. Biochem Soc Trans. 2006;34:594–599. doi: 10.1042/BST0340594. [DOI] [PubMed] [Google Scholar]
- Sato MM, Nashimoto M, Katagiri T, Yawaka Y, Tamura M. Bone morphogenetic protein-2 down-regulates miR-206 expression by blocking its maturation process. Biochem Biophys Res Commun. 2009;383:125–129. doi: 10.1016/j.bbrc.2009.03.142. [DOI] [PubMed] [Google Scholar]
- Timmusk T, Palm K, Metsis M, Reintam T, Paalme V, Saarma M, Persson H. Multiple promoters direct tissue-specific expression of the rat BDNF gene. Neuron. 1993;10:475–489. doi: 10.1016/0896-6273(93)90335-o. [DOI] [PubMed] [Google Scholar]
- Tollet J, Everett AW, Sparrow MP. Spatial and temporal distribution of nerves, ganglia, and smooth muscle during the early pseudoglandular stage of fetal mouse lung development. Dev Dyn. 2001;221:48–60. doi: 10.1002/dvdy.1124. [DOI] [PubMed] [Google Scholar]
- Tollet J, Everett AW, Sparrow MP. Development of neural tissue and airway smooth muscle in fetal mouse lung explants: a role for glial-derived neurotrophic factor in lung innervation. Am J Respir Cell Mol Biol. 2002;26:420–429. doi: 10.1165/ajrcmb.26.4.4713. [DOI] [PubMed] [Google Scholar]
- Tortorolo L, Langer A, Polidori G, Vento G, Stampachiacchere B, Aloe L, Piedimonte G. Neurotrophin overexpression in lower airways of infants with respiratory syncytial virus infection. Am J Respir Crit Care Med. 2005;172:233–237. doi: 10.1164/rccm.200412-1693OC. [DOI] [PubMed] [Google Scholar]
- Tucker KL, Meyer M, Barde YA. Neurotrophins are required for nerve growth during development. Nat Neurosci. 2001;4:29–37. doi: 10.1038/82868. [DOI] [PubMed] [Google Scholar]
- Undem BJ, Myers AC, Barthlow H, Weinreich D. Vagal innervation of guinea pig bronchial smooth muscle. J Appl Physiol. 1990;69:1336–1346. doi: 10.1152/jappl.1990.69.4.1336. [DOI] [PubMed] [Google Scholar]
- Undem BJ, Kajekar R, Hunter DD, Myers AC. Neural integration and allergic disease. J Allergy Clin Immunol. 2000;106:S213–S220. doi: 10.1067/mai.2000.110153. [DOI] [PubMed] [Google Scholar]
- Weaver M, Batts L, Hogan BL. Tissue interactions pattern the mesenchyme of the embryonic mouse lung. Dev Biol. 2003;258:169–184. doi: 10.1016/s0012-1606(03)00117-9. [DOI] [PubMed] [Google Scholar]
- Weichselbaum M, Sparrow MP. A confocal microscopic study of the formation of ganglia in the airways of fetal pig lung. Am J Respir Cell Mol Biol. 1999;21:607–620. doi: 10.1165/ajrcmb.21.5.3721. [DOI] [PubMed] [Google Scholar]
- Weigand LA, Myers AC. Synaptic and membrane properties of parasympathetic ganglionic neurons innervating mouse trachea and bronchi. Am J Physiol Lung Cell Mol Physiol. 2010;298:L593–L599. doi: 10.1152/ajplung.00386.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams AH, Valdez G, Moresi V, Qi X, McAnally J, Elliott JL, Bassel-Duby R, Sanes JR, Olson EN. MicroRNA-206 delays ALS progression and promotes regeneration of neuromuscular synapses in mice. Science. 2009;326:1549–1554. doi: 10.1126/science.1181046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winbanks CE, Wang B, Beyer C, Koh P, White L, Kantharidis P, Gregorevic P. TGF-β regulates miR-206 and miR-29 to control myogenic differentiation through regulation of HDAC4. J Biol Chem. 2011;286:13805–13814. doi: 10.1074/jbc.M110.192625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, White AC, Huh SH, Hilton MJ, Kanazawa H, Long F, Ornitz DM. An FGF-WNT gene regulatory network controls lung mesenchyme development. Dev Biol. 2008;319:426–436. doi: 10.1016/j.ydbio.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]