

Abstract
(R)-BINOL•SnCl4 was found to catalyze a formal [3+2] cycloaddition reaction between C(3)-substituted indoles and 2-amidoacrylates to provide pyrroloindolines. A variety of pyrroloindolines were prepared with high enantioselectivity in one step from simple precursors. This methodology is expected to facilitate the total synthesis of pyrroloindoline alkaloids, an important class of biologically active natural products.
Pyrroloindoline alkaloids are an important class of natural products1 that exhibit an impressive array of promising biological properties, including anticholinesterase,2 antiinflammatory,3 and anticancer activities.4 Owing to their medicinal relevance and structural complexity, pyrroloindoline alkaloids have served as a fertile area for the discovery and development of new chemical reactions.5,6 As part of a program targeting new methods for the total synthesis of alkaloid natural products, we sought to develop a convergent method to prepare enantioenriched pyrroloindolines.
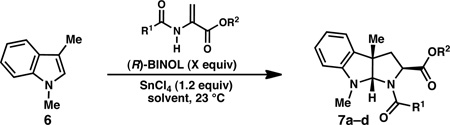
From a design perspective, it was hypothesized that pyrroloindolines (5) could be assembled through the union of a C(3)-substituted indole (1) and a 2-amidoacrylate (2) in what would constitute a formal [3+2] cycloaddition reaction (Scheme 1). It was envisioned that the reaction would proceed through a stepwise mechanism in which a Lewis acid activates 2-amidoacrylate 2, promoting conjugate addition by the indole to give iminium ion 4. Subsequent intramolecular attack of the nucleophilic amide would provide pyrroloindoline 5.
Scheme 1.

Proposed reaction to prepare pyrroloindolines.
The proposed reaction would harness the intrinsic C(3)-nucleophilicity of the indole substrate and, in principle, could provide rapid access to a variety of pyrroloindolines from simple C(3)-substituted indole precursors.7,8 Although 2-amidoacrylates are typically poor electrophiles for conjugate addition reactions due to the electron donating effects of the nitrogen lone pair, Piersanti and coworkers recently reported that EtAlCl2 promotes the reaction of indole (1, R=R1=R2=H) and methyl 2-acetamidoacrylate to provide the C(3)-Friedel-Crafts type product 3 (R=R2=H, R3=R4=Me, Scheme 1).9
Preliminary experiments were conducted in which equimolar mixtures of commercially available methyl 2-acetamidoacrylate (8) and 1,3-dimethylindole (6) were exposed to various Lewis acids. We were pleased to find that strong Lewis acids such as EtAlCl2, TiCl4, and SnCl4 provided isolable quantities of the desired pyrroloindoline 7a, as well as varying amounts of the C(2)-Friedel-Crafts type product analogous to 3 (not shown). After a preliminary survey of reaction parameters, the use of SnCl4 (1.2 equivalents) in dichloroethane at room temperature was found to provide pyrroloindoline 7a in 64% yield as a 6:1 mixture of exo and endo diastereomers (Table 1, entry 1; exo diastereomer is shown).
Table 1.
Optimization studies.
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | R1,R2 | pdt | BINOL (equiv) |
solvent | yield (%)a |
d.r.b | ee (%)c,d |
| 1 | Me, Me (8) | 7a | 0.0 | DCE | 64 | 6:1 | -- |
| 2 | Me, Me (8) | 7a | 1.1e | DCE | 86 | 4:1 | 64/83 |
| 3 | Me, Me (8) | 7a | 0.3 | DCE | 96 | 5:1 | 62/81 |
| 4 | Me, Me (8) | 7a | 0.2 | DCE | 94 | 5:1 | 63/83 |
| 5 | Me, Me (8) | 7a | 0.1 | DCE | 93 | 5:1 | 61/79 |
| 6 | Me, Me (8) | 7a | 0.05 | DCE | 82 | 5:1 | 51/72 |
| 7 | CF3, Me (9) | 7b | 0.2 | DCE | 77 | 6:1 | 86/nd |
| 8 | Me, Bn (10) | 7c | 0.2 | DCE | 81 | 2:1 | 74/82 |
| 9 | CF3, Bn (11) | 7d | 0.2 | DCE | 81 | 3:1 | 91/90 |
| 10 | CF3, Bn (11) | 7d | 0.2 | DCM | 86 | 4:1 | 94/91 |
| 11 | CF3, Bn (11) | 7d | 0.2 | CHCl3 | 58 | 3:1 | 88/89 |
| 12 | CF3, Bn (11) | 7d | 0.2 | CCl4 | 0 | -- | -- |
Isolated yield of combined diastereomers.
Determined by 1H NMR analysis of crude reaction mixture.
Determined by chiral stationary phase SFC.
ee of exo/endo diastereomers.
1.0 equiv of SnCl4 was used. nd = not determined.
At this stage we wished to determine the feasibility of accessing enantioenriched pyrroloindolines using this formal [3+2] cycloaddition reaction. To this end, a screen of chiral diol additives anticipated to form chiral Lewis or Brønsted acid complexes with SnCl4 was conducted.10 Of the conditions evaluated, it was found that utilization of a 1.1:1 mixture of (R)-BINOL and SnCl4 provided pyrroloindoline 7a in 86% yield as a 4:1 diastereomeric mixture favoring exo-7a, which was formed in 64% ee (Table 1, entry 2).11,12 Interestingly, the minor, endo diastereomer was formed in 83% ee. In follow-up studies, a side-by-side comparison of two reactions varying only by the presence or absence of (R)-BINOL showed that it accelerates the rate of 7a formation and increases the overall conversion.13
Based on these observations, it was hypothesized that catalytic quantities of (R)-BINOL may still provide pyrroloindoline 7a with good levels of enantioselectivity. Indeed, use of as low as 10 mol % (R)-BINOL furnished 7a without significantly affecting the observed enantioselectivity (Table 1, entries 3–5). Whereas 10 mol % (R)-BINOL provided comparable selectivities for the formation of 7a, 20 mol % (R)-BINOL imparted consistently higher enantioselectivities for more functionalized substrates (vide infra), and was therefore utilized in subsequent experiments. Control experiments confirmed that no reaction occurred in the absence of SnCl4, and that 1.2 equivalents SnCl4 were required to drive the reaction to high conversions.14
In an effort to improve the enantioselectivity of the reaction, the amide and ester groups of the acrylate were modified.15,16 An increase in ee was observed when methyl 2-trifluoroacetamidoacrylate (9) was employed (Table 1, entry 7). A similar increase was observed using benzyl 2-acetamidoacrylate (10, entry 8). Gratifyingly, these effects proved to be additive: use of benzyl 2-trifluoroacetamidoacrylate (11) provided pyrroloindoline 7d in 81% yield and 91% ee. Preliminary studies indicated that chlorinated solvents provided the best combination of yields and selectivities. A further screen identified methylene chloride as the solvent of choice. Under our optimized conditions, pyrroloindoline 7d was isolated in 86% yield as a 4:1 mixture of exo and endo diastereomers, which are formed in 94% and 91% ee, respectively (entry 10).17
Having identified conditions to prepare 7d in high yields and enantioselectivities, a survey of indole substrates was conducted. Substrates substituted at C(5) with electron-donating and electron-withdrawing substituents provided uniformly high ee’s, although a moderate decrease in yield was observed with electron poor substrates (Scheme 2, 13a–d). 1,3,6-Trimethylindole reacts smoothly to give a 4:1 mixture of exo- and endo-13e in 91% yield (94% and 90% ee, respectively).
Scheme 2.

Substrate scope of pyrroloindoline formation.
More functionalized substituents are also tolerated at C(3) of the indole substrate. Pyrroloindolines bearing t-butyldimethylsiloxyethyl (13f) and phenylethyl (13h) substituents are prepared in moderate to good yield and high enantioselectivity (Scheme 2). Most notably, use of N-methyl-1,2,3,4-tetrahydrocarbazole provided a single diastereomer of pyrroloindoline 13g in 65% yield and 86% ee. Using this reaction, the aza-propellane core of natural products such as vincorine18 and minfiensine19 was prepared in only two steps from commercially available 1,2,3,4-tetrahydrocarbazole.
Whereas the exo diastereomer predominates in the (R)-BINOL•SnCl4-catalyzed formation of the pyrroloindolines shown in Scheme 2, it is known that the endo diastereomer of similar compounds is favored thermodynamically.20,21 Accordingly, treatment of a 4:1 mixture of exo-7d and endo-7d diastereomers (94% and 91% ee, respectively) with excess DBU in CD2Cl2 resulted in epimerization to give a greater than 10:1 endo:exo mixture of products after 72 h (Scheme 3). Interestingly, the endo-7d product was recovered in 56% ee, favoring the opposite enantiomer.22 In a subsequent experiment, exposure of diastereomerically pure exo-7d to DBU (10 equiv) provided ent-endo-7d in 94% ee. Exposure of diastereomerically pure endo-7d to the epimerization conditions returned endo-7d without significant erosion of enantiomeric excess. Taken together, these studies suggest that the initially formed exo and endo diastereomers of 7d must be of opposite enantiomeric series. From a synthetic perspective, the diastereomers can be separated prior to subsequent functionalization to avoid erosion of optical activity through this epimerization mechanism.
Scheme 3.

Epimerization studies.
aDetermined by 1H NMR analysis of mixture. bDetermined by chiral stationary phase SFC or HPLC. c1.6 equiv SnCl4 was employed.
At this time the mechanism of the reaction remains to be elucidated. As exo- and endo-7d have opposite configurations at C(3), analysis of the diastereomeric and enantiomeric ratios suggests that the first step of the reaction (conjugate addition) occurs with modest levels of catalyst control, whereas the enolate protonation step occurs with high levels of catalyst control.23 In effect, the catalyst-controlled protonation step serves to resolve the mixture of enantiomeric intermediates. Yamamoto and coworkers have demonstrated that BINOL•SnCl4 behaves as a Lewis acid-assisted Brønsted acid that promotes a number of enantioselective protonation reactions.11a–c It is unusual, however, for high enantioselectivities to be observed when employing an excess of SnCl4 relative to BINOL. The observation that at least 1 equivalent SnCl4 is required for high conversions, coupled with the fact that use of strong Brønsted acids24 in the absence of SnCl4 fail to provide 7d, may suggest that this reaction benefits from cooperative Lewis acid-Brønsted acid activation.
In conclusion, a convergent method to prepare enantioenriched pyrroloindolines is described. The optimal conditions employ (R)-BINOL as a catalyst in the presence of stoichiometric SnCl4, and provide access to functionalized pyrroloindolines in uniformly high ee’s. The application of this methodology to the total synthesis of pyrroloindoline natural products, as well as studies aimed at understanding the mechanism of this new transformation are the focus of ongoing research in our laboratory.25
Supplementary Material
Acknowledgment
We thank Dr. Scott Virgil for insightful discussions; Drs. Michael Day and Larry Henling for X-ray crystallographic structural determination; and Prof. Brian Stoltz and the Caltech Center for Catalysis and Chemical Synthesis for access to analytical equipment. HRMS and X-ray crystallographic data were obtained on instruments purchased through awards to the California Institute of Technology by the NSF CRIF program (CHE-0639094, CHE-0541745). NMR spectra were obtained on a spectrometer funded by the NIH (RR027690). Financial support from the California Institute of Technology and the Baxter Foundation is gratefully acknowledged.
Footnotes
Supporting Information Available: Experimental details, characterization data, and NMR spectral charts. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Anthoni U, Christophersen C, Nielsen PH. Naturally Occurring Cyclotryptophans and Cyclotryptamines. In: Pelletier SW, editor. Alkaloids: Chemical & Biological Perspectives. Vol. 13. Oxford: Pergamon; 1999. pp. 163–236. [Google Scholar]
- 2.Physostigmine, a reversible cholinesterase inhibitor: Shaw KTY, Utsuki T, Rogers J, Yu QS, Sambamurti K, Brossi A, Ge YW, Lahiri DK, Greig NH. Proc. Natl. Acad. Sci. 2001;98:7605. doi: 10.1073/pnas.131152998.
- 3. Lansai B. an anti-inflammatory agent: Tuntiwachwuttikul P, Taechowisan T, Wanbanjob A, Thadaniti S, Taylor WC. Tetrahedron. 2008;64:7583. Taechowisan T, Wanbanjob A, Tuntiwachwuttikul P, Liu J. Food Agri. Immun. 2009;20:67.
- 4.(a) Chaetocin, an HDAC inhibitor: Isham CR, Tibodeau JD, Jin W, Xu R, Timm MM, Bible KC. Blood. 2007:2579. doi: 10.1182/blood-2006-07-027326. Leptosins: DNA topoisomerase inhibitors. Yanagihara M, Sasaki-Takahashi N, Sugahara T, Yamamoto S, Shinomi M, Yamashita I, Hayashida M, Yamanoha B, Numata A, Yamori T, Andoh T. Cancer Sci. 2005;96:816. doi: 10.1111/j.1349-7006.2005.00117.x.
- 5.Reviews: Crich D, Banerjee A. Acc. Chem. Res. 2007;40:151. doi: 10.1021/ar050175j. Steven A, Overman LE. Angew. Chem., Int. Ed. 2007;46:5488. doi: 10.1002/anie.200700612. Kim J, Movassaghi M. Chem. Soc. Rev. 2009;38:3035. doi: 10.1039/b819925f.
- 6.Selected references: Taniguchi M, Hino T. Tetrahedron. 1981;37:1487. Marsden SP, Depew KM, Danishefsky SJ. J. Am. Chem. Soc. 1994;116:11143. Overman LE, Paone DV, Stearns BA. J. Am. Chem. Soc. 1999;121:7702. Austin JF, Kim SG, Sinz CJ, Xiao WJ, MacMillan DWC. Proc. Natl. Acad. Sci. 2004;101:5482. doi: 10.1073/pnas.0308177101. Trost BM, Quancard J. J. Am. Chem. Soc. 2006;128:6314. doi: 10.1021/ja0608139. Lindel T, Bräuchle L, Golz G, Böhrer P. Org. Lett. 2007;9:283. doi: 10.1021/ol0627348. Kim J, Ashenhurst JA, Movassaghi M. Science. 2009;324:238. doi: 10.1126/science.1170777. Newhouse T, Baran PS. J. Am. Chem. Soc. 2008;130:10886. doi: 10.1021/ja8042307. Espejo VR, Li X-B, Rainier JD. J. Am. Chem. Soc. 2010;132:8282. doi: 10.1021/ja103428y.
- 7.For the direct arylation of indoles to provide racemic dehydroindolines: Barton DHR, Bhatnagar NY, Blazejewski JC, Charpiot B, Fibet JP, Lester DJ, Motherwell WB, Papoula MTB, Stanforth SP. J. Chem. Soc., Perk. Tran. 1. 1985:2657. Eastman K, Baran PS. Tetrahedron. 2009;65:3149.
- 8.For the direct preparation of enantioenriched pyrroloindolines from indoles see references 6e and 6f.
- 9.Angelini E, Balsamini C, Bartoccini F, Lucarini S, Piersanti G. J. Org. Chem. 2008;73:5654. doi: 10.1021/jo800881u. [DOI] [PubMed] [Google Scholar]
- 10.See Supporting Information.
- 11.For reports of asymmetric catalysis by BINOL•SnCl4: Ishihara K, Nakamura S, Kaneeda M, Yamamoto H. J. Am. Chem. Soc. 1996;118:12854. Ishihara K, Kurihara H, Yamamoto H. J. Am. Chem. Soc. 1996;118:3049. Yu SH, Ferguson MJ, McDonald R, Hall DG. J. Am. Chem. Soc. 2005;127:12808. doi: 10.1021/ja054171l. Rauniyar V, Zhai HM, Hall DG. J. Am. Chem. Soc. 2008;130:8481. doi: 10.1021/ja8016076. For a review, see: Yamamoto H, Futatsugi K. Angew. Chem., Int. Ed. 2005;44:1924. doi: 10.1002/anie.200460394.
- 12.The relative stereochemistry of exo-7b was confirmed by single crystal X-ray diffraction; the relative stereochemistry of exo-7a and the remaining compounds were assigned by analogy. The absolute stereochemistry has not been confirmed at this time.
- 13.BINOL catalyzes the addition of allyl and alkynyl boronates to aldehydes. Wu TR, Chong JM. J. Am. Chem. Soc. 2005;127:3244. doi: 10.1021/ja043001q. Lou S, Moquist PN, Schaus SE. J. Am. Chem. Soc. 2006;128:12660. doi: 10.1021/ja0651308. Wu TR, Chong JM. J. Am. Chem. Soc. 2007;129:4908. doi: 10.1021/ja0713734. Lou S, Moquist PN, Schaus SE. J. Am. Chem. Soc. 2007;129:15398. doi: 10.1021/ja075204v. Lou S, Schaus SE. J. Am. Chem. Soc. 2008;130:6922. doi: 10.1021/ja8018934.
- 14.Whereas organotin reagents are known for their toxicity, inorganic tin(IV) compounds are relatively non-toxic: Howe P, Watts P. Tin and Inorganic Tin Compounds. Switzerland: World Health Organization; 2005. SnCl4 is available from Sigma-Aldrich for $30/mol, and (R)-BINOL is approximately $10,000/mol.
- 15.Acrylates 9, 10, and 11 are prepared in one step from the commercially available materials. See Supporting Information for details.
- 16.A small screen of BINOL derivatives was also conducted, in which several 3,3’-difunctionalized-(R)-BINOL catalysts were found to provide diminished rates and selectivities.
- 17.Use of 3-methylindole provided the corresponding pyrroloindoline in 18% yield as a 8:1 exo:endo mixture. The exo diastereomer was formed in 95% ee.
- 18.Das BC, Cosson JP, Lukacs G, Potier P. Tetrahedron Lett. 1974;15:4299. [Google Scholar]
- 19.Massiot G, Thepenier P, Jacquier M-J, Le Men-Olivier L, Delaude C. Heterocycles. 1989;29:1435. [Google Scholar]
- 20.See reference 6a.
- 21.Crich D, Bruncko M, Natarajan S, Teo BK, Tocher DA. Tetrahedron. 1995;51:2215. [Google Scholar]
- 22.As determined by chiral stationary phase SFC. See Supporting Information for details.
- 23.A reversible, non-selective conjugate addition followed by a moderately diastereoselective cyclization cannot be ruled out at this time. Diastereomerically pure solutions of endo-7d or exo-7d do not equilibrate upon re-exposure to SnCl4 (1.2 equiv) and (R)-BINOL (0.2 equiv) in CD2Cl2.
- 24.For example, use of HCl (1.2 equiv) or diphenylphosphate (1.2 equiv) failed to promote the formal [3+2] reaction.
- 25.As we were preparing to submit this manuscript, similar findings for the preparation of racemic pyrroloindolines were reported: Lucarini S, Bartoccini F, Battistoni F, Diamantini G, Piersanti G, Righi M, Spadoni G. Org. Lett. 2010;12:3844. doi: 10.1021/ol101527j.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.