

Abstract
In mice, liver-restricted expression of lysosomal enzymes from adeno-associated viral serotype 8 (AAV8) vectors results in reduced antibodies to the expressed proteins. To ask whether this result might translate to patients, nonhuman primates (NHPs) were injected systemically with AAV8 encoding α-galactosidase A (α-gal). As in mice, sustained expression in monkeys attenuated antibody responses to α-gal. However, this effect was not robust, and sustained α-gal levels were 1–2 logs lower than those achieved in male mice at the same vector dose. Because our mouse studies had shown that antibody levels were directly related to expression levels, several strategies were evaluated to increase expression in monkeys. Unlike mice, expression in monkeys did not respond to androgens. Local delivery to the liver, immune suppression, a self-complementary vector and pharmacologic approaches similarly failed to increase expression. While equivalent vector copies reached mouse and primate liver and there were no apparent differences in vector form, methylation or deamination, transgene expression was limited at the mRNA level in monkeys. These results suggest that compared to mice, transcription from an AAV8 vector in monkeys can be significantly reduced. They also suggest some current limits on achieving clinically useful antibody reduction and therapeutic benefit for lysosomal storage diseases using a systemic AAV8-based approach.
Introduction
One of the potential obstacles to clinically successful gene or protein therapy for genetic diseases is the generation of host immune responses against the new protein antigen. For example, a host humoral response against the therapeutic protein could result in diminished therapeutic efficacy. Preclinical studies in mice and nonhuman primates (NHPs) with factor IX have shown that using an adeno-associated viral (AAV) vector to transduce the liver can result in expression levels of the transgene that are not only therapeutic but also lead to humoral tolerance, i.e., antibody generation against the transgene product is suppressed.1,2,3,4,5
Unlike factor IX however, the products of lysosomal storage disease (LSD) genes, such as α-galactosidase A (α-gal), are not normally secreted proteins—most is trafficked to the lysosome using mannose-6 phosphate targeting. Because only a relatively small proportion of the overexpressed protein produced from a gene therapy vector has the mannose-6 phosphate modification required for uptake by distant target tissues, relatively high serum levels of the secreted enzyme are required for therapeutic efficacy. For α-gal, serum levels approaching 1 µg/ml are required for significant substrate reduction in a Fabry mouse model that lacks α-gal activity.6 Despite these somewhat higher hurdles for efficacy and humoral tolerance, we have shown in Fabry mice that humoral immune tolerance to human α-gal can be generated after systemic administration of an AAV8 vector encoding human α-gal, and that the time required to induce this tolerance is inversely related to the α-gal serum level attained.7
The current studies were designed to determine whether the AAV-induced humoral immune tolerance seen in a mouse model of Fabry disease could be translated to NHPs. For this purpose, an AAV8 vector bearing human α-gal under the control of a hepatocyte-restricted promoter was constructed, purified, and characterized as a single preparation to evaluate its ability to reduce the antibody response to human α-gal in monkeys. To evaluate tolerance induction, both rhesus and cynomolgus macaques were challenged with purified α-gal protein in complete Freund's adjuvant (CFA) at different times after vector administration and the resultant humoral immune response characterized. In attempts to improve the immune tolerization achieved, several hypothesis-driven strategies were implemented to increase expression in NHPs, including those that (i) suppressed potential innate and adaptive immune responses, (ii) bypassed second strand synthesis, (iii) locally delivered vector to a hepatic lobe, and (iv) pharmacologically modulated pathways known to affect the intracellular fate of AAV genomes.
Results
Despite relatively low α-gal expression in NHPs, humoral immune tolerance can be achieved
Our goal was to evaluate the extent to which we could generate humoral immune tolerance to α-gal in NHPs by systemic administration of an AAV8 vector. Ours as well as other studies in (male) mice have shown that humoral immune tolerance (defined here as the lack of an antibody response to a subcutaneous challenge with the expressed protein in CFA) can be a consequence of liver-directed gene transfer with AAV vectors.4,7,8 Higher expression led to humoral immune tolerance relatively sooner, while lower expression required longer to achieve the same degree of tolerization.7
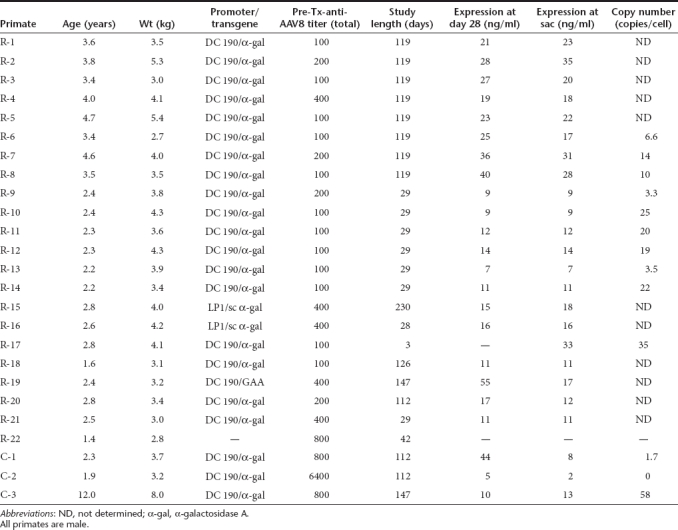
For three juvenile male rhesus macaques dosed with AAV8-αgal, Figure 1 illustrates α-gal expression levels (Figure 1a) and anti-α-gal antibody titers (Figure 1b) before and after challenges with α-gal protein in CFA. A naive animal generated a robust anti-α-gal response when challenged with α-gal in CFA (Figure 1b), illustrating that despite the high homology between human and rhesus α-gal, human α-gal can nonetheless be seen as a foreign protein in the NHP. A second NHP that received the AAV8-αgal vector also responded to a challenge at day 84 after vector administration with a significant anti-α-gal titer; this animal had only transient α-gal expression (Figure 1a). A third animal with higher and sustained expression was challenged at a later time point (day 112) and then rechallenged at day 202, but responded with only transient, low anti-α-gal titers, suggesting that at least partial humoral immune tolerization to human α-gal had been achieved in this animal.
Figure 1.

Despite the relatively low expression levels generated in primates, humoral immune tolerance to human α-galactosidase can be achieved after prolonged expression. Two juvenile male rhesus macaques (R-15, R-18) were injected intravenously with 2 × 1013 drp/kg AAV8-αgal; one juvenile male rhesus macaque (R-22) was a naive (non-transduced) control. (a) Serum α-galactosidase A (α-gal) and (b) anti-α-gal titers were determined over time. Day 0 levels represent background for each animal, i.e., before vector administration. All three monkeys were challenged with α-gal in complete Freund's adjuvant (CFA; arrows): R-22 at day 0, R-18 at day 84, and R-15 at day 112. Monkey R-15 was rechallenged with α-gal in incomplete Freund's adjuvant (IFA; arrows) at day 200. Three juvenile male cynomolgus macaques (C-1, C-2, C-3) were injected intravenously with 2 × 1013 drp/kg AAV8-αgal. (c) Serum α-gal and (d) anti-α-gal titers were determined over time. Day 0 levels represent background for each animal, i.e., before vector administration. One monkey (C-2) was challenged with α-gal in CFA at day 28, and the remaining two were challenged at day 84 (arrows).
We also evaluated antibody responses to α-gal in three cynomolgus macaques given vector. Figure 1c shows that the serum α-gal levels for two of these monkeys (C-1,C-3) are similar to each other and to that of rhesus R-18 (Figure 1a), while expression for the third (C-2) was consistently lower. It should be noted that the total anti-AAV8 titer for this latter monkey at the time of vector administration was 1:6400, and the vector copy number in the liver was negligible (Table 1) as predicted.9 Figure 1d shows that for this animal (C-2), the corresponding anti-α-gal titer after challenge increased to a level similar to that of the naive rhesus (Figure 1a). The remaining two animals were challenged at a later time point (day 84) and did not respond with increased anti-α-gal titers, suggesting that they were tolerized to α-gal. These results in rhesus and cynomolgus macaques are consistent with the earlier mouse studies, namely, humoral immune tolerization to a foreign protein can be achieved in NHPs when that protein is expressed from the liver, and the ability to achieve humoral immune tolerization is directly related to the expression level and duration prior to challenge. Thus, even though the sustained α-gal expression levels achieved in these experiments were significantly lower than those seen in NHP experiments with human factor IX, viz., tens of ng/ml for α-gal compared to hundreds of ng/ml for human factor IX, they were sufficient to generate humoral immune tolerance to α-gal when expression was allowed to proceed for a sufficiently long time prior to challenge.
Table 1. Summary of primates used in studies.
Stable α-gal expression levels are significantly lower in NHPs than in mice
Figure 2 directly compares the serum α-gal levels over time resulting from intravenous administration of the same 2 × 1013 DNase resistant particles (drp)/kg dose of AAV8-αgal to male rhesus macaques and both male and female C57BL/6J mice. As expected, serum α-gal levels obtained from male mice were significantly greater than those from female mice. Although the initial (day 3) serum α-gal expression level from male NHPs was similar to that of female mice, the longer term (>1–2 weeks), stable level in the NHPs was significantly lower (but above background).
Figure 2.

Sustained α-galactosidase A (α-gal) serum levels are significantly lower in nonhuman primates (NHPs) than in mice. Juvenile male rhesus monkeys (R-9, R-10, R-11) and both male and female C57BL/6J mice were dosed intravenously with 2 × 1013 drp/kg AAV8-αgal. Serum samples were analyzed for α-gal over time. As expected, the sustained expression levels in male mice were significantly greater than those seen in female mice. Sustained levels in female mice were significantly greater than those seen in the juvenile male primates.
The α-gal expression profile in NHPs argues against the involvement of adaptive immune responses, i.e., the time frame in which expression decreased, namely the first week after vector administration (Figures 1 and 2), would appear to be too rapid to be accounted for by either a cellular or humoral response against α-gal. This conclusion is supported by the absence of any vector-induced elevation in a panel of liver enzymes and cytokines (see Materials and Methods section) and by quantitation of IgG and IgM antibodies against α-gal (Supplementary Figure S1), which showed no titer increase in the first four weeks after vector administration.
To ask whether the lower NHP expression when compared to mouse was specific for the α-gal transgene, 2 × 1013 drp/kg of an AAV8 vector encoding the cDNA for human acid α-glucosidase (GAA) were also injected systemically into both species. Serum GAA levels from this vector were again 1–2 logs lower in a juvenile rhesus macaque (R-19; Table 1) than in male mice, and the sustained levels in the NHP were a few tens of ng/ml (data not shown), i.e., comparable to those seen for α-gal. For identical vector doses delivered systemically to mice and NHPs these results collectively demonstrate that: (i) the sustained serum levels of α-gal and GAA are significantly lower in NHPs than in mice, and (ii) these lower levels are unlikely to be accounted for by antibodies against the transgene product or the identity of the transgene.
Unlike mice, α-gal expression levels in male NHPs do not respond to androgen
Based on our mouse studies, the increases in stable α-gal expression needed in the NHP for robust tolerance induction or therapeutic efficacy are 1–2 logs. Several hypotheses were evaluated in an attempt to increase α-gal expression in NHPs and thereby improve immune tolerization. Studies have shown that AAV transduction of the murine liver leads to higher expression in male than female mice, and that this expression is sensitive to androgens.10,11 Given the extremely low testosterone levels (<10 pg/ml) in the juvenile monkeys that were dosed with AAV8-αgal, we hypothesized that increasing these levels might increase α-gal expression significantly. We first asked whether this murine androgen-related phenomenon also translated to the AAV8-mediated expression of α-gal by (i) implanting male and female mice with slow-release dihydrotestosterone (DHT) pellets prior to vector administration (Supplementary Figure S2a), (ii) surgically castrating male mice after vector administration and later replenishing androgen with DHT pellets (Supplementary Figure S2b), and (iii) implanting DHT pellets in female mice before and after vector administration (Supplementary Figure S2c). Results confirmed that α-gal expression from AAV8-αgal responded positively to androgen supplementation, and indeed, required the ongoing presence of androgen (see Supplementary Data).
We next asked whether the α-gal expression levels seen in juvenile male rhesus macaques (Figure 2) would respond to androgens. Compared to adult male NHPs, in which average testosterone levels are ∼12,000 pg/ml12,13,14 the testosterone levels in the juvenile male rhesus macaques in these studies were below the lower limit of quantitation (<10 pg/ml). As one test of the androgen sensitivity of expression in NHPs, we implanted a 21-day DHT pellet (25 mg) into a juvenile male rhesus macaque 3 days before vector dosing, resulting in serum DHT levels of ∼25,000 pg/ml at the time of vector administration. Compared to a juvenile male rhesus macaque that received the same vector dose, Figure 3 shows that these DHT levels had at most a minor effect on stable serum α-gal levels. In a second test of androgen sensitivity, we compared the results of giving the same vector dose/kg to a juvenile male and an adult male cynomolgus macaque. At the time of dosing, the juvenile had a serum testosterone level below quantitation limits, i.e., <10 pg/ml, and the adult had a level of 5,300 pg/ml. Figure 3 shows that the α-gal serum level in the adult male was essentially the same as those seen in the juvenile male rhesus and cynomolgus macaques. Collectively, these results demonstrate that: (i) in contrast to mice, expression in NHPs does not respond significantly to prevailing serum androgen levels, and (ii) α-gal expression after AAV8-αgal delivery to the liver is independent of the NHP species.
Figure 3.

Expression in monkeys does not respond to androgen. Two approaches were taken to explore the potential effects of androgen on expression from male nonhuman primates (NHPs). To increase serum androgen levels a 21-day release dihydrotestosterone (DHT) pellet (25 mg) was implanted in a juvenile male rhesus macaque (R-20) 3 days before the animal received virus. Expression over time was comparable to that of a juvenile male rhesus (R-18) in the absence of added DHT. In a second approach, expression was compared from administration of vector (2 × 1013 drp/kg AAV8-αgal) to a juvenile (C-1) and an adult (C-3) male cynomolgus monkey.
Low sustained α-gal expression levels in NHPs are unaffected by multiple pharmacologic and delivery strategies
Because transgene expression in NHPs was unaffected by DHT, we next evaluated alternative hypotheses in an attempt to increase expression. To eliminate the possibility that local immune responses might be depressing expression from the liver, NHPs were immune suppressed and the vector delivered either intravenously or directly into the liver using a balloon catheter. Figure 4 demonstrates that compared to systemic delivery in nonimmune-suppressed NHPs, neither immune suppression nor local vector delivery resulted in improved stable α-gal serum levels. Consistent with these results, over the course of the immune suppression regime, and for 6 weeks after ending the immune suppression, there was no significant increase in anti-α-gal antibodies in any of the eight monkeys (Supplementary Figure S3). Since we had verified that NHPs are capable of mounting an antibody response against α-gal (Figure 1), this result suggests that some degree of immune suppression had been achieved. In contrast to this total lack of response to α-gal, four of eight monkeys exhibited a significant increase in titer against the AAV8 vector, with initial (≤1:800) titers increasing to ≤1:6,400 (Supplementary Figure S4). These titers are to be compared to those in nonimmune-suppressed animals, which reached titers of >1:200,000, (data not shown) and again suggest that a significant degree of immune suppression had been achieved with this protocol. It is important to note that these anti-AAV8 titers are total antibody titers, not neutralizing titers. In our hands, we have shown that total titers are a more sensitive measure of prior AAV8 exposure.9
Figure 4.

Low sustained expression levels in nonhuman primates are unaffected by multiple pharmacologic and delivery strategies. Several strategies were used in an attempt to increase the long-term, sustained expression levels generated from liver-based expression in rhesus macaques. AAV8-αgal vector (2 × 1013 drp/kg) was delivered to juvenile male rhesus macaques that were: (i) naive to treatment (AAV8; R-9, R-10, R-11), (ii) immune suppressed and dosed either systemically (IS-Sys; R-6, R-7, R-8) or locally (IS-Local; R-1, R-2, R-3, R-4, R-5), (iii) treated with an anti-inflammatory (Anti-Infl; R-12, R-13, R-14), or (iv) treated with inhibitors of DNA silencing (Dep/Dac; R-21). Two juvenile male monkeys also received 2 × 1013 drp/kg of a self-complementary AAV8-αgal vector (scAAV8; R-15, R-16). Sustained expression levels (after day 15) were essentially indistinguishable for all groups. N values for each group are shown in parentheses.
To counter any local inflammatory sequelae of vector administration that might result in suppression of expression from the liver, we also treated NHPs with the anti-inflammatory glucocorticosteroid methyl prednisolone before, during, and after dosing with vector. To evaluate the hypothesis that inefficient second strand synthesis was limiting expression, an NHP was treated with a self-complementary α-gal vector (see Materials and Methods section). Although expression from this vector trended toward a transient (<2 weeks) improvement, Figure 4 shows that neither of these strategies significantly impacted the sustained serum level of α-gal. Because the self-complementary vector was constructed using a different hepatocyte-restricted promoter [the human α1-antitrypsin promoter (LP-1) rather than the human serum albumin promoter used in AAV8-αgal], these data also suggest that primate-specific promoter effects may be an unlikely explanation for these relatively low (compared to mouse) expression levels.
Gene silencing has been recognized as a potential hurdle to the clinical success of gene therapy15 e.g., by DNA methylation or histone deacetylation and heterochromatin formation.16,17 To evaluate the effects of possible gene silencing, a juvenile rhesus macaque was treated with a combination of Depakote and Dacogen to counter DNA methylation and histone deacetylation prior to AAV administration and through the first 10 days postadministration. Figure 4 shows that this pharmacologic strategy also failed to improve expression levels, suggesting that these silencing mechanisms may not be playing a major role in the relatively low (compared to mice) α-gal expression levels seen in NHPs.
In NHPs, liver expression from AAV8-αgal is inhibited at the transcriptional level
Although all NHPs were prescreened to have minimal total anti-AAV8 antibody titers, and we have shown using passive transfer experiments in mice that these levels should not impact liver transduction at the 2 × 1013 drp/kg dose used,9 it was nonetheless formally possible that these low titers (see Table 1) were sufficient to reduce liver transduction significantly. To ask whether equivalent amounts of AAV8-αgal actually reached mouse and primate liver, vector copy numbers in the liver were determined. Figure 5a shows that vector copy numbers are essentially identical at day 3 for both mouse and NHP (1 × 107 copies/µg DNA, or ∼35 copies/cell), and then decline over the next 4 weeks. This decline was virtually identical in both male mice and NHPs. These results are thus entirely consistent with our previous results at this vector dose and anti-AAV8 titers indicating that equivalent vector copies are deposited in the liver of NHPs and mice.9
Figure 5.

Vector copy numbers in liver are similar for male mouse and monkey but mRNA levels are significantly lower in nonhuman primates (NHPs). (a) Adeno-associated viral (AAV) vector copies were determined by real time PCR using a primer/probe set spanning the vector-specific α-gal/BGH poly A junction. Day 3 copy numbers are essentially the same in both mice and monkey (R-17); day 28 copy numbers decline slightly but remain essentially equivalent in male mice and monkeys (R-9 to 11). (b) Vector-specific α-gal mRNA copy numbers were determined for the same mice and monkeys shown in a. mRNA copies in monkeys were 1–2 logs lower than those seen in male mice at both time points.
In contrast to these similar vector copy numbers in the livers of mice and NHPs over time, Figure 5b demonstrates that the α-gal mRNA copy number is significantly less in NHPs than mice. At all time points after vector delivery, the mRNA copy number is 1–2 logs lower in NHP liver, which is consistent with the 1–2 log lower serum (Figure 2) and liver NHP α-gal levels (data not shown). Together with the vector copy number data discussed above, these results suggest that for the same 2 × 1013 drp/kg input vector dose, equivalent amounts of vector reach the liver in both mouse and NHP, but that the resulting steady-state mRNA level in NHPs is significantly decreased relative to that in mice.
Vector CpG methylation and dC deamination do not differ between species
Given these results, it became important to investigate the molecular aspects of the vector in mouse and NHP liver. To evaluate possible silencing further, we used quantitative bisulfite pyrosequencing18 to ask whether vector deoxycytidines (dCs) in CpG dinucleotides were modified preferentially in NHP liver. For both species, CpG methylation levels within the promoter and hybrid intron and a region adjacent to the 5′ inverted terminal repeat (ITR) were examined. Figure 6a is a schematic of the AAV8-αgal vector showing the locations of the 29 CpGs analyzed. Figure 6b shows that vector CpG methylation was relatively low in mice and NHPs at day 28, and most CpGs were significantly more methylated in mice than NHPs. Figure 6c compares vector CpG methylation near the ITR and within the hybrid intron in mice at days 3 and 28 to NHPs at days 3 and 145. Low levels of methylation were detected in both species with generally highest levels seen in day 28 mice. Figure 6d depicts the average vector methylation status across all examined CpGs in mice and NHPs over time. In both species, the extent of CpG methylation increased between day 3 and later time points, and in NHPs appeared to plateau by day 28. Overall, vector CpG methylation levels did not parallel observed species-dependent differences in transgene expression. At days 3 and 28, where samples from both species were analyzed, significantly greater CpG methylation was observed in mice than primates (P < 0.0001).
Figure 6.

Vector methylation and deamination do not differ between mouse and primate. (a) Schematic of the AAV8-αgal vector. Shown are the locations of 29 CpG dinucleotides in three vector regions that were analyzed for methylation by quantitative bisulfite pyrosequencing. In all panels, error bars indicate SEM. (b) Percent methylation for each CpG at day 28 in nonhuman primates (NHPs) (R-9 to 13) and four male mice is compared. For specific CpGs in each region, significantly more methylation is observed in mice than NHPs. (c) Percent methylation for 27 CpG in two locations near the 5′ inverted terminal repeat (ITR) and within the hybrid intron at day 28 in mice from b is compared with four male mice and NHP R-17 at day 3, and two day 145 NHPs (C-1, C-3). At specific CpGs, significantly greater percent methylation is observed in day 28 mice than either day 3 mice or NHPs at either time point. (d) Summary of methylation for NHPs and mice across all analyzed CpG sites. In panels (e–h), portions of the (e) hybrid intron region and (f) near ITR region were analyzed for possible vector deoxycytidine (dC) deamination by pyrosequencing. Percent dC to deoxythymidine (dT) sequence conversion, indicative of dC deamination to uracil, is shown for sites meeting analysis criteria. Very low levels of dC to dT conversion consistent with possible deamination were observed in both regions without significant differences between mice and primate groups at any time point. For the (g) hybrid intron and (h) near ITR regions, the single existing CCA site of each was specifically assessed for dC to dT conversion consistent with vector deamination in animals from panels b and c. No significant difference is observed between species at either CCA site. As single vector dC signals in a single animal were analyzed, measurement error could not be determined for the day 3 NHP data in panels g and h.
In addition to possible CpG methylation, deamination of dCs to deoxyuridines by intracellular deaminases is another cellular antiviral strategy that could result in decreased protein production.19 To evaluate this possibility, a subset of vector dCs within regions analyzed for CpG methylation were assessed by quantitative pyrosequencing for deamination (which would lead to conversion of dC sequence to deoxythymidine (dT) after PCR amplification). Figure 6e,f show possible deamination at multiple dCs in the hybrid intron and ITR regions, respectively. These levels appear too low to account for a substantial decrease in expression, and no significant differences were observed between mice and NHPs across time points. In the promoter region, no significant dC deamination was observed in either species (data not shown). The cytidine deaminase activity of the innate defense protein APOBEC3 has been shown to target (T/C)CA sites in single-stranded DNA sequences preferentially.20,21 In Figure 6g,h, we examined dC deamination at two vector CCA sites present in the hybrid intron and ITR regions, respectively. At each site, possible low level deamination was detected. However, as with measures of general dC deamination, no meaningful difference was observed between species or across time points for dCs in this context.
Vector form in mouse and primate liver do not differ significantly
Given similar vector copy numbers in the liver and the apparent lack of covalent vector modification differences, we next evaluated the molecular form of the vector in mouse and NHP liver. Figure 7a shows liver DNA from a naive mouse and mice at days 3 and 28 post-transduction analyzed by Southern blot. The identity of vector form was interpreted from calculated and observed molecular weights and previously published studies.22 In all treated mice, multiple extrachromosomal vector forms are evident, from single-stranded monomers to large double-stranded concatameric structures. At each time point the pattern of vector forms was consistent across samples. Several low molecular weight (presumably single-stranded) forms present at day 3 were lost by day 28, as well as an unidentified ∼10.5 kb form and a ∼4.6 kb form consistent with linear double-stranded monomer. High molecular weight forms of 15 kb, 40 kb and above, and forms consistent in size with relaxed and supercoiled monomeric double-stranded circular forms were apparent at day 3. These forms were indistinguishable from forms present at day 28 and appear to persist. At day 28, a form consistent with a 9.2 kb double-stranded linear dimer appeared in all samples. Forms consistent with relaxed and supercoiled double-stranded circular monomers, both present at day 3, were of increased prevalence by day 28.
Figure 7.

Vector form does not differ between mouse and primate. Vector molecular form as a function of time was assessed in liver samples from mice and primates by Southern blot. Assignments of vector form are tentative as they are interpreted from molecular weights and published studies. (a) Vector form in five mice at days 3 and 28. Relative molecular weights are provided and the source animal for each sample listed. Liver DNA from a naive mouse was included as a control for probe specificity. At each time point, the molecular weight and relative prevalence of vector forms are consistent across animals. (b) Vector forms in mice at days 3 (M-130) and 28 (M-138) and NHPs at days 3 (R-17), 28 (R-9 to 14) and 145 (C-1 to 3) were compared. Vector copies per liver genome as determined for each sample by quantitative PCR are provided. C-2 was included as a control for probe specificity in nonhuman primates. In this animal, liver transduction failed due to its acquisition of high anti-AAV8 titer prior to vector administration.
In Figure 7b vector forms observed in mice at days 3 and 28 are compared with forms in NHPs at days 3, 28, and 145. Included as a control for probe specificity was animal C-2, which had been treated with vector but as a result of high anti-AAV8 titer had no resulting detectable vector copies in liver samples. For all samples, vector quantity as determined by Southern band intensity was consistent with copy number as measured by quantitative PCR. Vector forms identified in NHPs were consistent at each time point and all forms detected in NHPs at day 28 were evident at day 145, suggesting their long-term persistence. Interestingly, all vector forms identified in NHPs appeared identical to those in mice, were present in similar relative amounts, and exhibited nearly identical time-dependent prevalence patterns. Thus, it would appear that vector processing is essentially the same in both species and is therefore unlikely to account for the large differences observed in vector transcripts and expression between mice and NHPs.
Discussion
Enzyme replacement therapy is a well-established approach for treating LSDs.23,24,25 Treatment requires periodic infusions of recombinant enzyme, which in some cases can elicit neutralizing antibodies that may decrease therapeutic efficacy.26 Studies in mouse LSD models have consistently shown that systemic administration of an AAV8 vector bearing the cDNA for the appropriate enzyme induces humoral immune tolerance to that enzyme.6,27,28,29 In the mouse, the degree of antibody reduction has been found to be directly related to the serum level attained by hepatic production of the therapeutic protein.7 Our overall goal in these translational studies was to assess the merits of a systemic gene therapy approach for treating LSDs using an AAV8 vector in NHPs, both in terms of inducing humoral immune tolerance to the therapeutic protein and generating serum levels of the enzyme that would be therapeutic.
In NHPs, a relatively high dose of AAV8-αgal (2 × 1013 drp/kg) resulted in stable α-gal serum levels in the tens of ng/ml—levels likely too low to be therapeutic in patients based on our earlier mouse model studies.6,7 These serum α-gal levels are ∼1.5 logs lower than those obtained in male mice at the same dose (Figure 2). Despite these relatively low serum α-gal levels in NHPs, immune tolerization to α-gal could be generated provided that a sufficient duration of expression (months) preceded an antigen challenge (Figure 1). In this respect, these NHP results are very similar to those obtained in mice.7
To increase the robustness of humoral tolerance induction in NHPs, we sought to increase hepatic expression from the AAV8-αgal vector, and therefore examined hypotheses that might account for the difference in expression between mice and NHPs. Control experiments in which known amounts of α-gal were spiked into mouse and primate serum demonstrated that the α-gal enzyme-linked immunosorbent assay (ELISA) could faithfully quantify vector-generated α-gal in both species (data not shown). Second, although it was formally possible that vector administration led to antibodies against α-gal in NHPs that resulted in reduced serum levels, these low serum levels were observed in the first week after vector administration, i.e., before significant antibody production should have occurred (Figure 2). Indeed, quantifying IgG and IgM antibodies against α-gal (Supplementary Figure S1) showed no titer increase in the first 4 weeks after vector administration. The fact that immune suppressed NHPs (Figure 4) showed the same expression profiles as NHPs that were not immune suppressed (Figures 1, 2 and 4) also provides indirect evidence that antibodies to α-gal were not playing a role in the observed low α-gal serum levels. Third, at the vector dose used, equivalent vector copies reached the liver in both mouse and NHP (Figure 6). Given the low anti-AAV8 titers in the NHPs studied (Table 1), this result is entirely consistent with our earlier passive transfer results,9 and confirms that at this vector dose (2 × 1013 drp/kg), pre-existing antibody titers in this range are simply overwhelmed by the input vector. Fourth, a direct comparison of the promoter used here showed that it is equally effective in mouse and primate primary hepatocytes.9 Lastly, quantitation of liver α-gal levels revealed that they were directly related to serum levels, i.e., liver α-gal levels in NHPs were also 1–2 logs lower than those in (male) mice (data not shown).
Our results would seem to rule out many potential explanations for the large differences in expression between mice and monkeys, such as (i) differences in vector preparations, (ii) antibodies to AAV8 or α-gal, (iii) promoter effects (we used DC190 and LP-1), (iv) transgene (we used α-gal and GAA), (v) second strand synthesis, (vi) inflammatory effects, and (vii) immune responses. Although the liver-based expression of exogenous (and endogenous) genes in rodents is highly responsive to androgens (Supplementary Figure S2),30,31,32 the α-gal cDNA in NHP liver was essentially nonresponsive to circulating androgen (Figure 3). The form of the vector (monomer, concatamer, etc.) in the liver was also virtually identical in mice and NHPs (Figure 7). Thus, although the amount and form of stabilized vector appeared to be the same in mouse and NHP, the resulting expression (hepatic and blood) was significantly less in the NHP.
The origin of this difference in expression between mouse and NHP could be traced to transcription or mRNA half-life, since at the same input vector dose and vector copy number in the liver, liver α-gal mRNA copy number in NHPs was ∼2 logs less than that in male mice (Figure 5). These differences in transcript levels were mirrored by the differences in liver (data not shown) and serum α-gal levels (Figure 2), suggesting that the efficiency of protein production and secretion was very similar for both mice and NHPs. Supporting this conclusion, we have previously demonstrated that near equivalent levels of α-gal transgene expression led to similar levels of α-gal secretion in primary hepatocytes of both species.9 The in vivo results presented here suggest species-dependent differential synthesis and/or degradation of the α-gal mRNA. Some epigenetic mechanisms that might impact mRNA production from the vector, such as CpG methylation, dC deamination and incorporation of the vector into heterochromatin, would not appear to be responsible, since pharmacologic DNA methylase and histone deacetylase (HDAC) inhibition failed to alter the expression profile (Figure 4). The methylation results were confirmed by quantitative bisulfite sequencing (Figure 6), which also appeared to rule out dC deamination as a potential mechanism for decreased mRNA production in the NHP relative to the mouse.
Given the Southern results and the absence of species-specific covalent vector modification, several hypotheses can be put forward to account for the mRNA differences between mice and NHPs. If virion and vector genome fates are the same in both species, a mouse-specific androgen effect might account for the differences in mRNA levels. Indeed, androgen-dependent increases in α-gal mRNA levels in mice but not NHPs may account for a large part, if not all, of the resulting difference in expression between male mice and NHPs. Although it is formally possible that androgens may upregulate transcription factors that result in enhanced expression in mice30,31 but not NHPs, we are not aware of such species-specific effects. It is also possible that virion trafficking and localization differ between mice and NHPs, ultimately leading to differential mRNA expression. For example, it has been shown recently in vitro33 that recombinant AAV2 vectors can distribute into both the nucleoplasm and the nucleolus. The (nuclear) factors governing this distribution could be species dependent and in principal give rise to the observed differences in expression. However, based on our Southern results (Figure 7), such a scenario would seem unlikely.
It is also possible that the cellular factors capable of interacting with the vector genome differ between species. Candidates for such NHP-specific trans acting factors that could decrease transcription include both microRNA- and protein-mediated processes. The latter category may include AAV rep,9 the APOBEC3-like family of innate defense proteins, and HDACs. The APOBEC3 proteins appear to be able to inhibit AAV replication by deaminase independent mechanisms;18 HDACs could drive incorporation of the vector genome into transcriptionally silent heterochromatin. Indeed, HDAC1 has been implicated as a transcriptional repressor for integrated retroviral34 and AAV35 sequences, and suggestive evidence exists that even episomal AAV genomes are associated with histones.36 Although our pharmacologic attempt to inhibit HDACs and reactivate any heterochromatin-sequestered vector genomes showed no benefit in this regard (Figure 4), valproate is a relatively weak type 1 HDAC inhibitor. It may be possible to counteract such silencing (if it occurs) by incorporating elements into the vector that favor its maintenance in a transcriptionally competent state.37
In summary, despite the relatively low expression levels of α-gal generated in NHPs, it was possible to induce humoral immune tolerance to α-gal. Consistent with our previous mouse studies,7 tolerance induction was more likely with longer periods of expression. This result in NHPs suggests that AAV8-based liver transduction may be a viable strategy for immune tolerization in LSD patients that would allow subsequent protein-based therapy without the potential complication of neutralizing antibodies. However, current expression levels are unlikely to be sufficient for meaningful substrate reduction in patients. The large difference in secretion of LSD proteins between mice and NHPs has its origins at the message level and may be a consequence of mouse-specific androgen effects, but other possibilities exist. A more complete understanding of virion and vector genome fate and factors that influence vector mRNA levels in the NHP has the potential of significantly enhancing liver-based expression from AAV8 vectors.
Materials and Methods
Biochemicals. Biochemicals, reagents and their sources are: 5α-dihydrotestosterone slow-release pellets (5α-DHT; Innovative Research of America, Sarasota, FL), Dacogen (decitabine; MGI Pharma, Bloomington, IL), Depakote (valproic acid; Abbott Labs, Abbott Park, IL), methyl prednisolone (Henry Schein, Westwood, MA), human α-gal protein (Genzyme, Framingham, MA), CFA and incomplete Freund's adjuvant (Sigma-Aldrich, St Louis, MO), X-ray contrast agent (Optiray; Mallinckrodt, Hazelwood, MO).
Vectors. The human α-gal cDNA and the hepatocyte-specific vector construct AAV2/8-DC190-hα-gal (heretofore abbreviated as AAV8-αgal) used here have been described previously.7 The recombinant liver-specific DC190 promoter consists of tandem thrombin enhancers and a portion of the human serum albumin promoter flanked by a hybrid intron and a stuffer sequence derived from an intron of human α1-antitrypsin. Its preparation and characterization have been described recently.9 Importantly, a single pool of characterized virus was used for all mouse and monkey studies.9
Two additional control vectors were synthesized and purified by the same procedures to ask whether the results obtained with the AAV8-αgal vector were specific. An AAV8 vector containing a gene coding for an alternative lysosomal protein, namely human GAA, was also constructed using the same hepatocyte-specific expression cassette. A dose of 2 × 1013 drp/kg of this vector in (male) mice resulted in a serum expression level of >5,000 ng/ml (data not shown). A self-complementary α-gal vector was constructed using a CpG-reduced and codon-optimized version of the LP-1 backbone,38 which contains a truncated version of the human α1-antitrypsin promoter. This vector was designed to ask whether a different promoter or a self-complementary construct would significantly affect expression in primates. In mice, this vector demonstrated an initial two to threefold increase in α-gal expression relative to that obtained from AAV8-αgal, but similar sustained expression levels.
Animal experiments. Use of all animals in these experiments was governed by the rules set forth in the NRC Guide for the Care and Use of Laboratory Animals. All procedures were performed in accord with the guidelines of each institution's Institutional Animal Care and Use Committee.
Mice. Male and female C57BL/6J mice 5–6 weeks old were obtained from Jackson Laboratory (Bar Harbor, ME) and were dosed with vector by tail vein injection. For castration, an incision was made in the abdominal region of anesthetized mice and the gonads (testis, vas deferens and attached testicular fat pad) were exteriorized, clamped and excised. The cut tissue was cauterized, replaced in the abdomen and the incision closed in sutured layers.
Monkeys. Juvenile male rhesus macaques were housed at three facilities, Charles River Laboratories, Sparks, NV (of Chinese origin, designated in Table 1 as R-1 through R-8), the New England Primate Research Center, Southboro, MA (of Indian origin, R-9 through R-17), and Medicilon/MPI Preclinical Research, Shanghai, China (of Chinese origin, R-18 through R-22). Male cynomolgus macaques (C-1 through C-3) on study were at the Mannheimer Foundation, Homestead, FL. For study, rhesus and cynomolgus macaques from each facility were screened based on their total anti-AAV8 antibody titers (see below). We have correlated these total titers with neutralizing titers, and determined that the total titer (as measured herein) is a more sensitive measure of prior exposure to AAV8.9 All monkeys placed on study had total anti-AAV8 titers ≤1:800, with the exception of C-2, which had a low titer on initial screening but then had an AAV8 titer (retrospectively) on administration (day 0) of 1:6,400. Table 1 lists all the study monkeys with their corresponding total anti-AAV8 titers, weights and ages at the time of vector administration (day 0).
Drug administrations. A single 21-day slow-release pellet containing 5α-DHT [0.5 mg (mice) or 25 mg (primate)], was implanted in the scapular region to minimize animal access to the pellet. In mice, a pellet was implanted at time points between day −14 and day 21 relative to vector administration. A single primate received a DHT pellet at day −3 relative to vector administration and resulted in a DHT level at day 1 of ∼20 ng/ml; DHT reached ∼30 ng/ml at day 7 and remained stable for at least 3 weeks (data not shown). Normal primate DHT levels are ∼1.7 ng/ml (see below).
Several strategies were used to minimize immune responses and to promote maximal expression and duration of expression. To minimize any adaptive immune responses, eight rhesus monkeys were given an immune suppression regimen consisting of mycophenolate mofetil, rapamycin and daclizumab3 and were dosed with the AAV8 vector either locally (liver lobe; see below) or systemically. From day −7 through day +77 relative to vector administration (day 0), primates were gavaged twice daily with 12.5 mg/kg mycophenolate mofetil (MMF/CellCept/oral suspension). Rapamycin (2 mg/kg Rapamune/oral suspension) was gavaged once daily every day from day +3 through day +77. On day −1, a 2 mg/kg daclizumab (Zenapax) dose diluted to 50 ml in sterile saline was administered by intravenous infusion. On day +15, a 1 mg/kg daclizumab dose diluted to 50 ml in sterile saline was administered intravenously.
As an anti-inflammatory regime, monkeys received 2 mg/kg methyl prednisolone intravenously 1 hour prior to vector administration on day 0. This was followed by daily intramuscular 1 mg/kg methyl prednisolone during the first week after vector administration, and then reduced by 0.1 mg/kg on each subsequent day to zero by day 17. This dosing regime was chosen based on kidney transplant experiments in cynomolgus macaques.39
HDAC inhibitors have been identified and demonstrated to reactivate silenced viral genes.35,36 Depakote is an approved HDAC inhibitor that has been shown to enhance gene expression from AAV vectors,40 and was administered orally by capsule at a dose of 60 mg/kg twice a day from day −1 to day 10. Depakote dosing was chosen based on studies in rhesus macaques using a dose equivalent to the maximum effective dose in humans.41 Dacogen inhibits DNA methyltransferase,42 and was given subcutaneously at 1 mg/ml to achieve a final dose of 0.52 mg/kg/day from day −1 through day 10 relative to vector administration. Dacogen dosing was chosen based on baboon studies in which it was shown that CpG methylation could be reduced and the γ-globin promoter reactivated.43
Testosterone/DHT. For mice, serum testosterone concentrations were determined in pooled samples by Analytix (Manhattan, KS); lower limit of detection was 3 pg/ml. Normal testosterone levels for adult male C57BL/6J mice are ∼1,000 pg/ml.44,45 Serum testosterone levels in primates were determined by IDEXX (Grafton, MA); lower limit of detection was 10 pg/ml. Normal testosterone levels in adult male macaques are ∼12,000 pg/ml.12,13,14 Serum DHT levels for mice and monkeys were quantified by RIA (Diagnostic Systems Laboratories, Webster, TX); lower limit of detection was 4 pg/ml. Normal DHT levels for adult male C57BL/6J mice and macaques are ∼150 and ∼1,700 pg/ml, respectively.45,46
Local delivery of vector to primate liver. A local delivery technique47 was used together with the same 3-month immune suppression protocol described above to deliver the viral vector (2 × 1013 drp/kg; ∼1 × 1013 drp/ml) through a hepatic vein of primate liver. To evaluate vector activity after transport and freeze/thaw, residual vector from the procedure was refrozen and shipped from the test site back to Genzyme, where it was injected into mice and compared to vector that had not been shipped. The resulting α-gal expression levels were indistinguishable, confirming stability of the vector to shipping and freeze/thaw.
Prior to these experiments in monkeys, control experiments evaluated the potential loss of viral infectivity due to (i) contrast agent, and (ii) adsorption or inactivation by the balloon catheter. The potential effects of contrast agent were evaluated in mice. No effect on expression was seen at the concentration (10% v/v) used to dilute the viral bolus (data not shown). To evaluate potential effects of the catheter on viral loss or activity, catheters were placed (in duplicate) into the hearts of ∼4 kg New Zealand white rabbits using surgical procedures identical to those used in the monkeys. As with delivery in monkeys, blood was withdrawn into the catheter to coat the surface with blood components. The catheter was then removed from the rabbit, rinsed once with 1 ml saline, and then 2 ml virus at 2.4 × 1011 drp/ml was pushed through the catheter from a syringe followed by 1 ml saline and aliquots collected. A separate catheter was pretreated with saline alone for comparison. Total viral drps collected and the subsequent ability of the collected virus to elicit expression in mice were not significantly different from that of the input virus (data not shown).
Systemic vector delivery. For systemic delivery, vector (2 × 1013 drp/kg) at the same concentration (1 × 1013 drp/ml) was administered as a slow (50–100 seconds) bolus injection into the saphenous or cephalic vein of sedated monkeys.
Clinical pathology. There were no test article-related effects on serum chemistry parameters associated with either route of administration. Small infrequent elevations in serum enzyme levels (alanine transaminase, aspartate transaminase, alkaline phosphatase and/or lactate dehydrogenase) were observed for animals dosed either locally or systemically at various times during the studies. The slightly increased enzyme values observed were considered to be due to blood sampling (venipuncture) and/or other study-related procedures, including surgery, which cause release of these enzymes secondary to minor tissue trauma. Increases in γ-glutamyltransferase were present in animals after both local and systemic delivery, but were more prevalent in animals dosed systemically. The changes in γ-glutamyltransferase were also considered to be incidental as there were no concurrent increases in total bilirubin, alkaline phosphatase, alanine transaminase or aspartate transaminase, nor pathologic changes in the liver which could result in the release of this enzyme. Other minor fluctuations observed in serum chemistry parameters were typical of normal inter-animal variation over time and were not regarded as test article related. There was no test article-related effect on hematology parameters associated with either route of administration. For the eight animals given the immune suppression regime (R-1 through R-8), we also monitored blood levels (ELISA, Life Technologies/Invitrogen, Carlsbad, CA) of the cytokines interleukin-6, interleukin-10, interleukin-12, interferon-γ, and tumor necrosis factor-α for 1 week before and after vector delivery, including a time point of 4 hours after delivery. There were no significant vector-related changes in these parameters (data not shown).
Tolerization challenges. Primates were challenged with purified human α-gal (manufactured at Genzyme) mixed with either CFA or incomplete Freund's adjuvant. One hundred microliters of a 0.4 mg/ml emulsion was administered as a subcutaneous injection on the abdominal wall.
Anti-AAV8 titers. ELISA plates (Corning, Corning, NY) were coated overnight at 4 °C with AAV8-αgal. Plates were washed and blocked with 5% milk in TBST a minimum of 1 hour at 37 °C. After washing off the blocking solution, serum samples were diluted twofold serially in duplicate across the plate with a starting dilution of 1:100 and incubated for 1 hour at 37 °C. A 1:10,000 dilution of the secondary antibody, horseradish peroxidase-conjugated goat anti-monkey IgG antibody (Immunology Consultants Lab, Newberg, OR) or horseradish peroxidase-conjugated goat anti-monkey IgM (Fc) (Nordic Immunology, Tilburg, The Netherlands) was applied to the plates and incubated for 1 hour at 37 °C. The ELISA was developed using TMB One Component Microwell Substrate (BioFX Labs, Owings Mills, MD) in the dark for 30 minutes. The reaction was stopped with 450 nm Stop Reagent (BioFX Labs) and the plates read in a plate reader (Molecular Devices Spectra Max Plus) at 450 nm. Titers are expressed as the reciprocal of the minimum serum dilution giving an OD450 ≤0.1. For each animal, day 0 serum samples were obtained immediately prior to vector administration.
Anti-αgal titers. Primate serum samples were assayed for antibodies against α-gal with an ELISA following the protocol (above) for AAV8, except that the plates were coated with recombinant α-gal protein (Genzyme). For each animal, day 0 serum samples were obtained immediately prior to vector administration. As noted above, immune suppressed monkeys (R-1 through R-8) dosed with AAV8-αgal did not mount an anti-α-gal antibody response. Nonimmune-suppressed monkeys, both rhesus (e.g., R-15, R-18) and cynomolgus (C-1, C-3) macaques also failed to generate a significant humoral response to α-gal until they were challenged with α-gal in adjuvant.
α-Gal levels. The α-gal levels in serum and liver tissue lysates were measured by ELISA as previously described.48 Serum was collected from mice by retro-orbital bleed and from primates by standard phlebotomy. For each animal, day 0 serum samples were obtained immediately prior to vector administration.
Copy number determination. To determine vector copy number in liver, samples of mouse and primate liver were collected at necropsy, flash frozen on dry ice, and stored at −80 °C. For mouse studies, in which there were four to five mice per treatment group, one sample of liver was analyzed per mouse. For primates, two to three pieces of liver were analyzed for each animal. DNA and RNA were purified from liver lysates using the Qiagen AllPrep DNA/RNA Mini Kit according to the manufacturer's instructions. Approximately 20 mg fragments of frozen liver were placed into buffer RLT (supplied in the AllPrep kit) containing guanidine hydrochloride, and then immediately homogenized with stainless steel beads using a TissueLyser (Qiagen, Valencia, CA). The remainder of the purification procedure was carried out as recommended. For PCR assays, DNA concentration was adjusted to ∼100 ng/µl and 5 µl used per reaction. To prevent possible cross-reaction with primate genomic DNA, vector-specific primers were used that spanned the region between the human α-gal cDNA and the bovine growth hormone polyadenylation signal sequence. Vector copies per 500 ng were determined in duplicate reactions using a realtime TaqMan PCR (quantitative PCR) assay (ABI PRISM 7700; Applied Biosystems, Foster City, CA). Vector copy numbers were normalized to 1.0 µg total DNA. Vector copies per cell were calculated as previously indicated9 using an average haploid genome weight (C-value) determined from the genome size and published densitometry-based estimates.
RNA samples were assayed to measure vector-specific mRNA copy number. A reverse transcriptase primer was designed to overlap with the vector region amplified by the PCR reaction (described above). Reactions were carried out using Promega M-MLV RT, as described by the manufacturer, to generate vector-specific cDNA. Briefly, samples were adjusted to ∼200 ng/ml RNA, and 10 µl used per reaction (∼2 ng). Samples were mixed with 5× buffer, dNTP's, RNasin (as per Promega protocol), and primer (Integrated DNA Technologies, Coraville, IA) to a final volume of 48 µl. Sample mixes were spilt into two aliquots, and 1 µl M-MLV RT added to one of the aliquots. Both aliquots were then incubated sequentially at 42 °C (60 minutes) followed by 95 °C (2 minutes) for cDNA synthesis. A 5 µl portion of these reactions (representing 200 pg of the original RNA sample) was then amplified using the vector-specific PCR reaction described above. The aliquot that did not receive M-MLV RT served as a control for any residual vector DNA contamination of the RNA that might be amplified in the PCR reaction.
Analysis of vector methylation and deamination. We utilized quantitative pyrosequencing to assess vector DNA sequences for methylation of CpG dinucleotides and deamination of vector dC, which would result in dC sequence conversion to dT.22 To assess CpG methylation, 500 ng of sample DNA was bisulfite treated using the Zymo EZ DNA Methylation Kit (Zymo research, Orange, CA). Bisulfite treated DNA was eluted in 20 µl volume and 1 µl was used for each PCR. To convert PCR products to single-stranded DNA templates for pyrosequencing, PCR was performed with a biotinylated primer. Single-stranded templates (each 10 µl) were sequenced with a Pyrosequencing PSQ96 HS System (Qiagen Pyrosequencing) following the manufacturer's instructions. The methylation status of each locus was analyzed individually as a T/C single-nucleotide polymorphism using QCpG software (Qiagen Pyrosequencing). dC deamination to uracil, which would lead to sequence conversion of a vector dC to a dT after PCR, was also assessed by quantitative pyrosequencing analysis. Ten nanograms of genomic DNA was used for PCR followed by two pyrosequencing analyses without bisulfite pretreatment. The relative ratios of T to C light signal strength was used for calculating the percentage of deamination. For technical reasons, sequence conversion to dT was only analyzed for dC's with fewer than two flanking dT's in the original vector sequence. Three vector regions were focused on for CpG methylation analysis, including: (i) a 260 bp near-ITR region containing four CpGs (the first of which is 26 bps from the 5′ vector ITR), (ii) a 119 bp region containing two CpGs within the DC190 promoter, and (iii) a 220 bp region containing 23 CpGs in the vector hybrid intron. Analysis of dC deamination was limited to portions of the near ITR, promoter and hybrid intron regions described above. Analysis of the near ITR, promoter and hybrid intron regions used 5, 3, and 5 pyrosequencing assays, respectively, with separate assays designed for deamination and methylation studies. The vector target sequence covered by each assay is provided in Supplementary Table S1. Assays were performed by EpigenDx (Worcester, MA).
Southern analysis. Southern blot was performed on total DNA extracted from mouse and primate liver samples to assess vector fate and provide an independent assessment of vector quantity, following methods described previously.49 Ten micrograms of total DNA from study animals was digested overnight with the restriction enzyme Stu I (New England Biolabs, Ipswich, MA), a vector noncutter. DNA fragments were separated on a 0.7% agarose gel and, by alkaline transfer, covalently linked to a nylon membrane with a positive surface charge (BrightStar-Plus; Ambion, Carlsbad, CA). Vector genomes were detected by overnight hybridization with a 32P-labeled probe (PerkinElmer, Waltham, MA) complementary to its BGH poly A in ULTRAhybe (Applied Biosystems) and were visualized by autoradiography with Kodak BioMAX MS film (Carestream Health, Rochester, NY). Probe specificity was demonstrated through the use of negative controls, as indicated.
Statistics and error analysis. Values shown represent means, and error bars depict SEM. Differences between groups were evaluated using the Student's t-test, and were considered statistically significant when P < 0.05.
SUPPLEMENTARY MATERIAL Figure S1. NHPs treated with AAV8-αgal generate neither IgM nor IgG anti-α-gal antibodies. Figure S2. Expression responds to the presence of androgen in mice. Figure S3. Immune suppressed NHPs do not generate a humoral immune response to the expressed alpha-galactosidase transgene. Table S1. CpG methylation and dC deamination assays. Data.
Acknowledgments
The authors gratefully acknowledge David Waxman (Boston University) for insightful discussions. We thank Kathy High (Children's Hospital of Philadelphia) for sharing her 3-drug immune suppression protocol prior to publication. We also thank I-Huan Wu and Zhengyu Luo for their help with the castration experiments, Kristin Taylor and Cristin Crawley for performing the cytokine and α-gal ELISAs, respectively, Benjamin Wronowski for androgen RIAs, Keith Mansfield and Angela Carville of the New England Primate Research Center, and Julie Bell and Kelly Hopper of the Mannheimer Foundation.
Supplementary Material
NHPs treated with AAV8-αgal generate neither IgM nor IgG anti-α-gal antibodies.
Expression responds to the presence of androgen in mice.
Immune suppressed NHPs do not generate a humoral immune response to the expressed alpha-galactosidase transgene.
Immune suppressed NHPs generate only a modest humoral immune response to the AAV8 vector.
CpG methylation and dC deamination assays.
REFERENCES
- Nathwani AC, Gray JT, McIntosh J, Ng CY, Zhou J, Spence Y.et al. (2007Safe and efficient transduction of the liver after peripheral vein infusion of self-complementary AAV vector results in stable therapeutic expression of human FIX in nonhuman primates Blood 1091414–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Couto LB, Patarroyo-White S, Liu T, Nagy D, Vargas JA.et al. (2006Effects of transient immunosuppression on adenoassociated, virus-mediated, liver-directed gene transfer in rhesus macaques and implications for human gene therapy Blood 1083321–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mingozzi F, Hasbrouck NC, Basner-Tschakarjan E, Edmonson SA, Hui DJ, Sabatino DE.et al. (2007Modulation of tolerance to the transgene product in a nonhuman primate model of AAV-mediated gene transfer to liver Blood 1102334–2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao O, Dobrzynski E, Wang L, Nayak S, Mingle B, Terhorst C.et al. (2007Induction and role of regulatory CD4+CD25+ T cells in tolerance to the transgene product following hepatic in vivo gene transfer Blood 1101132–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y, Powell S, Van Roey M., and, McArthur JG. Factors influencing the development of an anti-factor IX (FIX) immune response following administration of adeno-associated virus-FIX. Blood. 2001;97:3733–3737. doi: 10.1182/blood.v97.12.3733. [DOI] [PubMed] [Google Scholar]
- Ziegler RJ, Lonning SM, Armentano D, Li C, Souza DW, Cherry M.et al. (2004AAV2 vector harboring a liver-restricted promoter facilitates sustained expression of therapeutic levels of alpha-galactosidase A and the induction of immune tolerance in Fabry mice Mol Ther 9231–240. [DOI] [PubMed] [Google Scholar]
- Ziegler RJ, Cherry M, Barbon CM, Li C, Bercury SD, Armentano D.et al. (2007Correction of the biochemical and functional deficits in fabry mice following AAV8-mediated hepatic expression of alpha-galactosidase A Mol Ther 15492–500. [DOI] [PubMed] [Google Scholar]
- Sun B, Bird A, Young SP, Kishnani PS, Chen YT., and, Koeberl DD. Enhanced response to enzyme replacement therapy in Pompe disease after the induction of immune tolerance. Am J Hum Genet. 2007;81:1042–1049. doi: 10.1086/522236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurlbut GD, Ziegler RJ, Nietupski JB, Foley JW, Woodworth LA, Meyers E.et al. (2010Preexisting immunity and low expression in primates highlight translational challenges for liver-directed AAV8-mediated gene therapy Mol Ther 181983–1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidoff AM, Ng CY, Zhou J, Spence Y., and, Nathwani AC. Sex significantly influences transduction of murine liver by recombinant adeno-associated viral vectors through an androgen-dependent pathway. Blood. 2003;102:480–488. doi: 10.1182/blood-2002-09-2889. [DOI] [PubMed] [Google Scholar]
- Pañeda A, Vanrell L, Mauleon I, Crettaz JS, Berraondo P, Timmermans EJ.et al. (2009Effect of adeno-associated virus serotype and genomic structure on liver transduction and biodistribution in mice of both genders Hum Gene Ther 20908–917. [DOI] [PubMed] [Google Scholar]
- Narula A, Gu YQ, O'Donnell L, Stanton PG, Robertson DM, McLachlan RI.et al. (2002Variability in sperm suppression during testosterone administration to adult monkeys is related to follicle stimulating hormone suppression and not to intratesticular androgens J Clin Endocrinol Metab 873399–3406. [DOI] [PubMed] [Google Scholar]
- Lue Y, Wang C, Liu YX, Hikim AP, Zhang XS, Ng CM.et al. (2006Transient testicular warming enhances the suppressive effect of testosterone on spermatogenesis in adult cynomolgus monkeys (Macaca fascicularis) J Clin Endocrinol Metab 91539–545. [DOI] [PubMed] [Google Scholar]
- Malaivijitnond S, Takenaka O, Sankai T, Yoshida T, Cho F., and, Yoshikawa Y. Effects of single and multiple injections of ketamine hydrochloride on serum hormone concentrations in male cynomolgus monkeys. Lab Anim Sci. 1998;48:270–274. [PubMed] [Google Scholar]
- Bestor TH. Gene silencing as a threat to the success of gene therapy. J Clin Invest. 2000;105:409–411. doi: 10.1172/JCI9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriquez M, Aquino M, Bruno I, De Martino G, Taddei M., and, Gomez-Paloma L. Chemistry and biology of chromatin remodeling agents: state of art and future perspectives of HDAC inhibitors. Curr Med Chem. 2006;13:1119–1139. doi: 10.2174/092986706776360905. [DOI] [PubMed] [Google Scholar]
- Goffin J., and, Eisenhauer E. DNA methyltransferase inhibitors-state of the art. Ann Oncol. 2002;13:1699–1716. doi: 10.1093/annonc/mdf314. [DOI] [PubMed] [Google Scholar]
- England R., and, Pettersson M. Pyro Q-CpG: quantitative analysis of methylation in multiple CpG sites by Pyrosequencing®. Nat Methods. 2005;2:i–ii. [Google Scholar]
- Narvaiza I, Linfesty DC, Greener BN, Hakata Y, Pintel DJ, Logue E.et al. (2009Deaminase-independent inhibition of parvoviruses by the APOBEC3A cytidine deaminase PLoS Pathog 5e1000439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RS, Bishop KN, Sheehy AM, Craig HM, Petersen-Mahrt SK, Watt IN.et al. (2003DNA deamination mediates innate immunity to retroviral infection Cell 113803–809. [DOI] [PubMed] [Google Scholar]
- Mangeat B, Turelli P, Caron G, Friedli M, Perrin L., and, Trono D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature. 2003;424:99–103. doi: 10.1038/nature01709. [DOI] [PubMed] [Google Scholar]
- Sun X, Lu Y, Bish LT, Calcedo R, Wilson JM., and, Gao G. Molecular analysis of vector genome structures after liver transduction by conventional and self-complementary adeno-associated viral serotype vectors in murine and nonhuman primate models. Hum Gene Ther. 2010;21:750–761. doi: 10.1089/hum.2009.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heese BA. Current strategies in the management of lysosomal storage diseases. Semin Pediatr Neurol. 2008;15:119–126. doi: 10.1016/j.spen.2008.05.005. [DOI] [PubMed] [Google Scholar]
- Charrow J. Enzyme replacement therapy for Gaucher disease. Expert Opin Biol Ther. 2009;9:121–131. doi: 10.1517/14712590802573395. [DOI] [PubMed] [Google Scholar]
- Lim-Melia ER., and, Kronn DF. Current enzyme replacement therapy for the treatment of lysosomal storage diseases. Pediatr Ann. 2009;38:448–455. doi: 10.3928/00904481-20090723-09. [DOI] [PubMed] [Google Scholar]
- Kishnani PS, Goldenberg PC, DeArmey SL, Heller J, Benjamin D, Young S.et al. (2010Cross-reactive immunologic material status affects treatment outcomes in Pompe disease infants Mol Genet Metab 9926–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passini MA, Bu J, Fidler JA, Ziegler RJ, Foley JW, Dodge JC.et al. (2007Combination brain and systemic injections of AAV provide maximal functional and survival benefits in the Niemann-Pick mouse Proc Natl Acad Sci USA 1049505–9510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEachern KA, Nietupski JB, Chuang WL, Armentano D, Johnson J, Hutto E.et al. (2006AAV8-mediated expression of glucocerebrosidase ameliorates the storage pathology in the visceral organs of a mouse model of Gaucher disease J Gene Med 8719–729. [DOI] [PubMed] [Google Scholar]
- Barbon CM, Ziegler RJ, Li C, Armentano D, Cherry M, Desnick RJ.et al. (2005AAV8-mediated hepatic expression of acid sphingomyelinase corrects the metabolic defect in the visceral organs of a mouse model of Niemann-Pick disease Mol Ther 12431–440. [DOI] [PubMed] [Google Scholar]
- Ling G, Sugathan A, Mazor T, Fraenkel E., and, Waxman DJ. Unbiased, genome-wide in vivo mapping of transcriptional regulatory elements reveals sex differences in chromatin structure associated with sex-specific liver gene expression. Mol Cell Biol. 2010;30:5531–5544. doi: 10.1128/MCB.00601-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman DJ., and, Celenza JL. Sexual dimorphism of hepatic gene expression: novel biological role of KRAB zinc finger repressors revealed. Genes Dev. 2003;17:2607–2613. doi: 10.1101/gad.1154603. [DOI] [PubMed] [Google Scholar]
- Sereemaspun A, Takeuchi K, Sato Y, Iwamoto S, Inakagi T, Ookawara S.et al. (2005Testosterone-dependent transgene expression in the liver of the CAG-lacZ transgenic rat Gene Expr 12305–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JS., and, Samulski RJ. Enhancement of adeno-associated virus infection by mobilizing capsids into and out of the nucleolus. J Virol. 2009;83:2632–2644. doi: 10.1128/JVI.02309-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poleshko A, Palagin I, Zhang R, Boimel P, Castagna C, Adams PD.et al. (2008Identification of cellular proteins that maintain retroviral epigenetic silencing: evidence for an antiviral response J Virol 822313–2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WY, Bailey EC, McCune SL, Dong JY., and, Townes TM. Reactivation of silenced, virally transduced genes by inhibitors of histone deacetylase. Proc Natl Acad Sci USA. 1997;94:5798–5803. doi: 10.1073/pnas.94.11.5798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada T, Uchibori R, Iwata-Okada M, Takahashi M, Nomoto T, Nonaka-Sarukawa M.et al. (2006A histone deacetylase inhibitor enhances recombinant adeno-associated virus-mediated gene expression in tumor cells Mol Ther 13738–746. [DOI] [PubMed] [Google Scholar]
- Antoniou M, Harland L, Mustoe T, Williams S, Holdstock J, Yague E.et al. (2003Transgenes encompassing dual-promoter CpG islands from the human TBP and HNRPA2B1 loci are resistant to heterochromatin-mediated silencing Genomics 82269–279. [DOI] [PubMed] [Google Scholar]
- Nathwani AC, Gray JT, Ng CY, Zhou J, Spence Y, Waddington SN.et al. (2006Self-complementary adeno-associated virus vectors containing a novel liver-specific human factor IX expression cassette enable highly efficient transduction of murine and nonhuman primate liver Blood 1072653–2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozzi E, Vial C, Ostlie D, Farah B, Chavez G, Smith KG.et al. (2003Maintenance triple immunosuppression with cyclosporin A, mycophenolate sodium and steroids allows prolonged survival of primate recipients of hDAF porcine renal xenografts Xenotransplantation 10300–310. [DOI] [PubMed] [Google Scholar]
- Fan S, Maguire CA, Ramirez SH, Bradel-Tretheway B, Sapinoro R, Sui Z.et al. (2005Valproic acid enhances gene expression from viral gene transfer vectors J Virol Methods 12523–33. [DOI] [PubMed] [Google Scholar]
- Ferin M, Morrell M, Xiao E, Kochan L, Qian F, Wright T.et al. (2003Endocrine and metabolic responses to long-term monotherapy with the antiepileptic drug valproate in the normally cycling rhesus monkey J Clin Endocrinol Metab 882908–2915. [DOI] [PubMed] [Google Scholar]
- Müller A., and, Florek M. 5-Azacytidine/Azacitidine. Recent Results Cancer Res. 2010;184:159–170. doi: 10.1007/978-3-642-01222-8_11. [DOI] [PubMed] [Google Scholar]
- Lavelle D, Singh M, Vaitkus K, Hankewych M, Wilson L., and, DeSimone J. Polymorphic PvuII sites in the stage-selector elements of the baboon (P. anubis) gamma-globin gene promoter may determine HbF levels by reducing DNA methylation density. Blood. 2004;104:343A. [Google Scholar]
- Nelson JF, Latham KR., and, Finch CE. Plasma testosterone levels in C57BL/6J male mice: effects of age and disease. Acta Endocrinol. 1975;80:744–752. doi: 10.1530/acta.0.0800744. [DOI] [PubMed] [Google Scholar]
- Brouillette J, Rivard K, Lizotte E., and, Fiset C. Sex and strain differences in adult mouse cardiac repolarization: importance of androgens. Cardiovasc Res. 2005;65:148–157. doi: 10.1016/j.cardiores.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Mishra RG, Hermsmeyer RK, Miyagawa K, Sarrel P, Uchida B, Stanczyk FZ.et al. (2005Medroxyprogesterone acetate and dihydrotestosterone induce coronary hyperreactivity in intact male rhesus monkeys J Clin Endocrinol Metab 903706–3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges BL, Taylor KM, Chu Q, Scull SE, Serriello RG, Anderson SC.et al. (2005Local delivery of a viral vector mitigates neutralization by antiviral antibodies and results in efficient transduction of rabbit liver Mol Ther 121043–1051. [DOI] [PubMed] [Google Scholar]
- Ziegler RJ, Yew NS, Li C, Cherry M, Berthelette P, Romanczuk H.et al. (1999Correction of enzymatic and lysosomal storage defects in Fabry mice by adenovirus-mediated gene transfer Hum Gene Ther 101667–1682. [DOI] [PubMed] [Google Scholar]
- Southern EM. Detection of specific sequences among DNA fragments separated by gel electrophoresis. J Mol Biol. 1975;98:503–517. doi: 10.1016/s0022-2836(75)80083-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
NHPs treated with AAV8-αgal generate neither IgM nor IgG anti-α-gal antibodies.
Expression responds to the presence of androgen in mice.
Immune suppressed NHPs do not generate a humoral immune response to the expressed alpha-galactosidase transgene.
Immune suppressed NHPs generate only a modest humoral immune response to the AAV8 vector.
CpG methylation and dC deamination assays.