

Abstract
End-directed mismatch-provoked excision has been reconstituted in several purified systems. While 3′-directed excision displays a mismatch dependence similar to that observed in nuclear extracts (≈ 20-fold), the mismatch dependence of 5′-directed excision is only 3 to 4-fold, significantly less than that in extracts (8 to 10-fold). Utilizing a fractionation-based approach, we have isolated a single polypeptide that enhances mismatch dependence of reconstituted 5′-directed excision and have shown it to be identical to poly[ADP-ribose] polymerase 1 (PARP-1). Titration of reconstituted excision reactions or PARP-1-depleted HeLa nuclear extract with purified PARP-1 showed that the protein specifically enhances mismatch dependence of 5′-directed excision. Analysis of a set of PARP-1 mutants revealed that the DNA binding domain and BRCT fold contribute to the regulation of excision specificity. Involvement of the catalytic domain is restricted to its ability to poly(ADP-ribosyl)ate PARP-1 in the presence of NAD+, likely through interference with DNA binding. Analysis of protein-protein interactions demonstrated that PARP-1 interacts with mismatch repair proteins MutSα, exonuclease 1, replication protein A (RPA), and as previously shown by others, replication factor C (RFC) and proliferating cell nuclear antigen (PCNA) as well. The BRCT fold plays an important role in the interaction of PARP-1 with the former three proteins.
Keywords: mismatch repair, poly[ADP-ribose] polymerase, MutSα, exonuclease
1. Introduction
Among the several genetic stabilization functions of mismatch repair, the best understood is its role in the correction of DNA biosynthetic errors, which requires that repair be directed to a newly synthesized DNA strand [1-5]. A strand-specific nick or gap, which may reside either 3′ or 5′ to the mismatch, is sufficient to direct mismatch repair in mammalian cell extracts [6-8]. Analysis of the extract reaction has shown that mismatch-provoked excision removes the bulk of the incised strand spanning the nick and the mismatch, followed by repair DNA synthesis to fill the resulting gap [7, 9, 10].
Several purified systems have been reconstituted using near-homogeneous human proteins and support 3′- and/or 5′-directed mismatch-provoked excision or repair [11-14]. The simplest of these depends on the mismatch recognition activities MutSα (MSH2•MSH6 heterodimer) or MutSβ (MSH2•MSH3 heterodimer), MutLα (MLH1•PMS2 heterodimer), the single-stranded DNA-binding protein RPA (replication protein A), and the 5′ to 3′ double-strand hydrolytic activity Exo1 (exonuclease 1). These four activities support mismatch-provoked excision directed by a 5′ strand break, and hydrolysis is attenuated upon mismatch removal. RPA plays a primary role in terminating excision by MutSα-activated Exo1 [11, 15]. Although MutLα is not essential for this reaction, it significantly enhances the mismatch dependence of excision [11, 15]. Supplementation of MutSα, MutLα, RPA and Exo1 with the replication clamp PCNA (proliferating cell nuclear antigen) and the clamp loader RFC (replication factor C) yields a system that supports mismatch-provoked excision directed by a 3′ or 5′ strand break [12]. In contrast to the simpler 5′ to 3′ reconstituted reaction, 3′-directed excision in this six-component system absolutely requires MutLα [12]. The nature of 3′-directed excision in this system was clarified by the demonstration that MutLα is a latent endonuclease that is activated in a manner that depends on a mismatch, MutSα, RFC, PCNA, ATP and a strand break [16]. While function of RFC in MutLα activation is apparently restricted to clamp loading, the PCNA loading orientation determines the strand direction of MutLα incision, targeting endonuclease action to the heteroduplex strand that contains a preexisting break [17]. Incision in this manner introduces additional breaks into the discontinuous heteroduplex strand, providing 5′ nick that serves as a loading site for MutSα-activated Exo1, which removes the mismatch [16].
The 4- and 6-component systems described above support robust excision reactions under conditions where each activity is present at a level similar to that found in 50 μg of nuclear extract, which is optimal for the extract reaction [11, 12]. While the two purified systems display features reminiscent of mismatch-provoked excision in nuclear extracts, there are some differences. Although the mismatch dependence of 3′-directed excision in the 6-component system is comparable to that of nuclear extracts (≈ 20-fold), 5′-directed excision in either the 4- or 6-component system displays only 3 to 4-fold mismatch dependence, significantly less than that observed in nuclear extracts (8 to 10-fold) [11, 12]. The reduced specificity of 5′-directed excision in the purified systems suggests deficiency of one or more activities. Utilizing a fractionation-based approach, we have isolated a single polypeptide that enhances mismatch dependence of this reaction. We show that it is identical to the poly[ADP-ribose] polymerase 1 (PARP-1), an abundant nuclear protein with multiple functions in genome stability, chromatin structure and transcription [18, 19].
2. Materials and Methods
2.1 Cell lines, cell culture, and extract preparation
HeLa S3 cells were grown and nuclear extracts were prepared by published methods [6]. PARP-1+/+ and PARP-1−/− mouse embryo fibroblasts [20, 21] were cultured in DMEM high-glucose medium supplemented with 10% FBS (HyClone). MEF whole cell extracts were prepared as described [22].
2.2 Mismatch repair proteins, DNAs, and excision assays
Human MutSα, MutLα, RPA, Exo1, PCNA and RFC were prepared as previously described [11, 12]. Nicked circular 5′G-T heteroduplex and control 5′A•T homoduplex contained a single-strand break 128 base pairs 5′ to the location of the mismatch as viewed along the shorter path between the two sites; for 3′G-T heteroduplex and 3′A•T homoduplex DNAs the nick was 141 bp 3′ to the site of the mispair [9, 12]. 5′-gapped G-T heteroduplex and A•T homoduplex were prepared in a similar manner from bacteriophage f1MR1 and f1MR3 [23], except that the DNAs contained a 220-nucleotide gap in the complementary strand 128 bp 5′ to the mispair.
Mismatch-provoked excision assays were performed as described in the presence of 0.12 M KCl [24]. For the 4-protein excision system, 20-μl reactions contained 24 fmol of DNA, 400 fmol of MutSα, 300 fmol of MutLα, 900 fmol of RPA, and 21 fmol of Exo1. Six-protein reactions also contained 300 fmol of PCNA and 80 fmol of RFC [12]. Unless specified otherwise, PARP-1 effects in these systems were evaluated using the protein isolated from HeLa cells as described below.
2.3 Fractionation of HeLa nuclear extract
All fractionation steps were performed at 0° to 4°C in the presence of 0.5 μg/ml aprotinin, 1 μg/ml E64, 1 μg/ml leupeptin, 5 μg/ml pepstatin, 100 μg/ml pefabloc, and 0.1% phenylmethylsulfonyl fluoride. Nuclear extract (300 mg) containing 0.15 M NaCl was loaded onto a 6-ml ssDNA-cellulose column equilibrated with buffer A (25 mM Hepes-KOH, pH 7.5, 0.1 mM EDTA) containing 0.15 M NaCl. The column was washed with 50 ml of this buffer at 0.3 ml/min and eluted stepwise with 25 ml of buffer A containing 0.4 M, 0.55 M, 1 M and 2 M NaCl. Fractions were scored for activity that enhanced mismatch dependence of 5′-directed excision as judged by hydrolysis occurring on heteroduplex versus homoduplex DNA in the 4-protein system in the absence or presence of column fraction samples. Active fractions, which eluted in the 1 M NaCl step, were diluted with 20 mM potassium phosphate (KPi), pH 7.4, to a conductivity equal to that of 20 mM KPi, pH 7.4, 200 mM KCl. The sample was loaded onto a CHT2-I ceramic hydroxylapatite column (Bio-Rad) at 0.5 ml/min and eluted with a 40-ml linear gradient of 0.02 – 0.4 M KPi, pH 7.4, containing 0.2 M KCl. Active fractions, which eluted at about 0.35 M KPi, were pooled, dialyzed against buffer A containing 0.15 M NaCl for 2 hours, and then loaded onto a 1-ml MonoS column (GE Healthcare). The column was developed with a 10-ml linear gradient of 0.15 – 0.6 M NaCl in buffer A at 0.25 ml/min. Active fractions, which eluted at about 0.21 M NaCl, were pooled, frozen in liquid nitrogen, and stored at −80°C. This purification yielded 80 μg of near homogeneous material with a specific activity increase of 770-fold relative to nuclear extract.
2.4 Depletion of PARP-1 from HeLa nuclear extract
Twenty mg of HeLa nuclear extract at 55 mg/ml was adjusted with 5 M NaCl to the conductivity of 25 mM Hepes-KOH, pH 7.5, 0.1 mM EDTA, 0.5 M NaCl. The extract was loaded onto a 4-ml ssDNA-cellulose column equilibrated with this buffer and washed with 20 ml of buffer by gravity flow. Flow-through fractions were collected, and protein concentrations checked by Bradford assay. Fractions exceeding 5 mg/ml were pooled and dialyzed against 25 mM Hepes-KOH, pH 7.5, 0.1 mM EDTA, 0.15 M NaCl for 1 hr. As control, an extract sample identical to the column load was subjected to the same dialysis procedure.
2.5 Purification of recombinant PARP-1 and mutant variants
Expression plasmids for His-tagged wild-type PARP-1 and mutant variants [25, 26] were kindly provided by Dr. W. Lee Kraus (University of Texas Southwestern Medical Center). Recombinant proteins were isolated from E. coli in the presence of 0.5 μg/ml aprotinin, 1 μg/ml E64, 1 μg/ml leupeptin, 5 μg/ml pepstatin, 100 μg/ml pefabloc, and 0.1% phenylmethylsulfonyl fluoride as described [25] and further purified as follows. Fractions containing the target proteins from the nickel-nitrilotriacetic acid column were diluted with 20 mM KPi, pH 7.4, to the conductivity of 20 mM KPi, pH 7.4, 0.2 M KCl. The sample was loaded onto a 15-ml Bio-Gel HTP hydroxylapatite column (Bio-Rad) at 1.5 ml/min, which was washed with 200 ml of 0.2 M KPi, pH 7.4, 0.2 M KCl, and eluted with 75 ml of 0.4 M KPi, pH 7.4, 0.2 M KCl. Fractions containing target proteins of interest were dialyzed against buffer B (25 mM Hepes-KOH, pH 7.5, 0.1 mM EDTA) containing 0.15 M NaCl for 2 hours. For the mutant DBD-NBD, the hydroxylapatite eluate was further purified by loading onto a 1-ml MonoQ column (GE Healthcare), which was eluted with a 20-ml linear gradient of 0.15 – 0.6 M NaCl in buffer B at 0.5 ml/min. Fractions containing homogeneous DBD-NBD were pooled, quick-frozen in liquid nitrogen, and stored at −80°C. For wild-type PARP-1 and other mutants, the hydroxylapatite eluate was loaded onto a 5-ml HiTrap Q FF column (GE Healthcare) equilibrated with buffer B containing 0.15 M NaCl. The flow-through was collected and loaded onto a 1-ml MonoS column (GE Healthcare) and eluted with a 10-ml linear gradient of 0.15 – 0.6 M NaCl in buffer B at 0.5 ml/min. Fractions containing the target proteins were pooled, quick-frozen in liquid nitrogen, and stored at −80°C.
The coding sequence of BRCT fold was amplified from the wild type PARP-1 plasmid using primers d(CAGTAGGGATCCGCTGCTGTGAACTCCTCTGC) and d(GTCAACCTCGAGGGACAAGATGTGCGCTAAGA). After digestion with BamHI and XhoI, the PCR product was inserted into the multiple cloning site of plasmid pGEX-6P-1 (GE Healthcare) to yield an in frame fusion to a 5′ glutathione S-transferase (GST) gene, resulting plasmid pGEX-6P-1-BRCT that expresses GST-BRCT. GST and GST-BRCT were expressed in Rosetta 2 E. coli cells (Novagen) after transformation of pGEX-6P-1 or pGEX-6P-1-BRCT. For GST purification, 10 g cell paste was resuspended in 100 ml of buffer C (25 mM Hepes-KOH, pH 7.5, 0.15 M NaCl and 1 mM EDTA) containing proteinase inhibitors as above. After sonication, the lysate was clarified by centrifugation at 26,800 g for 20 min, and then loaded onto a column containing 10-ml Glutathione Sepharose 4 Fast Flow (GE Healthcare) pre-equilibrated with buffer C. The column was washed with 100 ml of buffer C at 1.0 ml/min, followed by elution with 50 ml of 10 mM reduced glutathione in buffer C. Fractions containing GST were pooled, desalted using a HiPrep 26/10 Desalting column (GE Healthcare) with buffer C, and then loaded onto 5-ml HiTrap SP FF and HiTrap Q FF columns (GE Healthcare) connected in series. The follow-through containing GST was collected, quick-frozen in liquid nitrogen, and stored at −80°C. For the purification of GST-BRCT, 44 g cell paste was lysed in 440 ml of buffer C and purified using Glutathione Sepharose 4 Fast Flow as described above. Fractions containing GST-BRCT were pooled, loaded onto an 8-ml MonoS column (GE Healthcare), and eluted with a 100-ml linear gradient of 0.15 – 0.6 M NaCl at 1.0 ml/min. The fusion protein, which eluted at 0.21 M NaCl, was frozen in liquid nitrogen, and stored at −80°C.
2.6 Other methods
The NAD+ concentration in nuclear extracts was estimated using the EnzyChrom NAD/NADH assay kit (BioAssay Systems). Nuclear co-immunoprecipitation and Western blotting were done as previously described [27]. The antibodies used were control mouse IgG (Santa Cruz Biotechnology), and those specific for MSH6 (DB Biosciences), MSH2 (Santa Cruz Biotechnology), PMS2 (Santa Cruz Biotechnology), MLH1 (Santa Cruz Biotechnology), Exo1 [28], RPA70 (Kamiya Biomedical), PCNA (Abcam), RFC p140 (a gift from Ulrich Hübscher, University of Zürich), poly[ADP-ribose] (BD Biosciences), and PARP-1 (Alexis Biochemicals, BD Biosciences), and β-actin (Novagen). Two types of secondary antibodies were used in this study: one was labeled with horseradish peroxidase (Invitrogen) and the other with IRDye (LI-COR). In the former case, proteins were visualized using the ECL Western blotting system (GE Healthcare); the latter utilized an Odyssey Infrared Imaging System (LI-COR). Far Western analysis [12] and GST pulldown assays (500-μl reactions) [27] were done as described previously.
3. Results
3.1 PARP-1 enhances mismatch dependence of 5′-directed excision
Purified MutSα, MutLα, Exo1 and RPA are sufficient to support a mismatch-provoked excision reaction directed by a strand break located 5′ to the mispair [11, 13], while supplementation of this 4-protein system with PCNA and RFC yields a 6-component system that supports bidirectional mismatch-provoked excision [12]. Unlike 3′-directed excision in the latter system, which displays mismatch dependence comparable to that observed in nuclear extracts (≈ 20-fold), the mismatch dependence of 5′-directed reaction in the 4- or 6-protein systems is only 3 to 4-fold, less than that observed in nuclear extracts [11, 12]. We therefore sought factors that might enhance the mismatch dependence of 5′-directed excision. To this end, HeLa nuclear extract was fractionated by ssDNA-cellulose chromatography (Materials and Methods). As shown in Fig. 1A, the 1 M-NaCl eluate contains an activity that enhances mismatch dependence of 5′-directed excision to 11-fold. This activity was purified as a single polypeptide to apparent homogeneity (Fig. 1B). MALDI-TOF (matrix-assisted laser desorption-time of flight) mass spectrometry showed this protein to be poly(ADP-ribose) polymerase 1 (PARP-1) by mass analysis and database searches via GPS Explorer/Protein Pilot (UNC-Duke Michael Hooker Proteomics Center, data not shown). This identity was confirmed by Western blot analysis using an antibody specific for PARP-1 (Fig. 1C).
Fig. 1.

Identification of PARP-1 as a mismatch dependence enhancing activity. (A) The upper diagram illustrates the 5′-directed excision assay used in this work [28]. The 6.44 kb circular substrate contained a G-T mismatch (A•T bp in homoduplex controls) and a site-specific strand break 128 bp 5′ to the mismatch, as viewed along the shorter path between the two sites. A single NheI site, located 5 bp distal to the mismatch, is rendered cleavage-resistant by mismatch-provoked excision. Cleavage of the native substrates with NheI and ClaI yields two fragments that migrate rapidly on an agarose gel, while cleavage of the excision product yields a species that runs slightly faster than full-length linear duplex. The lower panel shows 4-protein (MutSα, MutLα, Exo1 and RPA) excision assays on heteroduplex and control homoduplex in the absence or presence of 5 μg of ssDNA-cellulose column load (LD) and flow-through (FT), and 100-ng samples of 0.4 M, 0.55 M, 1.0 M and 2.0 M NaCl eluate fractions. (B) Coomassie blue-stained SDS polyacrylamide gel of samples from purification. Lane 1, protein markers; lane 2, HeLa nuclear extract; lane 3, 1 M-NaCl fraction from ssDNA-cellulose column; lane 4, pooled active fractions from hydroxylapatite column; lanes 5-13, flow through and fractions 2 to 10 from Mono S column. (C) Western blot shows the isolated polypeptide is recognized by PARP-1 antibody. Lane 1, HeLa nuclear extract; lanes 2-4, loading sample, flow through and 1 M-NaCl fraction from ssDNA-cellulose column; lane 5, active fractions from hydroxylapatite column; lanes 6-9, fractions 5-8 from Mono S column.
As estimated by quantitative Western blot, 50 μg of HeLa nuclear extract, which is optimal for excision/repair in the extract system [11, 12, 14], contains about 66 ng (580 fmol) of PARP-1, corresponding to a concentration of 29 nM in a standard 20-μl excision/repair reaction (data not shown). To determine the dependence of specificity enhancement on PARP-1 concentration, 4- and 6-protein excision reactions were titrated with the isolated protein. For 5′-directed excision in 4- or 6-protein systems, maximal specificity enhancement was achieved at a PARP-1 concentration of about 10 nM resulting in an increase in mismatch dependence from about 3- to 10-fold (Fig. 2A, 2C and data not shown), comparable to that observed in nuclear extracts [28]. This effect is the result of preferential suppression of excision on the 5′A•T homoduplex DNA. For example, addition of 4 nM PARP-1 to the 4-protein reaction reduced the 5′-directed excision on the G-T heteroduplex from 84% to 77%, while that on A•T homoduplex was reduced from 24% to 11%, resulting in an increase in mismatch dependence from 3.6 to 7.2-fold (Fig. 2A and 2C).
Fig. 2.

PARP-1 effects on 5′- and 3′-directed excision reactions. (A) 5′-directed 4-protein (MutSα, MutLα, Exo1, and RPA) reactions were titrated with 0 – 35 nM PARP-1, and excision activity scored on 5′-heteroduplex (∎) or 5′-homoduplex (●). Results shown are from triplicate experiments and expressed as mean ± one standard deviation. The arrow indicates the amount of PARP-1 present in 50 μg of nuclear extract, which is optimal for in vitro mismatch excision/repair. (B) 3′-directed 6-protein (MutSα, MutLα, Exo1, RPA, RFC, and PCNA) reactions were supplemented with PARP-1 as indicated and excision scored by NheI assay. Data are presented as in panel A. (C) Mismatch specificity was calculated as the ratio of excision on heteroduplex to that on homoduplex for 5′-directed excision in the 4-protein system using data from panel A (●), 3′-directed excision in the 6-protein system using data from panel B (▴), or 5′-directed excision in the 6-protein system (∎; raw excision data not shown). Results are expressed as mean ± one standard deviation. For clarity, only plus or minus error bars are shown for each type of reaction. (D and E) 5′- or 3′-directed excision reactions containing 10 μM or 500 μM NAD+ were supplemented with PARP-1 as indicated. Specificity values were calculated from excision yields and are presented as in panel C.
In contrast to these effects on 5′-directed excision, PARP-1 concentrations of 9 nM or less had little effect on the mismatch dependence of 3′-directed excision in the 6-protein system, which remained in the range of 18 to 22-fold (Fig 2B and 2C). However, higher PARP-1 concentrations (18 - 35 nM) reduced mismatch dependence of 3′-directed excision to about 13-fold. This is an apparent reduction that results from inhibition of hydrolysis on the 3′-heteroduplex as compared to the absence of detectable PARP-1 effects on hydrolysis of the 3′A•T homoduplex control, which remained at 2 – 4% background levels throughout the 0 - 35 nM PARP-1 range tested (Fig 2B).
The endogenous NAD+ concentration in the HeLa nuclear extract reactions described here is about 10 μM (Materials and Methods). Because a major function of PARP-1 is poly(ADP-ribosyl)ation of itself or other acceptor proteins [18, 19], we examined mismatch dependence of excision in the purified systems in the presence of 10 μM NAD+. As shown in Fig. 2D, PARP-1 effects on the 5′- or 3′-directed reactions in the presence of 10 μM NAD+ were similar to those observed in the absence of the nucleotide except that higher PARP-1 concentrations are required to achieve maximal effects. We also evaluated PARP-1 effects in the presence of 500 μM NAD+, which approximates the intracellular concentration [29, 30]. Except for a shift to somewhat higher PARP-1 concentrations, results were similar to those obtained in the presence of 10 μM NAD+ (Fig. 2E). A possible basis for this nucleotide effect is considered below.
3.2 Functional involvement of PARP-1 domains
Three functional domains have been identified in PARP-1: an N-terminal DNA-binding domain (DBD) containing three zinc fingers, two of which mediate binding to DNA breaks, a central automodification domain containing a BRCT (BRCA1 C terminus) fold that mediates protein-protein interaction, and a C-terminal catalytic domain (CAT) containing a PARP signature motif that binds NAD+ [18, 19]. To assess involvement of these elements in excision regulation, we examined a set of PARP-1 mutants with defects in one or more of these functions (Fig. 3A): DBD-Mut (a protein with point mutations in the zinc fingers of DBD, rendering the protein unable to bind DNA and nucleosomes), ΔBRCT (deletion of the BRCT fold), ΔCAT (deletion of CAT), DBD-NDB (a fusion protein between the DBD and the NAD+-binding motif), and DBD [25, 26, 31, 32]. These proteins were over-expressed in E. coli and purified to apparent homogeneity, with the exception of ΔCAT which was ≈80% pure (Fig. S1).
Fig. 3.

Dependence of mismatch repair effects on PARP-1 functional elements. (A) Domain representation of wild-type and mutant forms of PARP-1 is adapted from Wacker et al. [25]. Amino acid substitution mutations within DBD-Mut (C21G in zinc finger FI, C125G and L139P in zinc finger FII) are indicated by the letter X. (B) 5′-directed 4-protein reactions were supplemented with 0 – 35 nM recombinant wild type or mutant PARP-1. Specificity values shown are the mean of three independent determinations. Standard deviation values, which are omitted for clarity, were less than 20% of the corresponding mean values. The arrow indicates the amount of PARP-1 present in 50 μg of nuclear extract. (C) 5′-directed 4-protein reactions were titrated with 0 – 40 nM ΔCAT in the presence or absence of 500 μM NAD+. Results shown are the mean of three determinations ± one standard deviation. For clarity, only plus or minus error bars are shown.
In the absence of NAD+, the ΔCAT variant modulated the mismatch dependence of 5′-directed excision in the 4-protein system in a manner essentially identical to that of wild-type PARP-1 (Fig. 3B). By contrast, amino acid substitutions within zinc fingers FI and FII of the DNA binding domain of full length PARP-1 (DBD-Mut) abolished enhancement of mismatch dependence, suggesting that the DNA binding activity of the protein is essential for the enhancement of excision specificity. Interestingly, the DBD, DBD-NBD and ΔBRCT mutants all displayed a similar partial defect in their ability to enhance excision mismatch dependence. Since the common feature of these mutants is lack of the BRCT fold, it seems likely that this element is required to achieve full specificity-modulation activity.
As discussed above, more PARP-1 is needed in the presence of NAD+ than in its absence to achieve maximal mismatch dependence of excision (Fig. 2). To determine whether this NAD+ effect depends on the PARP-1 catalytic domain, we compared specificity enhancement by ΔCAT in the 4-protein system in the absence or presence of this nucleotide. As described above (Fig. 3B), ΔCAT behaves like wild-type PARP-1 in the 4-protein system in the absence of NAD+. However, unlike wild-type enzyme, the concentration dependence of specificity enhancement by ΔCAT was unaffected by NAD+ (Fig. 3C), indicating that this nucleotide effect depends on integrity of the catalytic domain. Western blot analysis demonstrated that in the presence of NAD+, PARP-1 was subject to auto-poly(ADP-ribosyl)ation in the presence of the 4- or 6-protein excision system (Fig. S2 and data not shown). However, poly(ADP-ribosyl)ation of MutSα, MutLα, RPA, Exo1, RFC or PCNA was not detected in these experiments. These results are consistent with the finding that PARP-1 itself is the primary target of poly(ADP-ribosyl)ation, with greater than 90% of poly(ADP-ribose) found on this protein in vivo [33-35]. Because auto-poly(ADP-ribosyl)ation reduces the DNA affinity of PARP-1 [18, 19, 36] and because integrity of the PARP-1 DNA binding domain is required for the enhancement of mismatch dependence, we attribute NAD+ modulation of this effect to PARP-1 auto-poly(ADP-ribosyl)ation.
3.3 Interaction of PARP-1 with components of the mismatch repair system
The BRCT fold has been implicated in the interaction of PARP-1 with other proteins [18, 19, 36]. PARP-1 variants lacking this motif display reduced specificity enhancement of mismatch-provoked 5′-directed excision (Fig. 3B), suggesting potential interaction of the protein with components of the repair system. PARP-1 has been previously shown to be capable of interaction with PCNA and RFC [37-39], but its interaction with other components of the mismatch repair system has not been addressed. Far Western analysis using purified proteins demonstrated direct interaction of PARP-1 with MutSα, Exo1, RPA and RFC (Fig. 4A, upper left panel), but interaction with MutLα and PCNA was not detected by this method. Co-immunoprecipitation using HeLa nuclear extract also showed interaction of PARP-1 with MutSα, RPA, RFC and PCNA, although Exo1 interaction was not demonstrable due to its low level in nuclear extract (data not shown). Since PCNA-PARP-1 interaction was not detected by Far Western analysis, we tested this interaction by co-immunoprecipitation using purified proteins. As shown in Fig 4B, immunoprecipitation with anti-PARP-1 resulted in robust co-precipitation of PCNA, confirming the finding that these two proteins directly interact [37].
Fig. 4.

Interactions of wild type and mutant forms of PARP-1 with mismatch repair activities. (A) Far Western analyses: bovine serum albumin (BSA), MutSα, MutLα, RPA, Exo1, RFC and PCNA were applied to a nitrocellulose membrane, which was then incubated with recombinant wild type PARP-1, ΔCAT, ΔBRCT, or DBD-NBD variants. Membrane-bound PARP-1 or mutant protein was visualized immunochemically by an antibody directed against the DNA binding domain. (B) Co-immunoprecipitation of purified PARP-1 and PCNA was done as described previously [27]. PCNA (20 nM) was incubated with PARP-1 (20 nM) and pulled down using anti-PARP-1 bound to protein G beads. The lane marked as Input (1/20) indicates that 5% of total reaction protein was loaded. Controls included PCNA incubation with protein G beads (beads only), or PCNA incubation with protein G beads and antibody (α-PARP-1). (C) For GST pulldown (Materials and Methods), 100 nM GST-BRCT was incubated with 100 nM MutSα, RPA or Exo1 as indicated. Negative controls included incubation of MutSα, RPA or Exo1 with glutathione-Sepharose beads alone (beads only), and substitution of GST for GST-BRCT (GST). The lane marked as Input (1/20) indicates that 5% of total reaction protein was loaded; for this lane equivalent samples from GST and GST-BRCT reactions were mixed prior to gel loading.
A PARP-1 variant lacking the catalytic domain (ΔCAT) displayed the same interaction profile as the wild-type protein (Fig. 4A, upper right panel). However, ΔBRCT and DBD-NDB variants, which lack the BRCT fold, were found to interact only with RFC (Fig. 4A, lower panels), suggesting that this PARP-1 structural motif is required for interaction with MutSα, RPA and Exo1. In fact, GST pulldown assays using purified GST or GST fused to the PARP-1 BRCT motif demonstrated direct interaction of the BRCT fold with MutSα, RPA and Exo1 (Fig. 4C).
3.4 Mismatch-provoked excision in PARP-1 depleted extracts
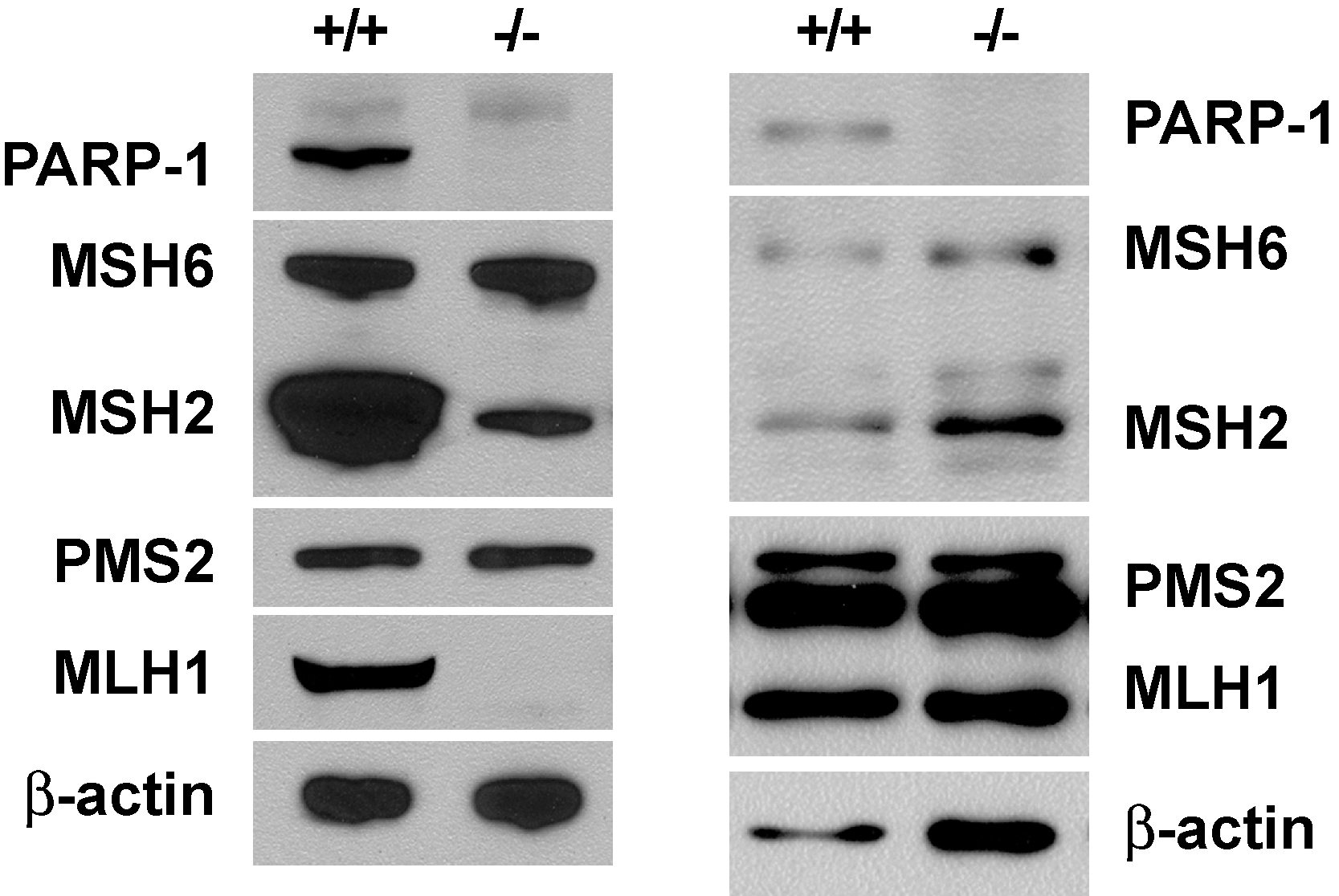
PARP-1 deficient mice and mouse embryo fibroblast (MEF) cells have been described by several laboratories [20, 21]. Although mismatch repair and mismatch-provoked excision were readily demonstrable in extracts of PARP-1+/+ MEFs from both laboratories, we have been unable to detect these reactions in PARP-1−/− MEF extracts from either source (not shown). This effect does not appear to be due to presence of a diffusible inhibitor in PARP-1−/− extracts because repair was demonstrable in mixed PARP-1−/− and PARP-1+/+ extracts at the levels expected based on wild type input. Western blot analysis revealed substantial reduction of MSH2 and MLH1 levels in the PARP-1−/− MEF cells originating from the study of de Murcia et al. [21], while the two proteins were present at near normal levels in PARP-1 deficient cells constructed by Wang et al. [20] (Fig. S3). The basis of these effects is unclear, but such observations could be indicative of down-regulation of mismatch repair under conditions of PARP-1 deficiency due to selection, regulatory effects, or other factors.
We used extract depletion as an alternate approach to evaluation of PARP-1 involvement in mismatch repair in crude fractions. Passage of HeLa nuclear extract through ssDNA-cellulose in the presence of 0.5 M NaCl (Materials and Methods) effectively removed PARP-1 from the extract without significantly affecting MutSα, MutLα or PCNA levels (Fig. 5A). However, amounts of RPA and RFC were reduced by 67% and 85%, respectively; analysis of reconstituted systems has indicated that RPA plays a regulatory role in 5′-directed mismatch-provoked excision while RFC is required for 3′- but not 5′-directed excision [11-13, 15].
Fig. 5.

Mismatch excision in HeLa nuclear extract with PARP-1 depletion. (A) PARP-1 was depleted from HeLa nuclear extract by ssDNA-cellulose chromatography (Materials and Methods). Samples (12.5 μg protein) of the column load (LD) and flow-through (FT) were resolved by SDS-PAGE and subjected to Western blotting. Proteins were visualized using an Odyssey Infrared Imaging System (LI-COR). Non-specific bands that cross-react with RFC p140 antibody are marked with an asterisk. (B) Load and flow-through samples were assayed for their ability to support 5′-directed excision. The heteroduplex and homoduplex used contained a 220-nucleotide gap located 128 bp 5′ to the G-T mismatch or the A•T bp in control DNA. The flow-through fraction was supplemented with RFC (17 ng or 68 fmol) and RPA (70 ng or 630 fmol) as indicated. (C) Load and flow-through fractions were scored for 3′-directed excision using nicked heteroduplex/homoduplex DNAs in the absence or precence of RFC and RPA as described for panel B. The mismatch dependence of 3′-directed excision in load and RPA- and RFC-supplemented flow-through fractions (≈ 12-fold) is significantly less than that found in unmanipulated nuclear extract (≈ 20-fold) ([11, 12]; data not shown).
5′- and 3′-directed excision was evaluated in column load and flow-through fractions, in the latter case with and without addition of exogenous RPA and RFC. Extract passage through DNA cellulose had no significant effect on the extent of 5′-directed heteroduplex excision. However, mismatch dependence of the reaction was reduced from 8- to 3-fold, and supplementation of the flow-through fraction with purified RPA and RFC had little effect (Fig. 5B). Reduced mismatch specificity observed with the flow-through fraction was due to increased excision on homoduplex DNA (Fig. 5B), and supplementation of this fraction with exogenous PARP-1 restored mismatch dependence to about 8-fold by preferentially suppressing homoduplex hydrolysis (Fig. 6). These results mirror those described above for 5′ heteroduplex excision in the purified system in the absence or presence of PARP-1. In contrast to these results with a 5′ heteroduplex, excision on a 3′ heteroduplex was dramatically reduced by passage through DNA cellulose, although 3′-directed hydrolysis was largely restored by addition of purified RPA and RFC (Fig. 5C). Previous work in a reconstituted system has demonstrated a selective requirement for RFC in 3′-directed mismatch-provoked excision [12].
Fig. 6.

PARP-1 titration of 5′-directed excision in PARP-1-depleted HeLa nuclear extract. Reactions contained 5′-gapped G-T heteroduplex or A•T homoduplex (Materials and Methods) and PARP-1 depleted HeLa extract supplemented with PARP-1 as indicated. Excision on 5′ heterduplex (∎) and 5′ homoduplex (●) are shown in (A), and mismatch dependence of excision in (B). Results are from triplicate experiments and expressed as mean ± one standard deviation. Arrows indicate the amount of PARP-1 present in 50 μg of nuclear extract, which is the optimal amount of extract for in vitro mismatch excision/repair.
4. Discussion
Utilizing in vitro complementation and a fractionation-based approach, we have identified PARP-1 as a factor that enhances mismatch dependence of 5′-directed excision in a purified system and in crude nuclear fractions. This effect is due to preferential suppression by PARP-1 of Exo1 hydrolysis on homoduplex DNA. We have previously shown that mismatch dependence of hydrolysis by MutSα-activated Exo1 is also modestly but significantly enhanced by MutLα [11, 15]. Like the PARP-1 results described here, this MutLα enhancement also results from suppression of hydrolysis on homoduplex DNA, an effect that may involve interaction of the MutLα MLH1 subunit with the exonuclease [40, 41].
PARP-1 has been implicated in strand break and base excision repair [42-45]. Our results suggest that its DNA repair functions may extend to mismatch repair as well. PARP-1 binds tightly to DNA strand breaks, which leads to activation of its catalytic center. The protein itself is a primary target of poly(ADP-ribosyl)ation, and this auto-modification reduces its affinity for DNA by a mechanism that involves electrostatic repulsion between the poly(ADP-ribose) polymer and DNA [42, 46]. By functioning in this manner PARP-1 is postulated to stabilize and protect strand breaks by controlling access of other activities, including repair enzymes, to the break.
The results we describe here can be understood in terms of a variation on this sort of mechanism. The mismatch dependence effects that we have observed are due to preferential suppression by PARP-1 of hydrolysis on homoduplex DNA as compared to that on heteroduplex DNA. Based on previous work [11], we presume that hydrolysis on homoduplex DNA reflects nonspecific Exo1 action whereas that on heteroduplex is mediated by the MutSα-activated form of the exonuclease. The fact that heteroduplex excision is much more resistant to PARP-1 inhibition than that occurring on homoduplex molecules suggests that the MutSα-Exo1 complex may be able to displace bound PARP-1 from a strand break, even in the absence of NAD+ (Fig. 2). Such an idea is consistent with our finding that MutSα and Exo1 are capable of interacting with PARP-1 via its BRCT domain and that PARP-1 variants lacking the BRCT domain are less effective in enhancing the mismatch dependence of 5′-directed heterouplex excision (Figs. 3 and 4).
These features of PARP-1 modulation of heteroduplex/homoduplex excision suggest that the protein is a bona fide participant in 5′-directed mismatch repair. Indeed, both MutSα and PARP-1 localize to replication foci [47, 48]. MutSα localization, which is mediated by interaction with PCNA, presumably reflects function of the mismatch repair protein in replication error correction [48]. PARP-1 localization may reflect its ability interact with PCNA [37], DNA polymerase α [49], or MutSα. Although the functional consequences of PARP-1 affiliation with the replication apparatus have not been established, association of the protein with the fork is consistent with the role we have proposed for its involvement in mismatch repair. Furthermore, both MutSα and PARP-1 have been implicated in the G2 checkpoint response to SN1 DNA methylators, a process that depends on the ATR kinase [21, 50]. Although it is not clear whether MutSα and PARP-1 are involved in coupled checkpoint events, it is interesting to note that in addition to interacting with each other, the two proteins are both capable of interaction with ATR [27, 51, 52].
During the course of this study we examined two independently constructed PARP-1−/− MEF cell lines from the Wagner and de Murcia laboratories [20, 21]. Gene disruption procedures for construction of both of these lines targeted DNA binding domain exons (exon 2 or exon 4), but analysis of the resulting knockouts have led different conclusions concerning phenotypic severity of PARP-1 deficiency. Sister chromatid exchange was found to be markedly elevated in PARP-1−/− cells from both laboratories [21, 53]. However, de Murcia et al found PARP-1-deficient animals to be hypersensitive to killing by γ-radiation while Wagner and colleagues observed similar survival for PARP-1-deficient and proficient animals (or cells) after radiation exposure [21, 53]. PARP-1−/− cells from the study of de Murcia et al. have also been shown to be hypersensitive to SN1 and SN2 alkylating agents and to display prolonged G2/M arrest after exposure to such agents [21, 54]. Although similar experiments have not been reported for PARP-1-deficient cells from the Wagner lab, these authors concluded that UV and SN1 methylator lesions are subject to efficient repair in the absence of PARP-1 [20].
We have also observed similarities and differences with MEF cell lines from the two laboratories. We have been unable to detect significant levels of mismatch repair in extracts derived from either cell line. We have also observed reduced levels of MSH2 and MLH1 in PARP-1−/− MEFs generated by de Murcia et al. [21] but near normal levels of the two proteins in PARP-1−/− cells from the Wagner group [20] (Fig. S3). The basis for these differences in repair protein levels is not clear, but involvement of cell passage and/or selection effects cannot be excluded, an issue of concern because PARP-1−/− cells have been reported to display a proliferative disadvantage [53]. To our knowledge, the question of microsatellite instability, which is generally regarded as diagnostic for mismatch repair deficiency, has not been examined in PARP-1 deficient cells. In view of the possible involvement of selection effects, such studies would presumably require early passage cells or a newly constructed knockout.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Highlights.
PARP-1 enhances mismatch dependence of 5′-directed excision by MutSα-activated Exo1
Mismatch specificity enhancement depends on PARP-1 DNA-binding and BRCT domains.
PARP-1 BRCT domain interacts with MutSα, Exo1, and RPA
Acknowledgements
We thank W. Lee Kraus (University of Texas Southwestern Medical Center) for wild type and mutant PARP-1 expression plasmids; Sam Wilson (NIEHS), Wenbin Deng (University of California-Davis) and Cynthia M. Simbulan-Rosenthal (Georgetown University) for PARP-1+/+ and PARP-1−/− MEF cell lines; Ulrich Hübscher (University of Zürich) for RFC antibody; and Elisabeth Penland for cell culture.
Funding This work was supported in part by National Institutes of Health Grant GM45190. P.M. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest The authors declare the absence of any conflicts of interest.
References
- [1].Kunkel TA, Erie DA. DNA Mismatch Repair. Annu. Rev. Biochem. 2005;74:681–710. doi: 10.1146/annurev.biochem.74.082803.133243. [DOI] [PubMed] [Google Scholar]
- [2].Iyer RR, Pluciennik A, Burdett V, Modrich PL. DNA mismatch repair: functions and mechanisms. Chem. Rev. 2006;106:302–323. doi: 10.1021/cr0404794. [DOI] [PubMed] [Google Scholar]
- [3].Jiricny J. The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell. Biol. 2006;7:335–346. doi: 10.1038/nrm1907. [DOI] [PubMed] [Google Scholar]
- [4].Modrich P. Mechanisms in eukaryotic mismatch repair. J. Biol. Chem. 2006;281:30305–30309. doi: 10.1074/jbc.R600022200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Li GM. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008;18:85–98. doi: 10.1038/cr.2007.115. [DOI] [PubMed] [Google Scholar]
- [6].Holmes J, Clark S, Modrich P. Strand-specific mismatch correction in nuclear extracts of human and Drosophila melanogaster cell lines. Proc. Natl. Acad. Sci. U. S. A. 1990;87:5837–5841. doi: 10.1073/pnas.87.15.5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Thomas DC, Roberts JD, Kunkel TA. Heteroduplex repair in extracts of human HeLa cells. J. Biol. Chem. 1991;266:3744–3751. [PubMed] [Google Scholar]
- [8].Iams K, Larson ED, Drummond JT. DNA template requirements for human mismatch repair in vitro. J. Biol. Chem. 2002;277:30805–30814. doi: 10.1074/jbc.M200846200. [DOI] [PubMed] [Google Scholar]
- [9].Fang W.-h., Modrich P. Human strand-specific mismatch repair occurs by a bidirectional mechanism similar to that of the bacterial reaction. J. Biol. Chem. 1993;268:11838–11844. [PubMed] [Google Scholar]
- [10].Wang H, Hays JB. Mismatch repair in human nuclear extracts. Quantitative analyses of excision of nicked circular mismatched DNA substrates, constructed by a new technique employing synthetic oligonucleotides. J. Biol. Chem. 2002;277:26136–26142. doi: 10.1074/jbc.M200357200. [DOI] [PubMed] [Google Scholar]
- [11].Genschel J, Modrich P. Mechanism of 5′-directed excision in human mismatch repair. Mol. Cell. 2003;12:1077–1086. doi: 10.1016/s1097-2765(03)00428-3. [DOI] [PubMed] [Google Scholar]
- [12].Dzantiev L, Constantin N, Genschel J, Iyer RR, Burgers PM, Modrich P. A defined human system that supports bidirectional mismatch-provoked excision. Mol. Cell. 2004;15:31–41. doi: 10.1016/j.molcel.2004.06.016. [DOI] [PubMed] [Google Scholar]
- [13].Zhang Y, Yuan F, Presnell SR, Tian K, Gao Y, Tomkinson AE, Gu L, Li GM. Reconstitution of 5′-directed human mismatch repair in a purified system. Cell. 2005;122:693–705. doi: 10.1016/j.cell.2005.06.027. [DOI] [PubMed] [Google Scholar]
- [14].Constantin N, Dzantiev L, Kadyrov FA, Modrich P. Human mismatch repair: Reconstitution of a nick-directed bidirectional reaction. J. Biol. Chem. 2005;280:39752–39761. doi: 10.1074/jbc.M509701200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Genschel J, Modrich P. Functions of MutL{alpha}, RPA, and HMGB1 in 5′-directed mismatch repair. J. Biol. Chem. 2009;284:21536–21544. doi: 10.1074/jbc.M109.021287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kadyrov FA, Dzantiev L, Constantin N, Modrich P. Endonucleolytic function of MutLalpha in human mismatch repair. Cell. 2006;126:297–308. doi: 10.1016/j.cell.2006.05.039. [DOI] [PubMed] [Google Scholar]
- [17].Pluciennik A, Dzantiev L, Iyer RR, Constantin N, Kadyrov FA, Modrich P. PCNA function in the activation and strand direction of MutLalpha endonuclease in mismatch repair. Proc. Natl. Acad. Sci. U. S. A. 2010;107:16066–16071. doi: 10.1073/pnas.1010662107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Krishnakumar R, Kraus WL. The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Mol. Cell. 2010;39:8–24. doi: 10.1016/j.molcel.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. PARP inhibition: PARP1 and beyond. Nat. Rev. Cancer. 2010;10:293–301. doi: 10.1038/nrc2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wang ZQ, Auer B, Stingl L, Berghammer H, Haidacher D, Schweiger M, Wagner EF. Mice lacking ADPRT and poly(ADP-ribosyl)ation develop normally but are susceptible to skin disease. Genes Dev. 1995;9:509–520. doi: 10.1101/gad.9.5.509. [DOI] [PubMed] [Google Scholar]
- [21].de Murcia JM, Niedergang C, Trucco C, Ricoul M, Dutrillaux B, Mark M, Oliver FJ, Masson M, Dierich A, LeMeur M, Walztinger C, Chambon P, de Murcia G. Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc. Natl. Acad. Sci. U. S. A. 1997;94:7303–7307. doi: 10.1073/pnas.94.14.7303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kadyrov FA, Genschel J, Fang Y, Penland E, Edelmann W, Modrich P. A possible mechanism for exonuclease 1-independent eukaryotic mismatch repair. Proc. Natl. Acad. Sci. U. S. A. 2009;106:8495–8500. doi: 10.1073/pnas.0903654106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Su S-S, Lahue RS, Au KG, Modrich P. Mispair specificity of methyl-directed DNA mismatch correction in vitro. J. Biol. Chem. 1988;263:6829–6835. [PubMed] [Google Scholar]
- [24].Genschel J, Modrich P. Analysis of the excision step in human DNA mismatch repair. Methods in enzymology. 2006;408:273–284. doi: 10.1016/S0076-6879(06)08017-7. [DOI] [PubMed] [Google Scholar]
- [25].Wacker DA, Ruhl DD, Balagamwala EH, Hope KM, Zhang T, Kraus WL. The DNA binding and catalytic domains of poly(ADP-ribose) polymerase 1 cooperate in the regulation of chromatin structure and transcription. Mol. Cell. Biol. 2007;27:7475–7485. doi: 10.1128/MCB.01314-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kim MY, Mauro S, Gevry N, Lis JT, Kraus WL. NAD+-dependent modulation of chromatin structure and transcription by nucleosome binding properties of PARP-1. Cell. 2004;119:803–814. doi: 10.1016/j.cell.2004.11.002. [DOI] [PubMed] [Google Scholar]
- [27].Liu Y, Fang Y, Shao H, Lindsey-Boltz L, Sancar A, Modrich P. Interactions of human mismatch repair proteins MutSalpha and MutLalpha with proteins of the ATR-Chk1 pathway. J. Biol. Chem. 2010;285:5974–5982. doi: 10.1074/jbc.M109.076109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Genschel J, Bazemore LR, Modrich P. Human exonuclease I is required for 5′ and 3′ mismatch repair. J. Biol. Chem. 2002;277:13302–13311. doi: 10.1074/jbc.M111854200. [DOI] [PubMed] [Google Scholar]
- [29].Loetscher P, Alvarez-Gonzalez R, Althaus FR. Poly(ADP-ribose) may signal changing metabolic conditions to the chromatin of mammalian cells. Proc. Natl. Acad. Sci. U. S. A. 1987;84:1286–1289. doi: 10.1073/pnas.84.5.1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Williams GT, Lau KM, Coote JM, Johnstone AP. NAD metabolism and mitogen stimulation of human lymphocytes. Exp. Cell Res. 1985;160:419–426. doi: 10.1016/0014-4827(85)90189-2. [DOI] [PubMed] [Google Scholar]
- [31].Gradwohl G, de Murcia J.M. Menissier, Molinete M, Simonin F, Koken M, Hoeijmakers JH, de Murcia G. The second zinc-finger domain of poly(ADP-ribose) polymerase determines specificity for single-stranded breaks in DNA. Proc. Natl. Acad. Sci. U. S .A. 1990;87:2990–2994. doi: 10.1073/pnas.87.8.2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hassa PO, Covic M, Hasan S, Imhof R, Hottiger MO. The enzymatic and DNA binding activity of PARP-1 are not required for NF-kappa B coactivator function. J. Biol. Chem. 2001;276:45588–45597. doi: 10.1074/jbc.M106528200. [DOI] [PubMed] [Google Scholar]
- [33].D’Amours D, Desnoyers S, D’Silva I, Poirier GG. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 1999;342(Pt 2):249–268. [PMC free article] [PubMed] [Google Scholar]
- [34].Huletsky A, de Murcia G, Muller S, Hengartner M, Menard L, Lamarre D, Poirier GG. The effect of poly(ADP-ribosyl)ation on native and H1-depleted chromatin. A role of poly(ADP-ribosyl)ation on core nucleosome structure. J. Biol. Chem. 1989;264:8878–8886. [PubMed] [Google Scholar]
- [35].Ogata N, Ueda K, Kawaichi M, Hayaishi O. Poly(ADP-ribose) synthetase, a main acceptor of poly(ADP-ribose) in isolated nuclei. J. Biol. Chem. 1981;256:4135–4137. [PubMed] [Google Scholar]
- [36].Woodhouse BC, Dianov GL. Poly ADP-ribose polymerase-1: an international molecule of mystery. DNA Repair (Amst) 2008;7:1077–1086. doi: 10.1016/j.dnarep.2008.03.009. [DOI] [PubMed] [Google Scholar]
- [37].Frouin I, Maga G, Denegri M, Riva F, Savio M, Spadari S, Prosperi E, Scovassi AI. Human proliferating cell nuclear antigen, poly(ADP-ribose) polymerase-1, and p21waf1/cip1. A dynamic exchange of partners. J. Biol. Chem. 2003;278:39265–39268. doi: 10.1074/jbc.C300098200. [DOI] [PubMed] [Google Scholar]
- [38].Droit A, Hunter JM, Rouleau M, Ethier C, Picard-Cloutier A, Bourgais D, Poirier GG. PARPs database: a LIMS systems for protein-protein interaction data mining or laboratory information management system. BMC Bioinformatics. 2007;8:483. doi: 10.1186/1471-2105-8-483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Isabelle M, Moreel X, Gagne JP, Rouleau M, Ethier C, Gagne P, Hendzel MJ, Poirier GG. Investigation of PARP-1, PARP-2, and PARG interactomes by affinity-purification mass spectrometry. Proteome Sci. 2010;8:22. doi: 10.1186/1477-5956-8-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Tran PT, Simon JA, Liskay RM. Interactions of Exo1p with components of MutLalpha in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U. S. A. 2001;98:9760–9765. doi: 10.1073/pnas.161175998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Nielsen FC, Jager AC, Lutzen A, Bundgaard JR, Rasmussen LJ. Characterization of human exonuclease 1 in complex with mismatch repair proteins, subcellular localization and association with PCNA. Oncogene. 2004;23:1457–1468. doi: 10.1038/sj.onc.1207265. [DOI] [PubMed] [Google Scholar]
- [42].Satoh MS, Lindahl T. Role of poly(ADP-ribose) formation in DNA repair. Nature. 1992;356:356–358. doi: 10.1038/356356a0. [DOI] [PubMed] [Google Scholar]
- [43].Dantzer F, de La Rubia G, Menissier-De Murcia J, Hostomsky Z, de Murcia G, Schreiber V. Base excision repair is impaired in mammalian cells lacking Poly(ADP-ribose) polymerase-1. Biochemistry. 2000;39:7559–7569. doi: 10.1021/bi0003442. [DOI] [PubMed] [Google Scholar]
- [44].Cistulli C, Lavrik OI, Prasad R, Hou E, Wilson SH. AP endonuclease and poly(ADP-ribose) polymerase-1 interact with the same base excision repair intermediate. DNA repair. 2004;3:581–591. doi: 10.1016/j.dnarep.2003.09.012. [DOI] [PubMed] [Google Scholar]
- [45].Parsons JL, Dianova, Allinson SL, Dianov GL. Poly(ADP-ribose) polymerase-1 protects excessive DNA strand breaks from deterioration during repair in human cell extracts. FEBS J. 2005;272:2012–2021. doi: 10.1111/j.1742-4658.2005.04628.x. [DOI] [PubMed] [Google Scholar]
- [46].Ferro AM, Olivera BM. Poly(ADP-ribosylation) in vitro. Reaction parameters and enzyme mechanism. J. Biol. Chem. 1982;257:7808–7813. [PubMed] [Google Scholar]
- [47].Simbulan-Rosenthal CM, Rosenthal DS, Hilz H, Hickey R, Malkas L, Applegren N, Wu Y, Bers G, Smulson ME. The expression of poly(ADP-ribose) polymerase during differentiation-linked DNA replication reveals that it is a component of the multiprotein DNA replication complex. Biochemistry. 1996;35:11622–11633. doi: 10.1021/bi953010z. [DOI] [PubMed] [Google Scholar]
- [48].Kleczkowska HE, Marra G, Lettieri T, Jiricny J. hMSH3 and hMSH6 interact with PCNA and colocalize with it to replication foci. Genes Dev. 2001;15:724–736. doi: 10.1101/gad.191201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Simbulan CM, Suzuki M, Izuta S, Sakurai T, Savoysky E, Kojima K, Miyahara K, Shizuta Y, Yoshida S. Poly(ADP-ribose) polymerase stimulates DNA polymerase alpha by physical association. J. Biol. Chem. 1993;268:93–99. [PubMed] [Google Scholar]
- [50].Stojic L, Brun R, Jiricny J. Mismatch repair and DNA damage signalling. DNA Repair (Amst) 2004;3:1091–1101. doi: 10.1016/j.dnarep.2004.06.006. [DOI] [PubMed] [Google Scholar]
- [51].Wang Y, Qin J. MSH2 and ATR form a signaling module and regulate two branches of the damage response to DNA methylation. Proc. Natl. Acad. Sci. U. S. A. 2003;100:15387–15392. doi: 10.1073/pnas.2536810100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Kedar PS, Stefanick DF, Horton JK, Wilson SH. Interaction between PARP-1 and ATR in mouse fibroblasts is blocked by PARP inhibition. DNA repair. 2008;7:1787–1798. doi: 10.1016/j.dnarep.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Wang ZQ, Stingl L, Morrison C, Jantsch M, Los M, Schulze-Osthoff K, Wagner EF. PARP is important for genomic stability but dispensable in apoptosis. Genes Dev. 1997;11:2347–2358. doi: 10.1101/gad.11.18.2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Trucco C, Oliver FJ, de Murcia G, Menissier-de Murcia J. DNA repair defect in poly(ADP-ribose) polymerase-deficient cell lines. Nucleic Acids Res. 1998;26:2644–2649. doi: 10.1093/nar/26.11.2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.