

Pseudouridine synthases catalyze formation of the most abundant modification of functional RNAs by site-specifically isomerizing uridines to pseudouridines. While the structure and substrate specificity of these enzymes has been studied in detail, the kinetic and the catalytic mechanism of pseudouridine synthases remain unknown. This paper presents the first pre-steady-state kinetic analysis of three Escherichia coli pseudouridine synthases. All three showed very slow and rate-limiting catalytic steps. Thus, uniformly slow catalysis might be a general feature of pseudouridine synthases.
Keywords: pseudouridine, pseudouridine synthase, kinetic mechanism, RNA modification, pre-steady-state kinetic analysis
Abstract
Pseudouridine synthases catalyze formation of the most abundant modification of functional RNAs by site-specifically isomerizing uridines to pseudouridines. While the structure and substrate specificity of these enzymes have been studied in detail, the kinetic and the catalytic mechanism of pseudouridine synthases remain unknown. Here, the first pre-steady-state kinetic analysis of three Escherichia coli pseudouridine synthases is presented. A novel stopped-flow absorbance assay revealed that substrate tRNA binding by TruB takes place in two steps with an overall rate of 6 sec−1. In order to observe catalysis of pseudouridine formation directly, the traditional tritium release assay was adapted for the quench-flow technique, allowing, for the first time, observation of a single round of pseudouridine formation. Thereby, the single-round rate constant of pseudouridylation (kΨ) by TruB was determined to be 0.5 sec−1. This rate constant is similar to the kcat obtained under multiple-turnover conditions in steady-state experiments, indicating that catalysis is the rate-limiting step for TruB. In order to investigate if pseudouridine synthases are characterized by slow catalysis in general, the rapid kinetic quench-flow analysis was also performed with two other E. coli enzymes, RluA and TruA, which displayed rate constants of pseudouridine formation of 0.7 and 0.35 sec−1, respectively. Hence, uniformly slow catalysis might be a general feature of pseudouridine synthases that share a conserved catalytic domain and supposedly use the same catalytic mechanism.
INTRODUCTION
Functional RNAs are typically modified within the cell to enhance their structural and functional properties. Numerous different modifications of the ribose and the nucleobases have been identified (Limbach et al. 1994), of which pseudouridines (Ψ) are the most abundant modifications found in all major functional RNAs such as tRNA, rRNA, snRNA, and snoRNA (Charette and Gray 2000). Pseudouridines are isomers of uridines differing only in the glycosidic bond, which is a C5-glycosidic bond in pseudouridine instead of the canonical N1-glycosidic bond (Fig. 1A). While pseudouridines can form the same base-pairing interactions as uridines, the extra hydrogen bond donor in the nitrogenous base, the N1H imino group, can form additional interactions. For example, it has been shown that the N1H imino group can stabilize a water molecule between the nucleobase and the phosphate backbone, thereby rigidifying the local RNA structure (Arnez and Steitz 1994). Besides their widely accepted structural role, pseudouridines may also play additional functional roles for rRNA and snRNA. Most pseudouridines are located close to the functional centers of the RNAs, such as the ribosomal peptidyltransferase center, the ribosomal decoding center, or the spliceosomal branch site (Charette and Gray 2000; Decatur and Fournier 2002). Specifically, pseudouridines have been proposed to play a role in translation termination (Ejby et al. 2007; Liang et al. 2007) and in the first step of splicing by positioning the branch site adenosine (Newby and Greenbaum 2002; Yang et al. 2005). In accordance with their importance for RNA structure and function, the abundance of pseudouridines increases with the evolutionary complexity of an organism; while Escherichia coli rRNA contains 11 pseudouridines, about 100 of these modifications have been identified in human rRNA (Ofengand 2002).
FIGURE 1.

Structure of pseudouridine and modification sites in E. coli tRNAPhe. (A) Uridines in RNA are isomerized to pseudouridines (Ψ) by enzymes called pseudouridine synthases. The N1 and C5 atoms are indicated, which are part of the glycosidic bond in uridines and pseudouridine, respectively. Also, the tritium label at C5 is shown, which is released upon pseudouridine formation in the tritium release assay. (B) The positions of the three pseudouridines found in E. coli tRNAPhe are depicted in black, and the responsible pseudouridine synthase (RluA, TruA, or TruB) is indicated for each modification site. Note that the structure of yeast tRNAPhe shown here does not contain a pseudouridine at position 32 (Protein Data Bank ID code 4TRA [Westhof et al. 1988]). This figure was generated with Pymol (www.pymol.org).
The formation of pseudouridines is catalyzed by enzymes called pseudouridine synthases, which can be grouped in five families found in bacteria plus one additional pseudouridine synthase found in archaea and some eukaryotes (Hamma and Ferré-D'Amaré 2006; McCleverty et al. 2007). Each bacterial pseudouridine synthase catalyzes pseudouridine formation at one or a few distinct sites within cellular RNA. In addition, eukaryotes and archaea use H/ACA small (nucleolar) ribonucleoproteins which catalyze pseudouridylation at many different sites within cellular RNA with the help of many different box H/ACA guide RNAs (Ye 2007). Within the last decade, crystal structures of pseudouridine synthases from all six families have been determined (Hoang and Ferré-D'Amaré 2001, 2004; Ericsson et al. 2004; Kaya et al. 2004; Hoang et al. 2006; Hur and Stroud 2007; McCleverty et al. 2007; Alian et al. 2009). Despite substantial differences in the primary sequences, all pseudouridine synthases share the same fold in the catalytic domain and very similar active sites containing an essential aspartate residue (Hamma and Ferré-D'Amaré 2006). After a long debate on the inhibition mode of 5-fluorouracil for different pseudouridine synthases, it has recently been shown that all carefully analyzed pseudouridine synthases handle RNA containing 5-fluorouracil identically (McDonald et al. 2011). Based on all these findings, it is very likely that all pseudouridine synthases evolved from one common ancestor and share the same catalytic mechanism (Mueller 2002). However, this catalytic mechanism is still not unambiguously identified, as two alternatives have been proposed which are both in agreement with all data reported so far (McDonald et al. 2011); the catalytic aspartate could either attack the uracil base or the ribose to form a covalent intermediate (Kammen et al. 1988; Huang et al. 1998). Beyond the catalytic domain, pseudouridine synthases differ substantially from each other; many of these enzymes contain additional domains that contribute to the specificity of substrate RNA binding (Hamma and Ferré-D'Amaré 2006).
One of the best-studied pseudouridine synthases is E. coli TruB which catalyzes pseudouridylation at position 55 in the TΨC arm of all elongator tRNAs (Fig. 1B; Nurse et al. 1995). TruB was the first pseudouridine synthase to be crystallized in complex with its substrate RNA (Hoang and Ferré-D'Amaré 2001), and numerous biochemical investigations have been performed to investigate its substrate specificity and catalytic mechanism (Gu et al. 1998; Spedaliere and Mueller 2004; Spedaliere et al. 2004; Hamilton et al. 2005; Hoang et al. 2005; Phannachet et al. 2005). Furthermore, TruB is of general interest as it is a close homolog and structurally very similar to the eukaryotic pseudouridine synthase Cbf5, the catalytic subunit of H/ACA small nucleolar ribonucleoproteins (Koonin 1996). Two other well-studied bacterial pseudouridine synthases are TruA and RluA, which represent two enzyme families different from TruB. TruA introduces pseudouridine modifications in the anticodon arm at positions 38, 39, and 40 of many tRNAs (Singer et al. 1972; Cortese et al. 1974; Kammen et al. 1988), whereas RluA is the only bacterial dual-specific pseudouridine synthase which modifies position 32 in several tRNAs as well as position 746 in 23S rRNA (Fig. 1B; Wrzesinski et al. 1995; Raychaudhuri et al. 1999; Hoang et al. 2006). While the basic steady-state kinetic parameters of many pseudouridine synthases have been determined (Gu et al. 1998; Huang et al. 1998; Ramamurthy et al. 1999b), not much insight has been gained into the mechanism of pseudouridine synthases, which has been suggested to consist of up to six steps (Arluison et al. 1999). So far, only some information on the kinetics of substrate tRNA binding by the Saccharomyces cerevisiae pseudouridine synthase Pus1 has been reported (Arluison et al. 1999).
Here, we have analyzed for the first time the kinetic mechanism of pseudouridine formation using the model enzyme TruB with the aim of identifying the rate-limiting step during pseudouridylation. For this purpose, two novel rapid kinetic assays were established, enabling us to monitor in real time the binding step as well as the catalytic step under pre-steady-state conditions. Interestingly, the absorbance stopped-flow measurements revealed a rapid, two-step binding mechanism for TruB. Using the quench-flow technique, we have demonstrated that the catalytic step of pseudouridine formation is slow and rate-limiting for TruB. Notably, two other pseudouridine synthases, RluA and TruA, were also shown to have very similar rate constants for pseudouridine formation, suggesting that catalysis is a uniformly slow step for bacterial pseudouridine synthases. This study represents the first detailed, pre-steady-state kinetic analysis of pseudouridine synthases shedding light on the kinetic mechanism of these enzymes.
RESULTS AND DISCUSSION
Absorbance assay for tRNA binding to TruB
In order to dissect the kinetic mechanism of pseudouridylation, the well-studied E. coli pseudouridine synthase TruB was used as a model enzyme (Nurse et al. 1995). TruB was overexpressed containing an N-terminal histidine-tag and purified by affinity and size-exclusion chromatography, similarly to previous reports (Nurse et al. 1995; Hoang and Ferré-D'Amaré 2001). For binding measurements, the active site residue aspartate 48 in TruB was mutated to asparagine (Ramamurthy et al. 1999a; Hoang et al. 2005). This substitution completely abolishes any catalytic activity as confirmed by a tritium release assay detecting pseudouridine formation (data not shown) and thus prevents tRNA modification and product release while retaining the ability to bind tRNA. First, the interaction of E. coli tRNAPhe with TruB D48N was characterized by nitrocellulose filtration. For this purpose, [3H]-labeled tRNAPhe was generated by in vitro transcription, purified by anion exchange chromatography, and incubated with increasing amounts of TruB D48N. tRNAPhe bound to the enzyme remained on the nitrocellulose membrane during filtration and was quantified by scintillation counting (Fig. 2A). Fitting of the binding curve to a hyperbolic equation revealed a dissociation constant, KD, for the interaction of TruB D48N with tRNAPhe of 1.4 ± 0.3 μM. This value is in excellent agreement with previously published results for TruB D48A and TruB D48C determined by nitrocellulose filtration (Ramamurthy et al. 1999a), thus confirming the quality of our protein and tRNA preparations.
FIGURE 2.

Detecting the interaction of tRNAPhe with TruB by nitrocellulose filtration and absorbance measurements. (A) Nitrocellulose filtration to determine the dissociation constant of [3H]-labeled tRNAPhe to catalytically inactive TruB D48N (KD = 1.4 ± 0.3 μM). (B) Determination of the dissociation constant for the tRNAPhe-TruB D48N interaction by measuring the change in absorbance at 260 nm (the increase in absorbance due to the increasing TruB D48N concentration was subtracted). Hyperbolic fitting yielded a KD of 0.34 ± 0.06 μM. (C) Stopped-flow experiments detecting the change in absorbance at 260 nm upon rapidly mixing tRNAPhe (0.75 μM final concentration) with TruB D48N (10 μM), TruB WT (10 μM), or no TruB (labeled tRNA). The time courses shown are averages of 8–12 individual time courses and were fitted to a one-exponential equation (smooth lines) to determine the apparent rate constants: kapp(WT) = 5.2 sec−1; kapp(D48N) = 12.7 sec−1. Note the logarithmic time scale of the time courses. (D) TruB WT concentration dependence of the apparent rates of individual time courses recorded by stopped-flow measurements as in C. The average apparent rate over all measured concentrations is 6.0 ± 1.8 sec−1, as indicated by the straight line.
In order to study the kinetic mechanism of the TruB–tRNA interaction, we developed a new absorbance-based assay for subsequent use in rapid-kinetic stopped-flow measurements. As in the nitrocellulose filtration assay, unlabeled, in vitro transcribed and purified tRNAPhe was incubated with increasing concentrations of TruB D48N. This time, the absorbance of the reaction mixture was recorded at 260 nm. In parallel, TruB D48N was titrated into buffer; the increasing absorbance of this solution was also monitored and subtracted from the absorbance of the reaction mixture. Thereby, only the absorbance change due to tRNA binding to TruB D48N was observed. The absorbance of tRNAPhe increases upon binding to TruB D48N (Fig. 2B); this hyperchromic effect might be due to a conformational change in the tRNA upon binding TruB, resulting in a larger proportion of unstacked bases in the tRNA. As in the nitrocellulose filtration assay, a hyperbolic binding curve is observed, and fitting yielded a dissociation constant of 0.34 ± 0.06 μM. Interestingly, this value is significantly lower than the KD determined by nitrocellulose filtration, which can be explained by the different nature of these assays. In contrast to the nitrocellulose filtration assay, the absorbance assay is an equilibrium method where both bound and unbound tRNAPhe are present in the analyzed sample. This assay should, therefore, provide the true dissociation constant. In the nonequilibrium filtration assay, however, the washing step separates bound from unbound tRNAPhe. While washing was performed fast and with precooled buffer, it is likely that some TruB D48N-tRNA complex dissociates during this step, thus resulting in a slightly higher KD. This is also consistent with our observation, as well as with previous reports, that only ∼60% binding could be observed in the filter binding assay (Ramamurthy et al. 1999a).
Rapid kinetic stopped-flow analysis of tRNA binding to TruB
To analyze the kinetics of tRNA binding to TruB, 0.75 μM tRNA was rapidly mixed with at least a threefold excess of TruB in a stopped-flow apparatus, and the absorbance at 260 nm was monitored on a msec-to-second time scale. Based on the determined dissociation constant, relatively high tRNA and TruB concentrations were used in order to ensure binding of each tRNA by TruB such that a maximal signal is obtained. Furthermore, due to the excess of enzyme under these conditions, TruB wild-type (WT) can undergo only a single round of catalysis which simplifies the kinetic analysis. As expected from the equilibrium measurements, rapid mixing of tRNAPhe with TruB D48N resulted in an absorbance increase which took place within 0.5 sec (Fig. 2C). The observed absorbance change could be fitted to a one-exponential equation resulting in an apparent rate, kapp, of 12.7 sec−1. When tRNAPhe is rapidly mixed with buffer instead of TruB, no absorbance change is observed (Fig. 2C). Next, we performed the same experiment with TruB WT, and again an increase in absorbance was observed within 0.5 sec. Interestingly, the absorbance remained high for >60 sec (data not shown), suggesting that the tRNA may not dissociate from TruB wt after formation of pseudouridine under these conditions. This is in agreement with the comparatively high concentrations used in this assay and with previous findings indicating that TruB WT has a rather high affinity for its product (Ramamurthy et al. 1999a). The time course for tRNA binding to TruB WT closely resembled the time courses observed with TruB D48N, which shows that the absorbance change is a result of an early event in the interaction of TruB and tRNA which takes place before catalysis. Fitting to a one-exponential equation revealed an apparent rate, kapp, of 5.2 sec−1 for TruB WT, which is in a similar order of magnitude as the rate observed with TruB D48N. Interestingly, no change in absorbance was observed upon binding of tRNAPhe to the pseudouridine synthase RluA, which targets the anticodon loop (data not shown).
In order to further characterize the kinetics of TruB's interaction with tRNAPhe, similar stopped-flow experiments were carried out using increasing concentrations of TruB WT (2.5–10.0 μM final concentrations). Very similar time courses were obtained as before (data not shown). The amplitudes of the absorbance increases remained constant (data not shown), which is expected as complete binding of all tRNAs occurs at these TruB concentrations, given a KD for the interaction of ∼0.34 μM (vide supra). As before, the time courses were fitted to a one-exponential equation to obtain the apparent rates, kapp. Interestingly, the apparent rates stayed constant over the TruB concentration range tested, with an average rate of 6.0 ± 1.8 sec−1 (Fig. 2D). This lack of a concentration dependence is in contrast to the linear concentration dependence expected for a bimolecular binding reaction: a faster rate of binding is expected at higher concentrations. Therefore, the absorbance assay apparently monitors a different step than the initial contact of tRNA and TruB. This finding suggests that the interaction of TruB and tRNA occurs in a two-step binding mechanism (Fersht 1998).
These observations are compatible with two different two-step kinetic mechanisms describing the interaction of TruB with its substrate RNA. In a two-step equilibrium reaction, the apparent rate can either increase hyperbolically or decrease hyperbolically with the enzyme concentration, depending on whether the first or the second step is fast, respectively (Fersht 1998). In other words, the absorbance change could result from a slow and rate-limiting conformational change in the unbound tRNA which has to precede rapid binding to TruB. Alternatively (and maybe more likely), the initial and fast encounter of TruB and tRNA could occur without a change in absorbance but be followed by a slower conformational change in the tRNA, which is reflected in the increased absorbance. Since no concentration dependence of the apparent rate of binding is observed for TruB, both models are in accordance with the data presented here. No concentration dependence was observed due to the experimental conditions which result in saturated apparent rates at the TruB concentrations used. To distinguish between these models, measurements at lower concentrations of TruB would be necessary; however, this is difficult since the enzyme has to be in excess over tRNA to maintain pseudo-first-order conditions. Accordingly, the tRNA concentration would also have to be reduced, which is not feasible since the absorbance change is small and cannot be detected at lower tRNA concentrations. In summary, we conclude from the presented experiments that substrate binding to TruB takes places with an overall rate of 6 sec−1 without assigning this rate to a specific step.
It is tempting to speculate that the observed absorbance increase is a result of conformational changes in the tRNA resulting in unstacking of bases. By comparing the crystal structures of tRNA alone with the reported TruB-RNA complex (Hoang and Ferré-D'Amaré 2001; Pan et al. 2003), it becomes evident that several conformational changes of the tRNA are required in order to allow it to bind productively to TruB. First, the interaction of the D and TΨC arm in the elbow region of tRNA has to be disrupted in order to bind the TΨC arm to TruB. Furthermore, three bases flip out of the TΨC loop into the catalytic pocket of TruB. Either of these two conformational changes or other tRNA rearrangements could contribute to the absorbance increase observed upon the interaction of tRNA with TruB. However it is more likely that the partial unfolding of the D and TΨC arm contributes mostly to the absorbance change, since RluA also flips out bases into its catalytic pocket (Hoang et al. 2006), but no absorbance change could be recorded for this interaction.
Kinetic analysis of pseudouridine formation by TruB
In order to analyze the kinetics of TruB-catalyzed pseudouridylation, we conducted steady-state and pre-steady-state kinetic experiments. For these studies, full-length E. coli tRNAPhe transcripts were used that contained tritium labels at position C5 of all uracils allowing for the detection of pseudouridylation by the release of tritium upon formation of the C5-glycosidic bond in pseudouridine (Fig. 1A; Cortese et al. 1974). First, we confirmed that our purified components are fully active by assessing the steady-state kinetics of pseudouridine formation by measuring the initial rates of pseudouridine formation by TruB at different substrate tRNA concentrations (Fig. 3A). Our experimental conditions are similar to previous studies, and TruB displayed a kcat value of 0.7 ± 0.1 sec−1 and a KM value of 550 ± 150 × 10−9 M, similar to published results (Gu et al. 1998; Ramamurthy et al. 1999b). Next, the tritium release assay was adapted to allow for detection of the pre-steady-state kinetics of pseudouridine formation by the quench-flow technique. Upon rapid mixing of tritium-labeled tRNA with an excess of TruB, i.e., under single-turnover conditions, the time courses of pseudouridine formation were followed at increasing TruB concentrations (Fig. 3B). Under these conditions, the tritium release assay monitors the appearance of enzyme-product complex as the tritium is released during the last catalytic step, the formation of the new C-C glycosidic bond. The TruB active site is accessible to water (Hoang and Ferré-D'Amaré 2001), and therefore, it is conceivable that the released tritium can easily escape the enzyme-product complex before product release occurs. Furthermore, the enzyme is denatured by quenching the reaction with 0.1 M HCl, which further facilitates release of tritium into the supernatant. Therefore, this assay detects the product as soon as it appears in the enzyme-product complex. In the quench-flow experiments, close to 100% pseudouridine formation was detected within 5 sec for all TruB concentrations tested (2.5–10 μM). Single-exponential fitting yielded the apparent rate of pseudouridylation by TruB (Fig. 3C). In the analyzed concentration range, the apparent rate of pseudouridylation was independent of the TruB concentration with an average rate of 0.5 ± 0.2 sec−1. The absence of a concentration dependence suggests that pseudouridine formation by TruB is not limited by tRNA binding under these conditions. Moreover, the rate of substrate binding at 20°C is 6 sec−1 and will be even higher at 37°C, where the tritium release assays were conducted, thus further supporting the finding that tRNA binding is not rate-limiting for TruB. In conclusion, the rate of pseudouridine formation measured here directly reflects the rate constant of pseudouridine catalysis (kΨ = 0.5 ± 0.2 sec−1). Here, pseudouridine catalysis is understood as the overall process comprising glycosidic bond cleavage, base rotation, and new C-C glycosidic bond formation, as these steps cannot be distinguished by the tritium release assay. It cannot be excluded that kΨ also comprises flipping of the target U55 into the active site of TruB if this flipping would be substantially slower than the conformational changes observed in the absorbance experiments.
FIGURE 3.

Michaelis-Menten and quench-flow analysis of pseudouridine formation by TruB. (A) Michaelis-Menten experiment of TruB. Ten nM of enzyme was mixed with increasing concentrations of [3H]-labeled tRNAPhe, and pseudouridine formation was detected using the tritium release assay. The dependence of the initial rates v0 on the tRNA concentration was fitted to the Michaelis-Menten equation yielding a Michaelis constant, KM, of 550 ± 150 nM and a catalytic constant, kcat, of 0.7 ± 0.2 sec−1 for TruB. (B) Time courses of pseudouridine formation by TruB under single-round, pre-steady-state conditions. [3H]-labeled tRNAPhe (1 μM final concentration) was rapidly mixed with TruB (circles: 2.5 μM, squares: 10 μM final concentration) in a quench-flow apparatus. The percentage of pseudouridine formed at a certain time point was determined using the modified tritium release assay. The apparent rate of pseudouridine formation for each TruB concentration was determined by fitting the time courses to a one-exponential function (smooth lines). (C) Dependence of the apparent rate of pseudouridine formation under single-round conditions on the enzyme concentration. The average apparent rate is 0.5 ± 0.2 sec−1 for TruB, as indicated by the horizontal line.
Notably, this rate constant of pseudouridine formation, kΨ, determined under single-round conditions is very similar, within the experimental error, to the kcat obtained from steady-state experiments under multiple-turnover conditions. The main difference between these experimental conditions is the fact that, in the steady-state experiments, each enzyme has to release the product RNA prior to catalyzing pseudouridine formation in a new tRNA substrate. Therefore, the steady-state experiments in conjunction with the pre-steady-state quench-flow experiments show that the rate-limiting step within the kinetic mechanism of pseudouridine formation by TruB is the catalytic step, and not product release. This is the first time that a detailed kinetic study of a pseudouridine synthase provides important insight into several steps of the kinetic mechanism, i.e., into substrate binding, catalysis, and product release.
Kinetic analysis of pseudouridylation by RluA and TruA
In order to address the question whether catalysis of pseudouridylation is generally a slow step for pseudouridine synthases, we also performed a pre-steady-state kinetic analysis of the E. coli pseudouridine synthases RluA and TruA, representing two pseudouridine synthase families different from TruB. Both enzymes catalyze pseudouridylation in the anticodon arm of many tRNAs, including tRNAPhe. Specifically, RluA modifies position 32, and TruA targets position 39, in tRNAPhe (Fig. 1B; Turnbough et al. 1979; Kammen et al. 1988; Wrzesinski et al. 1995). Therefore, the same tritium-labeled tRNAPhe used in the TruB studies could also serve as a substrate for RluA and TruA. Time courses of pseudouridine formation were determined under single-turnover conditions by quench-flow measurements, as described for TruB. Again, 100% pseudouridine formation was observed after 5–10 sec for both enzymes (Fig. 4A,B). As before, the apparent rates were determined by one-exponential fitting and plotted against the enzyme concentration (Fig. 4C,D). Interestingly, the apparent rate of pseudouridine formation increased hyperbolically with increasing RluA concentration. This finding indicates that at low RluA concentrations substrate binding is limiting, which is overcome at higher RluA concentrations where catalysis is limiting. Therefore, we fit the concentration-dependence of the apparent rates, kapp, to a hyperbolic function in order to obtain the maximal rate at high RluA concentrations (kmax). This maximal rate corresponds to the rate constants of pseudouridine formation, kΨ, and was determined to be 0.7 ± 0.15 sec−1. For TruA, the apparent rate of pseudouridine formation, kapp, was not dependent on the enzyme concentration in the range tested, with an average rate of 0.35 ± 0.2 sec−1 (Fig. 4D). This finding indicates that substrate binding is not rate-limiting for TruA under these experimental conditions, as previously observed for TruB. Hence, the apparent rate for pseudouridine formation by TruA reflects the rate constant of catalysis, kΨ.
FIGURE 4.

Quench-flow titrations of pseudouridine formation by RluA and TruA. Time courses of pseudouridine formation by RluA (A) and TruA (B) under single-round, pre-steady conditions. In a quench-flow apparatus, [3H]-labeled tRNAPhe (1 μM final concentration) was rapidly mixed, in A, with 2.5 μM (circles), 5.0 μM (triangles), or 10.0 μM (squares) RluA, or, in B, with 2.5 μM (circles) or 10.0 μM (squares) TruA. The percentage of pseudouridine formed at a certain time point was determined using the modified tritium release assay. Smooth lines are the result of fitting the time courses to a one-exponential equation. (C) Dependence of the apparent rate, kapp, of RluA-catalyzed pseudouridine formation under single-round conditions on the enzyme concentration. Apparent rates were determined by single-exponential fitting of quench-flow time courses at increasing RluA concentrations. The data were fit to a hyperbolic equation with a maximal apparent rate of 0.7 ± 0.15 sec−1 (smooth line). (D) Dependence of the apparent rate, kapp, of TruA-catalyzed pseudouridine formation under single-round conditions on the enzyme concentration. The average apparent rate is 0.35 ± 0.2 sec−1 for TruA as indicated by the horizontal line.
Uniform, slow catalysis of pseudouridine formation
The rate constants determined here for catalysis of pseudouridine formation, kΨ, by RluA, TruA, and TruB are remarkably similar (Table 1). Given the precision of our measurements, the rate constants are almost identical and differ at most by a factor of two. The observation that catalysis is uniformly slow for all three pseudouridine synthases tested here raises the question whether catalysis is the rate-limiting step for these pseudouridine synthases. Since the apparent rates of pseudouridine formation reported here for TruB and TruA are independent of the enzyme concentration (Figs. 3C, 4D), substrate binding is clearly not a limiting factor for TruB and TruA. Furthermore, the concentration dependence of pseudouridine formation by RluA (Fig. 4C) indicates that substrate binding is not limiting at concentrations above ∼7.5 μM for this enzyme. This finding is in accordance with a previous report that binding is not the kinetically limiting step for yeast Pus1 (Arluison et al. 1999). The next question is whether product release could be limiting, which can be answered by comparing the elementary rate constant kΨ and the catalytic constant kcat determined under multiple-turnover conditions. The catalytic constant of TruA has been reported as 0.18 sec−1 (Huang et al. 1998), and under our experimental conditions an even higher value could be obtained by steady-state experiments (kcat = 0.7 ± 0.2 sec−1, data not shown). Thus, kcat (0.18–0.7 sec−1) and kΨ (0.35 sec−1, vide supra) are comparable for TruA, indicating that product release is not overall rate-limiting, but that catalysis itself is the limiting step for TruA, as explained above for TruB. Thus, TruA and TruB resemble each other in this property. The catalytic constant kcat for RluA has previously been measured to be ∼0.1 sec−1 (Ramamurthy et al. 1999b), which is significantly lower than the rate constant for pseudouridine formation under single-turnover conditions determined here (kΨ = 0.7 sec−1). This indicates that, potentially, product release could be a rate-limiting step for RluA. It is not surprising that the enzymes might differ substantially in product release (and substrate binding) as they all display a different mode of specifically recognizing and interacting with their substrate RNA (Hoang and Ferré-D'Amaré 2001; Hoang et al. 2006; Hur and Stroud 2007). Therefore, it will be interesting in the future to study the kinetic mechanism of RluA, TruA, and also TruB in greater detail using other stopped-flow techniques in order to analyze the mechanism of substrate recognition and product release by these pseudouridine synthases. Based on the data presented here, the mechanism of pseudouridine synthases consists of at least four steps: (1) substrate binding, (2) some conformational change such as tRNA unfolding and/or movement of the target uridine into the catalytic site, (3) catalysis consisting of several sub-steps (vide infra), and (4) product release. Further pre-steady-state kinetic analysis will reveal whether additional steps exist, as speculated previously (Arluison et al. 1999).
TABLE 1.
Rate constants for pseudouridine formation, kΨ, by the three E. coli enzymes RluA, TruA, and TruB

As the three pseudouridine synthases analyzed here are characterized by a uniformly slow rate constant for pseudouridylation (Table 1), it is conceivable that all pseudouridine synthases could have similar rate constants of ∼0.5 sec−1. All six families of pseudouridine synthases contain the same fold in the catalytic domain and very similar active sites including a catalytic aspartate (Hamma and Ferré-D'Amaré 2006). Therefore, it has been proposed that all pseudouridine synthases share a common catalytic mechanism (Hamma and Ferré-D'Amaré 2006); however, the catalytic mechanism is still not identified. Since RluA, TruA, and TruB represent three different families of pseudouridine synthases, it is rather likely that also the other enzyme families share a rather low rate constant for pseudouridine formation. Interestingly, a catalytic rate constant of ∼0.5 sec−1 is rather small compared to many other enzymes which often achieve kcat values of 102–106 sec−1 (Voet and Voet 2011). The rate enhancements by pseudouridine synthases cannot be quantitatively determined since no data exist on the rate of the uncatalyzed reaction, maybe because this reaction would not occur without catalysis. Assuming that the uncatalyzed reaction is very slow or not occurring at all, the rate enhancement by pseudouridine synthases might be significant despite the relatively low catalytic rate constant kΨ.
Three reasons can be envisioned to explain the low rate constant of pseudouridine formation. First, the low rate constant of pseudouridine formation might be due to the absence of evolutionary pressure to further increase the rate of pseudouridine formation. While pseudouridines are the most common RNA modifications and supposed to enhance RNA structure and function, many pseudouridines and, in turn, many pseudouridine synthases, are not essential for the cell (Raychaudhuri et al. 1999; Gutgsell et al. 2000; Del Campo et al. 2001; Kinghorn et al. 2002). Second, it might even be envisioned that pseudouridine synthases have been selected to be slow. Such a selection could arise if another function in addition to pseudouridylation is important for this enzyme family, such as a role in RNA folding which has been suggested previously (Hoang and Ferré-D'Amaré 2001). Third, it might not be possible to further increase the rate constant of catalysis of pseudouridine formation due to the actual chemistry of the reaction. Pseudouridine synthases catalyze a challenging chemical reaction consisting of multiple steps including cleavage of the N1-glycosidic bond, rotation of the uracil base, and formation of a new C5-glycosidic bond. To the best of our knowledge, this complex isomerization of uridine to pseudouridine is irreversible. It is remarkable that these enzymes efficiently catalyze these three different chemical reactions in the same catalytic pocket. These restrictions might impose an upper limit on the achievable rate constant for catalysis. In this case, all pseudouridine synthases would be limited by the same chemical difficulty and most likely display the same rate constants for catalysis—as observed here for RluA, TruA, and TruB. More investigations are necessary to distinguish among the three chemical reactions in pseudouridine catalysis and to identify the rate-limiting step within these reactions. Importantly, the quench-flow technique used here might help to isolate and characterize transient intermediates on the reaction pathway (Barman et al. 2006). Interestingly, the first substep, cleavage of the N-glycosidic bond, resembles the reaction catalyzed by uracil-DNA glycosylases, which display kcat values of 4–200 sec−1 (Duraffour et al. 2007; Liu et al. 2007), at least 10-fold higher than the catalytic rate constant of pseudouridine formation reported here. Therefore, N-glycosidic bond cleavage can be fast in principle; and the subsequent steps of base rotation or C-C bond formation are more likely the limiting steps during pseudouridine formation.
In conclusion, we present here the first pre-steady-state rapid kinetic analysis of pseudouridine synthases. Thereby, important insight into the kinetic mechanism of TruB has been obtained revealing a two-step substrate binding and slow, rate-limiting catalysis of pseudouridylation. This two-step binding mechanism might be common among RNA modification enzymes and might contribute to the specific recognition of selected target sites. Furthermore, our kinetic analysis of RluA, TruA, and TruB representing three different families of pseudouridine synthases demonstrated that catalysis of pseudouridine formation is a uniformly slow step (Table 1), which is most likely a general feature of all pseudouridine synthases. These findings are pivotal for the analysis and dissection of the catalytic mechanism and the kinetics of the three chemical substeps taking place during pseudouridine formation. Moreover, it will be interesting to compare the presented kinetic mechanism of bacterial, stand-alone pseudouridine synthases with eukaryotic homologs, as well as with H/ACA small ribonucleoproteins, which employ a different approach to substrate RNA binding by base-pairing to an H/ACA guide RNA and might, therefore, have a different rate-limiting step such as product release (Li 2008). In summary, the presented pre-steady-state analysis of the basic kinetic mechanism of pseudouridylation identified for the first time catalysis as a slow step in three pseudouridine synthase families and lays the groundwork for future investigations on the detailed kinetic mechanism of H/ACA small ribonucleoproteins, on the mechanism of substrate binding by TruB and other stand-alone pseudouridine synthases, and on the catalytic mechanism of pseudouridine formation in general.
MATERIALS AND METHODS
Buffers and reagents
Buffer TAKEM4: 50 mM Tris-HCl pH 7.5, 70 mM NH4Cl, 30 mM KCl, 1 mM EDTA, 4 mM MgCl2. Nucleotide triphosphates and guanine monophosphate for in vitro transcription, DNaseI, and inorganic pyrophosphatase were from Sigma; all other enzymes were from Fermentas. Chemicals were purchased from VWR. DNA oligos were obtained from IDT, and radioactive UTP was from Moraveck.
Molecular cloning and mutagenesis
The open reading frame of E. coli truB was ligated into pET28(+) vector encoding an N-terminal histidine-tag using restriction sites NheI and BamHI to generate the plasmid pET28a-TruB (similar to Nurse et al. [1995]). The QuickChange method (Strategene) was used for site-directed mutagenesis generating plasmid pET28a-TruBD48N. Gene sequences were confirmed by DNA sequencing (Macrogen).
Protein expression and purification
For protein expression, pET28a-TruB and pET28a-TruBD48N were transformed into BL21(DE3) competent E. coli cells (EMD Bioscience) which were grown in LB medium supplemented with 50 μg/mL kanamycin at 37°C. TruA and RluA were expressed using the plasmids pCA24N(GFP minus)-JW2315 and pCA24N(GFP minus)-JW0057 in AG1(ME5305) E. coli cells (Kitagawa et al. 2005) (obtained from the National BioResource Project, NIG, Japan), which were grown in LB medium with 50 μg/mL chloramphenicol at 37°C. At an OD600 of ∼0.6, protein expression was induced by the addition of isopropyl β-D-1-thiogalactopyranoside (IPTG) to a final concentration of 1 mM. Cells were harvested 3 h after induction by centrifugation at 5000g for 15 min, flash frozen, and stored at −80°C.
For purification of TruB, TruA, or RluA, cells were resuspended in 5 mL/g cells Buffer A [20 mM Tris-HCl pH 8.1, 400 mM KCl, 1 mM β-mercaptoethanol, 30 mM imidazole, 0.5 mM phenylmethylsulfonyl fluoride (PMSF), 5% (v/v) glycerol] and lysed for 30 min on ice by adding lysozyme (1 mg/mL final concentration), followed by addition of sodium deoxycholate (12.5 mg/g cells) and further incubation for 30 min on ice. The solution was then sonicated five times for 1 min each (intensity level 6, duty cycle 60%, Branson Sonifier) and centrifuged for 30 min at 30,000g, 4°C. The cleared lysate was loaded onto a 5 mL Ni2+ Sepharose column (GE Healthcare) with a flow rate of 0.5 mL/min and washed with ∼60 mL Buffer A at a flow rate of 1 mL/min until the A280 returned to baseline (BioLogic LP chromatography system). Protein was subsequently eluted with a linear gradient (50 mL, 1 mL/min) to Buffer B (same as A except for 500 mM imidazole and no PMSF). Peak fractions were analyzed by 12% SDS-PAGE, pooled, and concentrated by ultrafiltration (Vivaspin MWCO 10,000). The protein was further purified by size exclusion chromatography using a Superdex 75 column (XK26/100 column, GE Healthcare) in Buffer C (20 mM HEPES-KOH pH 7.5, 150 mM KCl, 1mM β-mercaptoethanol, 0.5 mM EDTA, 5 mM MgCl2, 20% (v/v) glycerol) at a flow rate of 1 mL/min (BioLogic DuoFlow chromatography system). Peak fractions were concentrated as before, flash frozen, and stored in aliquots at −80°C. Protein concentration was determined photometrically at 280 nm using a molar extinction coefficient of 20,860 M−1 cm−1 for TruB, 29,910 M−1 cm−1 for RluA, and 45,380 M−1 cm−1 for TruA (calculated using ProtParam [Gill and von Hippel 1989]).
In vitro transcription
The template for the in vitro transcription of E. coli tRNAPhe was generated by PCR amplification from the plasmid pCFO (Sampson et al. 1989) (kind gift of O. Uhlenbeck) using the following primers:
5′-GCTGCAGTAATACGACTCACTATAG-3′ and
5′-mUmGGTGCCCGGACTCG-3′.
Subsequently, the in vitro transcriptions were performed using the PCR template [10% (v/v)] in transcription buffer (40 mM Tris-HCl pH ∼7.5, 15 mM MgCl2, 2 mM spermidine, 10 mM NaCl, 10 mM DTT) with 3 mM NTPs (ATP, CTP, GTP, and UTP), 5 mM GMP, 0.01 U/μL inorganic pyrophosphatase, 0.3 μM T7 RNA Polymerase, and 0.12 U/μL RNase inhibitor at 37°C for 16 h. For generation of [3H]-labeled tRNA, the in vitro transcriptions were performed for 4 h using 3 mM ATP, CTP, and GTP each, and 0.1 mM [5-3H]UTP (0.46 Ci/mmole). Following the in vitro transcription step, the template was digested with DNaseI for 1 h at 37°C. The nonradioactive RNA was purified by DEAE anion exchange chromatography (Easton et al. 2010). [3H]-labeled RNA was purified with a Nucleobond AX20 column (Macherey-Nagel) using equilibration buffer R0 [100 mM Tris-acetate pH 6.3, 10 mM MgCl2, 15% (v/v) ethanol], washing buffer R1 (R0 plus 300 mM KCl), and elution buffer R3 (R0 plus 1150 mM KCl). The obtained tRNA was concentrated by isopropanol precipitation and dissolved in H2O. The tRNA concentration was determined photometrically at 260 nm using the extinction coefficient 5 × 105 M−1 cm−1 (Peterson and Uhlenbeck 1992). The specific activity of the purified tRNA was determined by scintillation counting.
Nitrocellulose filtration
Prior to the experiment, folding of tRNAPhe was allowed to occur by heating 2 μM [3H]tRNAPhe in TAKEM4 buffer to 60°C for 5 min and subsequent slow cooling to 37°C (Hengesbach et al. 2010). To allow for tRNA binding to TruB D48N, 50 nM [3H]tRNAPhe were incubated with 0–30 μM TruB D48N in TAKEM4 buffer for 10 min at room temperature. The complete 50 μL reaction mixture was then filtered through a nitrocellulose membrane followed by washing of the membrane with 1 mL cold TAKEM4 buffer. Membranes were dissolved for 30 min in 10 mL EcoLite scintillation cocktail [EcoLite (+), MP Biomedical], and the amount of tRNA bound to TruB D48N retained on the membrane was determined by scintillation counting (Perkin-Elmer Tri-Carb 2800TR liquid scintillation analyzer). In order to obtain the dissociation constant (KD), the increase in the fraction of bound tRNA as a function of the TruB D48N concentration was analyzed by fitting to a hyperbolic equation:
Absorbance spectroscopy and stopped-flow measurements
Following folding of the tRNA as described above, the absorbance at 260 nm of a 0.8 μM tRNA solution in TAKEM4 was recorded. Increasing amounts of TruB D48N were added to the tRNA solution which was incubated at room temperature for 1 min, and the absorbance increase at 260 nm was monitored. The same titration of TruB D48N into TAKEM4 buffer was performed, and the resulting absorbance readings were subtracted from the data in the presence of tRNA, yielding the increase in absorbance at 260 nm due to the interaction of tRNAPhe and TruB D48N. This change in absorbance was then plotted against the TruB D48N concentration, and the data were subsequently analyzed by fitting with a quadratic function ([RNA] = 0.8 μM) to obtain the dissociation constant, KD:
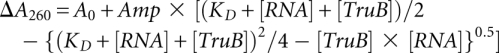 |
A quadratic function was used for fitting instead of a hyperbolic function since the RNA concentration was not significantly lower than the TruB concentration used.
Pre-steady-state kinetics of tRNA interaction with TruB were monitored in a KinTek SF-2004 stopped-flow apparatus. Twenty five μL folded tRNAPhe (final concentration 0.75 μM) were rapidly mixed with 25 μL TruB (final concentration 2.5–10 μM) at 20°C in TAKEM4, and the absorbance at 260 nm was recorded. The starting absorbance was subtracted, and the resulting time courses were analyzed by fitting with a one-exponential function to determine the apparent rate kapp:
The apparent rates kapp were then plotted against the enzyme concentration.
Tritium release assay to detect pseudouridylation
Prior to all experiments, [3H]tRNAPhe was allowed to fold as described above. For Michaelis-Menten experiments, different concentrations of [3H]tRNAPhe (100–2000 nM) were incubated with 10 nM enzyme in TAKEM4 buffer at 37°C, and samples (10.8 pmol [3H]tRNAPhe) were removed after 30 sec, 60 sec, and 120 sec. The reaction was stopped by adding the samples to 1 mL 5% (w/v) activated charcoal (Norit A) in 0.1 M HCl. Following centrifugation at 10,000 × g for 2 min, 0.8 mL of the supernatant was added to 0.5 mL fresh 5% Norit A (w/v) in 0.1 M HCl, mixed, and centrifuged again. One mL of the supernatant was filtered through glass wool plugged in a 1-mL micropipet tip, and 0.8 mL of the resulting filtrate was then used for scintillation counting in 4 mL EcoLite scintillation cocktail. The concentration of released tritium corresponding to the formed pseudouridine was calculated and divided by the respective incubation time, yielding the initial rate of the reaction. The dependence of the initial rates v0 on the tRNA concentration was fitted with a Michaelis-Menten equation
with  .
.
Pre-steady-state measurements were performed in a KinTek quench-flow apparatus by rapidly mixing 14.5 μL folded [3H]tRNAPhe (final concentration 1.0 μM) with 13 μL of enzyme (final concentration 2.5–15 μM) at 37°C in TAKEM4. The reaction was stopped at desired time points by 0.1 M HCl. The total [3H]tRNAPhe concentration was determined by liquid scintillation counting of 2 μL of the quenched sample. To measure the concentration of released tritium, a defined volume (120–220 μL) of the quenched sample was added to 1 mL 5% Norit A (w/v) in 0.1 M HCl, and processed as described above. The percentage of pseudouridine formation was calculated from the total [3H]tRNAPhe concentration and the concentration of released tritium for each time point. Fitting of the time courses with a one-exponential equation
yielded the apparent rate, kapp, of pseudouridine formation, which then was plotted against the enzyme concentration. For RluA, this concentration dependence was fit to a hyperbolic equation to obtain the maximal rate kmax:
ACKNOWLEDGMENTS
We thank Laura Hagstrom and Theron White for their help with cloning and initial purification of TruB; Olke Uhlenbeck (Northwestern University, Evanston, IL) for plasmid pCFO; the National BioResource Project (NIG, Japan) for the TruA and RluA expression plasmids; and Hans-Joachim Wieden for providing access to the quench-flow and stopped-flow apparatus as well as for critically reading the manuscript. This work was supported by the National Science and Engineering Research Council of Canada (NSERC), and the Canada Foundation for Innovation (CFI).
Footnotes
Abbreviations: Ψ, pseudouridine; A, alanine; C, cysteine; D, aspartate; IPTG, isopropyl β-D-1-thiogalactopyranoside; N, asparagine; PMSF, phenylmethylsulfonyl fluoride; WT, wild-type.
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.2905811.
REFERENCES
- Alian A, DeGiovanni A, Griner SL, Finer-Moore JS, Stroud RM 2009. Crystal structure of an RluF–RNA complex: A base-pair rearrangement is the key to selectivity of RluF for U2604 of the ribosome. J Mol Biol 388: 785–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arluison V, Buckle M, Grosjean H 1999. Pseudouridine synthetase Pus1 of Saccharomyces cerevisiae: Kinetic characterisation, tRNA structural requirement, and real-time analysis of its complex with tRNA. J Mol Biol 289: 491–502 [DOI] [PubMed] [Google Scholar]
- Arnez JG, Steitz TA 1994. Crystal structure of unmodified tRNA(Gln) complexed with glutaminyl-tRNA synthetase and ATP suggests a possible role for pseudo-uridines in stabilization of RNA structure. Biochemistry 33: 7560–7567 [DOI] [PubMed] [Google Scholar]
- Barman TE, Bellamy SR, Gutfreund H, Halford SE, Lionne C 2006. The identification of chemical intermediates in enzyme catalysis by the rapid quench-flow technique. Cell Mol Life Sci 63: 2571–2583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charette M, Gray MW 2000. Pseudouridine in RNA: What, where, how, and why. IUBMB Life 49: 341–351 [DOI] [PubMed] [Google Scholar]
- Cortese R, Kammen HO, Spengler SJ, Ames BN 1974. Biosynthesis of pseudouridine in transfer ribonucleic acid. J Biol Chem 249: 1103–1108 [PubMed] [Google Scholar]
- Decatur WA, Fournier MJ 2002. rRNA modifications and ribosome function. Trends Biochem Sci 27: 344–351 [DOI] [PubMed] [Google Scholar]
- Del Campo M, Kaya Y, Ofengand J 2001. Identification and site of action of the remaining four putative pseudouridine synthases in Escherichia coli. RNA 7: 1603–1615 [PMC free article] [PubMed] [Google Scholar]
- Duraffour S, Ishchenko AA, Saparbaev M, Crance JM, Garin D 2007. Substrate specificity of homogeneous monkeypox virus uracil-DNA glycosylase. Biochemistry 46: 11874–11881 [DOI] [PubMed] [Google Scholar]
- Easton LE, Shibata Y, Lukavsky PJ 2010. Rapid, nondenaturing RNA purification using weak anion-exchange fast performance liquid chromatography. RNA 16: 647–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ejby M, Sorensen MA, Pedersen S 2007. Pseudouridylation of helix 69 of 23S rRNA is necessary for an effective translation termination. Proc Natl Acad Sci 104: 19410–19415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ericsson UB, Nordlund P, Hallberg BM 2004. X-ray structure of tRNA pseudouridine synthase TruD reveals an inserted domain with a novel fold. FEBS Lett 565: 59–64 [DOI] [PubMed] [Google Scholar]
- Fersht A 1998. Structure and mechanism in protein science: A guide to enzyme catalysis and protein folding. W.H. Freeman and Co, New York [Google Scholar]
- Gill SC, von Hippel PH 1989. Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem 182: 319–326 [DOI] [PubMed] [Google Scholar]
- Gu X, Yu M, Ivanetich KM, Santi DV 1998. Molecular recognition of tRNA by tRNA pseudouridine 55 synthase. Biochemistry 37: 339–343 [DOI] [PubMed] [Google Scholar]
- Gutgsell N, Englund N, Niu L, Kaya Y, Lane BG, Ofengand J 2000. Deletion of the Escherichia coli pseudouridine synthase gene truB blocks formation of pseudouridine 55 in tRNA in vivo, does not affect exponential growth, but confers a strong selective disadvantage in competition with wild-type cells. RNA 6: 1870–1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton CS, Spedaliere CJ, Ginter JM, Johnston MV, Mueller EG 2005. The roles of the essential Asp-48 and highly conserved His-43 elucidated by the pH dependence of the pseudouridine synthase TruB. Arch Biochem Biophys 433: 322–334 [DOI] [PubMed] [Google Scholar]
- Hamma T, Ferré-D'Amaré AR 2006. Pseudouridine synthases. Chem Biol 13: 1125–1135 [DOI] [PubMed] [Google Scholar]
- Hengesbach M, Voigts-Hoffmann F, Hofmann B, Helm M 2010. Formation of a stalled early intermediate of pseudouridine synthesis monitored by real-time FRET. RNA 16: 610–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang C, Ferré-D'Amaré AR 2001. Cocrystal structure of a tRNA Ψ55 pseudouridine synthase: Nucleotide flipping by an RNA-modifying enzyme. Cell 107: 929–939 [DOI] [PubMed] [Google Scholar]
- Hoang C, Ferré-D'Amaré AR 2004. Crystal structure of the highly divergent pseudouridine synthase TruD reveals a circular permutation of a conserved fold. RNA 10: 1026–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang C, Hamilton CS, Mueller EG, Ferré-D'Amaré AR 2005. Precursor complex structure of pseudouridine synthase TruB suggests coupling of active site perturbations to an RNA-sequestering peripheral protein domain. Protein Sci 14: 2201–2206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang C, Chen J, Vizthum CA, Kandel JM, Hamilton CS, Mueller EG, Ferré-D'Amaré AR 2006. Crystal structure of pseudouridine synthase RluA: Indirect sequence readout through protein-induced RNA structure. Mol Cell 24: 535–545 [DOI] [PubMed] [Google Scholar]
- Huang L, Pookanjanatavip M, Gu X, Santi DV 1998. A conserved aspartate of tRNA pseudouridine synthase is essential for activity and a probable nucleophilic catalyst. Biochemistry 37: 344–351 [DOI] [PubMed] [Google Scholar]
- Hur S, Stroud RM 2007. How U38, 39, and 40 of many tRNAs become the targets for pseudouridylation by TruA. Mol Cell 26: 189–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kammen HO, Marvel CC, Hardy L, Penhoet EE 1988. Purification, structure, and properties of Escherichia coli tRNA pseudouridine synthase I. J Biol Chem 263: 2255–2263 [PubMed] [Google Scholar]
- Kaya Y, Del Campo M, Ofengand J, Malhotra A 2004. Crystal structure of TruD, a novel pseudouridine synthase with a new protein fold. J Biol Chem 279: 18107–18110 [DOI] [PubMed] [Google Scholar]
- Kinghorn SM, O'Byrne CP, Booth IR, Stansfield I 2002. Physiological analysis of the role of truB in Escherichia coli: A role for tRNA modification in extreme temperature resistance. Microbiology 148: 3511–3520 [DOI] [PubMed] [Google Scholar]
- Kitagawa M, Ara T, Arifuzzaman M, Ioka-Nakamichi T, Inamoto E, Toyonaga H, Mori H 2005. Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): Unique resources for biological research. DNA Res 12: 291–299 [DOI] [PubMed] [Google Scholar]
- Koonin EV 1996. Pseudouridine synthases: Four families of enzymes containing a putative uridine-binding motif also conserved in dUTPases and dCTP deaminases. Nucleic Acids Res 24: 2411–2415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H 2008. Unveiling substrate RNA binding to H/ACA RNPs: One side fits all. Curr Opin Struct Biol 18: 78–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang XH, Liu Q, Fournier MJ 2007. rRNA modifications in an intersubunit bridge of the ribosome strongly affect both ribosome biogenesis and activity. Mol Cell 28: 965–977 [DOI] [PubMed] [Google Scholar]
- Limbach PA, Crain PF, McCloskey JA 1994. Summary: The modified nucleosides of RNA. Nucleic Acids Res 22: 2183–2196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Yang X, Wang K, Tan W, Li H, Tang H 2007. Real-time monitoring of uracil removal by uracil-DNA glycosylase using fluorescent resonance energy transfer probes. Anal Biochem 366: 237–243 [DOI] [PubMed] [Google Scholar]
- McCleverty CJ, Hornsby M, Spraggon G, Kreusch A 2007. Crystal structure of human Pus10, a novel pseudouridine synthase. J Mol Biol 373: 1243–1254 [DOI] [PubMed] [Google Scholar]
- McDonald MK, Miracco EJ, Chen J, Xie Y, Mueller EG 2011. The handling of the mechanistic probe 5-fluorouridine by the pseudouridine synthase TruA and its consistency with the handling of the same probe by the pseudouridine synthases TruB and RluA. Biochemistry 50: 426–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller EG 2002. Chips off the old block. Nat Struct Biol 9: 320–322 [DOI] [PubMed] [Google Scholar]
- Newby MI, Greenbaum NL 2002. Sculpting of the spliceosomal branch site recognition motif by a conserved pseudouridine. Nat Struct Biol 9: 958–965 [DOI] [PubMed] [Google Scholar]
- Nurse K, Wrzesinski J, Bakin A, Lane BG, Ofengand J 1995. Purification, cloning, and properties of the tRNA Ψ55 synthase from Escherichia coli. RNA 1: 102–112 [PMC free article] [PubMed] [Google Scholar]
- Ofengand J 2002. Ribosomal RNA pseudouridines and pseudouridine synthases. FEBS Lett 514: 17–25 [DOI] [PubMed] [Google Scholar]
- Pan H, Agarwalla S, Moustakas DT, Finer-Moore J, Stroud RM 2003. Structure of tRNA pseudouridine synthase TruB and its RNA complex: RNA recognition through a combination of rigid docking and induced fit. Proc Natl Acad Sci 100: 12648–12653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson ET, Uhlenbeck OC 1992. Determination of recognition nucleotides for Escherichia coli phenylalanyl-tRNA synthetase. Biochemistry 31: 10380–10389 [DOI] [PubMed] [Google Scholar]
- Phannachet K, Elias Y, Huang RH 2005. Dissecting the roles of a strictly conserved tyrosine in substrate recognition and catalysis by pseudouridine 55 synthase. Biochemistry 44: 15488–15494 [DOI] [PubMed] [Google Scholar]
- Ramamurthy V, Swann SL, Paulson JL, Spedaliere CJ, Mueller EG 1999a. Critical aspartic acid residues in pseudouridine synthases. J Biol Chem 274: 22225–22230 [DOI] [PubMed] [Google Scholar]
- Ramamurthy V, Swann SL, Spedaliere CJ, Mueller EG 1999b. Role of cysteine residues in pseudouridine synthases of different families. Biochemistry 38: 13106–13111 [DOI] [PubMed] [Google Scholar]
- Raychaudhuri S, Niu L, Conrad J, Lane BG, Ofengand J 1999. Functional effect of deletion and mutation of the Escherichia coli ribosomal RNA and tRNA pseudouridine synthase RluA. J Biol Chem 274: 18880–18886 [DOI] [PubMed] [Google Scholar]
- Sampson JR, DiRenzo AB, Behlen LS, Uhlenbeck OC 1989. Nucleotides in yeast tRNAPhe required for the specific recognition by its cognate synthetase. Science 243: 1363–1366 [DOI] [PubMed] [Google Scholar]
- Singer CE, Smith GR, Cortese R, Ames BN 1972. Mutant tRNAHis ineffective in repression and lacking two pseudouridine modifications. Nat New Biol 238: 72–74 [DOI] [PubMed] [Google Scholar]
- Spedaliere CJ, Mueller EG 2004. Not all pseudouridine synthases are potently inhibited by RNA containing 5-fluorouridine. RNA 10: 192–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spedaliere CJ, Ginter JM, Johnston MV, Mueller EG 2004. The pseudouridine synthases: Revisiting a mechanism that seemed settled. J Am Chem Soc 126: 12758–12759 [DOI] [PubMed] [Google Scholar]
- Turnbough CL Jr, Neill RJ, Landsberg R, Ames BN 1979. Pseudouridylation of tRNAs and its role in regulation in Salmonella typhimurium. J Biol Chem 254: 5111–5119 [PubMed] [Google Scholar]
- Voet D, Voet JG 2011. Biochemistry. John Wiley & Sons, Inc, Hoboken, NJ [Google Scholar]
- Westhof E, Dumas P, Moras D 1988. Restrained refinement of two crystalline forms of yeast aspartic acid and phenylalanine transfer RNA crystals. Acta Crystallogr A 44: 112–123 [PubMed] [Google Scholar]
- Wrzesinski J, Nurse K, Bakin A, Lane BG, Ofengand J 1995. A dual-specificity pseudouridine synthase: An Escherichia coli synthase purified and cloned on the basis of its specificity for Ψ746 in 23S RNA is also specific for Ψ32 in tRNA(phe). RNA 1: 437–448 [PMC free article] [PubMed] [Google Scholar]
- Yang C, McPheeters DS, Yu YT 2005. Ψ35 in the branch site recognition region of U2 small nuclear RNA is important for pre-mRNA splicing in Saccharomyces cerevisiae. J Biol Chem 280: 6655–6662 [DOI] [PubMed] [Google Scholar]
- Ye K 2007. H/ACA guide RNAs, proteins and complexes. Curr Opin Struct Biol 17: 287–292 [DOI] [PubMed] [Google Scholar]