

Abstract
Vector-associated side effects in clinical gene therapy have provided insights into the molecular mechanisms of hematopoietic regulation in vivo. Surprisingly, many retrovirus insertion sites (RIS) present in engrafted cells have been found to cluster nonrandomly in close association with specific genes. Our data demonstrate that these genes directly influence the in vivo fate of hematopoietic cell clones. Analysis of insertions thus far has been limited to individual clinical studies. Here, we studied >7,000 insertions retrieved from various studies. More than 40% of all insertions found in engrafted gene-modified cells were clustered in the same genomic areas covering only 0.36% of the genome. Gene classification analyses displayed significant overrepresentation of genes associated with hematopoietic functions and relevance for cell growth and survival in vivo. The similarity of insertion distributions indicates that vector insertions in repopulating cells cluster in predictable patterns. Thus, insertion analyses of preclinical in vitro and murine in vivo studies as well as vector insertion repertoires in clinical trials yielded concerted results and mark a small number of interesting genomic loci and genes that warrants further investigation of the biological consequences of vector insertions.
Introduction
Integrating gammaretroviral vectors have demonstrated the ability to successfully treat life-threatening diseases, as convincingly shown by the correction of the genetic defect and amelioration of clinical manifestations in X-linked and ADA-deficient severe combined immunodeficiencies (SCID), in chronic granulomatous disease (X-CGD) and in Wiskott Aldrich syndrome.1,2,3,4,5,6,7 Unfortunately, five treated X-SCID patients developed clonal acute T-cell lymphoproliferative disorders,8,9,10 and two treated CGD patients developed myelodysplasia.11 In all cases, these complications were causally linked to insertional activation of proto-oncogenes, most strikingly LMO2 in X-SCID, and MDS1/EVI1 in X-CGD. As a result, there has been recent intense investigation of vector insertion patterns in an attempt to understand the influence of vector design, target cell properties, and patient variables that harbors an increased risk of genotoxicity, and to design safer vectors and clinical gene therapy protocols.
Integrated murine gammaretroviral proviruses contain transgene complementary DNA flanked by strong retrovirus promoter and enhancer elements that potentially activate adjacent cellular genes. Analysis of vector insertion patterns in the SCID-X1 trials clearly demonstrated nonrandom and potentially detrimental insertion effects.8,10,12 Clinical and experimental studies have further shown insertion mediated clonal selection resulting in in vitro immortalization of myeloid cells11,13 and in vivo clonal dominance3,14,15 or even subsequent leukemia in mice, nonhuman primates and human trial participants.8,16,17,18 We hypothesized that biological consequences of insertional mutagenesis are much more frequent than predicted, affecting neighboring genes in many transduced repopulating cell clones. Comparative large-scale retrovirus insertion site (RIS) analysis should enable investigators to assess probable vector mutagenic effects before or in the absence of overt clonal dominance in animal models and clinical trials.
The present report provides a comparative analysis of RIS profiles from five different clinical gene therapy studies and three preclinical models in toto.3,19,20,21,22,23,24,25 RIS were analyzed with the same bioinformatics tools and aligned to the identical human or animal genome using NCBI BLAST tools. Species and study specific features were defined in relation to (i) genomic distribution of RIS, (ii) relevance of common insertion sites (CIS), (iii) vector-targeted genes and their classifications using gene ontology (GO) and ingenuity databases, and (iv) analogy and predictive potential of preclinical models for clinical applications.
Results
Distribution of RIS among the different studies
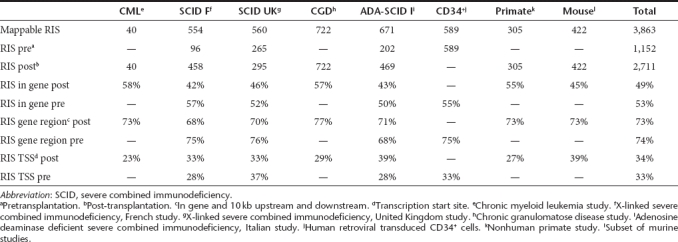
To compare the RIS of different studies referring to the same annotation of the human or mouse genome, all sequences were imported as raw FASTA formatted sequence data. Out of 3,863 exactly mappable RIS, 1,316 and 2,547 RIS were derived from preclinical and clinical samples, respectively. 2,711 RIS were determined in mature circulating blood cells or bone marrow samples after transplantation (1984 in clinical samples) and 1,152 were derived from CD34+ cells present after transduction, but before transplantation (563 in clinical samples). 49% (1,323 of 2,711) of all RIS of post-transplantation samples and 53% (616 of 1,152) of all pretransplantation samples were located in the transcribed region of a RefSeq gene. When including the 10 kb DNA region surrounding RefSeq genes, we found ~3/4th (73%; 1,979 out of 2,711) of all RIS in post-transplantation samples and 74% (850 out of 1,152) of RIS in pretransplantation samples in or near a RefSeq gene. As expected from prior studies, a third of these insertion sites were located within 10 kb around the transcription start site of the gene (Table 1).
Table 1. Distribution of retroviral insertion sites (RIS) detected in murine, nonhuman primate, and human preclinical and clinical studies.
Presence of CIS in human/primate pre- and post-transplantation samples
Comparative analysis of the 3,441 RIS of the human/primate dataset including pre- and post-transplantation samples revealed that 45% (1,547 RIS) were clustered in small genomic regions, termed CIS, compared to 6.5% expected under a uniform random distribution of the RIS (P < 10−5). The proportion of RIS involved in CIS post-transplant (37%, 839 of 2,289) was significantly increased compared to the corresponding proportion pretransplant (20%, 232 of 1,152), even after adjusting for the difference in RIS numbers between the pre- and post-transplant samples (P < 10−5). The degree of RIS clustering in the human/primate post-transplant samples showed a further substantial difference to the pretransplant cells. Out of all RIS involved in CIS post-transplant, 41% (340 of 839) were involved in a CIS of 4th or higher order whereas only 11% (25 of 232) of all RIS involved in CIS from pretransplantation samples were located in CIS of 4th or higher order even after adjusting for the difference in RIS numbers between the pre- and post-transplant samples (P < 10−4). Most CIS were found to be present in more than one study (Table 2). Even when eliminating the very redundant CIS at the EVI1/MDS1 locus from the CGD trial, we still observed that 32% (724 of 2,174) of all post-transplantation RIS were involved in CIS versus 20% of all pretransplantation RIS (P < 10−4).
Table 2. Common insertion sites (CIS) identified in post-transplantation samples of the murine, nonhuman primate, and clinical studies.
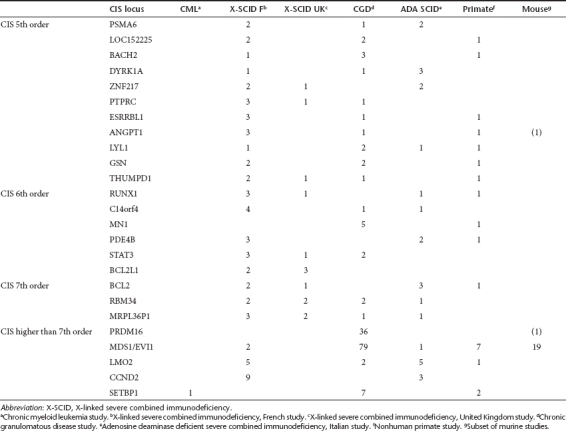
Genes most frequently involved in CIS
In pretransplantation samples, one CIS of 5th order could be identified (FLJ10597, a zinc finger protein). The other 22 RefSeq genes located in or near a CIS region of ≥5th order were found exclusively in post-transplantation samples. For 18 of these genes, evidence for direct involvement in tumor formation is available from previous mutagenesis studies.26 Of the eight genes comprising CIS locations of ≥7th order (BCL2, RBM34, PCBP1, PRDM16, MDS1/EVI1, LMO2, CCND2, and SETBP1), all are known as cancer promoting genes. Of these eight genes, five have been associated with overt clonal expansion in clinical trials.3,9,10 (Table 2). All human and nonhuman primate in vivo studies showed insertions affecting at least three CIS of 7th or higher order per 300 insertions studied.
Overrepresentation of distinct gene categories
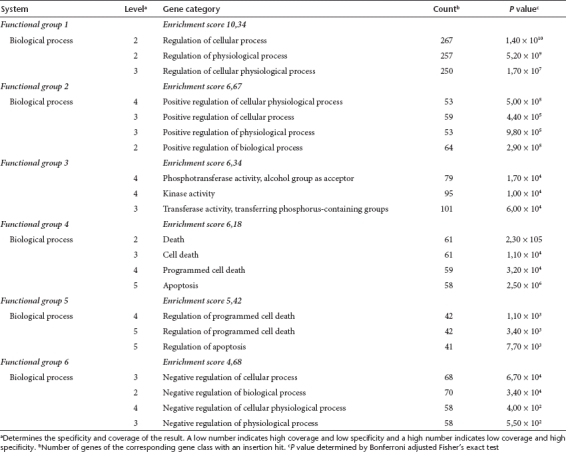
We used GO analyses to identify overrepresented functional gene categories within the RIS datasets. The combined GO analysis using all nine available post-transplantation datasets yielded significantly overrepresented specific gene categories by Fisher's exact test after Bonferroni correction, including regulation of cellular processes, protein kinase activities, and regulation of cell death (Table 3). In contrast, GO analysis of the individual pretransplantation datasets did not result in significantly overrepresented gene categories.
Table 3. Gene ontology (GO) analysis of genes with an insertion site within the gene or the neighboring 10 kb detected in post-transplantation samples.
Network analysis using Ingenuity Pathway Analysis
To define specific physiological functions and networks included in the RIS datasets, we performed comparative Ingenuity Pathway Analyses (IPA). Ingenuity analysis also offers further insights into gene data sets associated with specific diseases. In pre- and post-transplantation samples, RIS are mainly found in genes involved in hematological system development and functions. Hematopoiesis-related gene classes are the only physiological category significantly overrepresented in engrafted cells, i.e., post-transplantation samples (Figure 1).
Figure 1.

Ingenuity pathway analysis (IPA). Genes of post-transplantation samples of all studies with an insertion within the gene or in the neighboring 10 kb were classified according to physiological function. The x-axis indicates the category to which the analyzed genes contribute. We show only the significant categories. For each analyzed gene group the statistical significance (Bonferroni corrected Fisher's exact test) of overrepresented genes in a pathway is given on the y-axis.
Overrepresented “molecular function” gene categories in post-transplantation samples are “gene expression,” “molecular growth and proliferation,” “cell death,” “cell cycle,” and other cellular growth-related categories. In CIS genes, overrepresentation of these categories is most significant. The most significant disease gene categories in the RIS dataset is “cancer” followed by “immunological disease,” “hematological disease,” and “connective tissue disorders.”
Predictive value of nonhuman preclinical data for clinical studies
To study potential overlaps between human and mouse insertion sites and to determine possible CIS in homologous human and murine genomic regions, shared between both datasets, we translated the mouse gene names into human gene names using the NCBI Matchminer27 and the clone ID converter.28 One hundred and four RIS (25%) of murine insertions were located within a 100 kb region of human and nonhuman primate genes (pre- and post-) harboring RIS. When we determined the CIS separately in the mouse dataset we could show that 22% (92 RIS) of all insertion sites were located in CIS. 19 RIS in mice could be identified in the EVI1 gene locus (Supplementary Table S1). 44 (42%) of the 104 mouse RIS corresponding to a gene locus in the human/nonhuman primate dataset were located in a human/nonhuman primate CIS. This corresponds well to the findings in the human/nonhuman primate dataset.
Insertions from primate models were analyzed using the human database to allow the direct comparison of the insertion sites between human and primates. However, although the recent publication of the rhesus genome also allows sophisticated mapping of RIS to the actual species genome, the use of the human genomic sequence allows direct comparisons and more precise annotations of genes and other genomic features. The comparison of RIS between nonhuman preclinical and human clinical studies showed a large overlap: 77 RIS (25%) of the primate study could be localized in human CIS, including EVI1 as the only CIS locus ≥5th order. In primates, seven RIS were located in EVI1.
Human in vivo insertion inventories with pyrosequencing technology
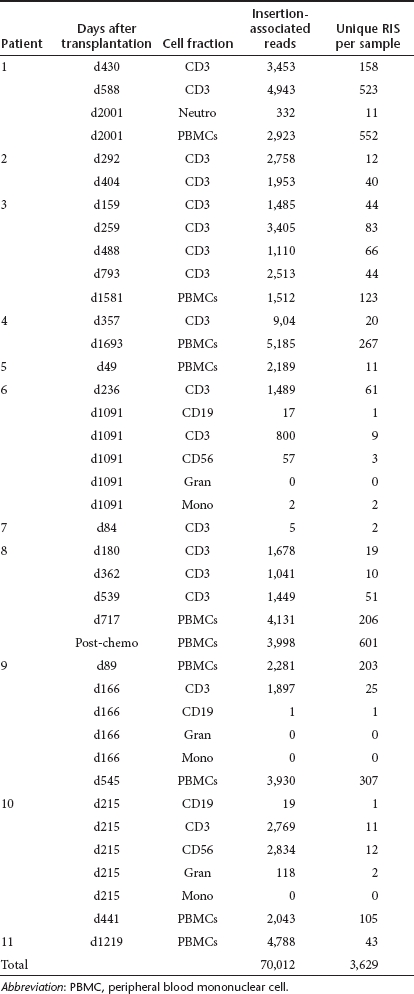
To investigate whether standard linear amplification mediated PCR (LAM-PCR) analysis followed by shotgun cloning of PCR products in bacteria and Sanger sequencing identifies the full spectrum of RIS efficiently, we reanalyzed materials from the British X-SCID study with next generation sequencing, namely pyrosequencing (GS FLX, 454/Roche) on samples derived from all 11 patients at different time points after transplantation. Sequencing reactions were carried out in five different runs with the 454 GS FLX platform (Roche, Mannheim, Germany). In total, we collected 224,457 reads. After aligning the sequences against the human genome assembly (Build 36.2) via NCBI BLAST and removing short sequences, sequences with incomplete or without LTR sequences and double sequences, we obtained 3,629 mappable RIS. The RIS distribution per patient, at specific time intervals relative to transplantation and in specific cell lineages is shown in Table 4. Of these RIS, 487 (13%) could be detected at more than one time point or in more than one lineage. If we subtract these double RIS we identified 3,142 unique RIS (Supplementary Table S2). We compared the 3,142 pyrosequenced RIS with the 295 RIS identified using Sanger sequencing on post-transplantation samples from the same patients. As not all of the original LAM-PCR products could be resequenced by 454 because of lack of material, we could not carry out a formal comparison of the relative efficiency of each method. However, 77 RIS (26%) identified by Sanger sequencing were also identified by 454 pyrosequencing technology, suggesting that the pyrosequencing approach yields similar results to Sanger sequencing. The large number of RIS identified via high throughput sequencing impressively demonstrates the extent of the clonal inventory. Four hundred and four RIS (13%) of the pyrosequencing samples and 230 (15%) RIS involved in CIS could be detected more than once at different time intervals.
Table 4. Pyrosequencing results of the British X-SCID study.
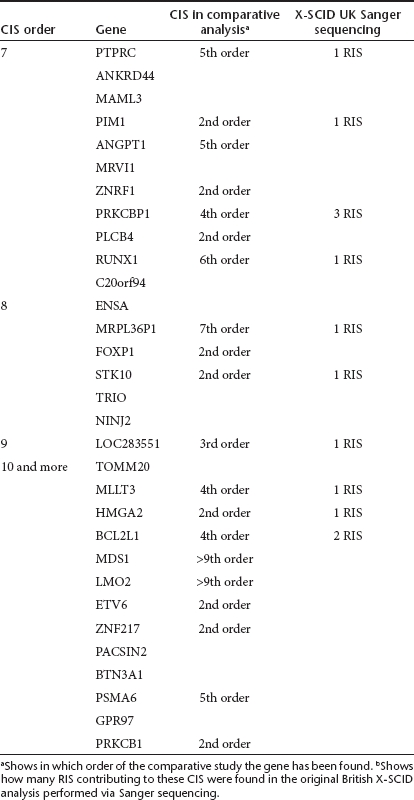
Concerning the occurrence of CIS in the pyrosequenced SCID-X1 samples, we detected 1,560 RIS (50%) in such CIS compared to 187 (6%) expected under a uniform distribution (P < 10−5). If we examined only the RIS located in CIS, we found 710 (46%) in CIS ≥4th order, 534 (34%) in CIS ≥5th order and 404 (26%) in CIS ≥6th order. Among the 505 genes involved in CIS, 34 (7%) are listed in the human cancer gene census (CGC) database29 and 192 (as much as 38%) in the mouse Retrovirus Tagged Cancer Gene Database (RTCGD).30 Higher order CIS have a higher percentage of genes listed in these two cancer gene databases. In CIS ≥7th order, 8 (26%) of the 31 genes are listed in the CGC and 20 (65%) in the RTCGD database (Table 5). The genes involved in CIS ≥7th order are listed in Table 6. Twenty of the 31 genes were CIS genes also in the comparative analysis, and 10 also occurred in the British Sanger sequenced samples (Table 6). Ingenuity analysis of the pyrosequencing samples confirmed the results obtained with the comparative meta-analysis based on Sanger sequencing.
Table 5. Presence of cancer genes near insertion sites.
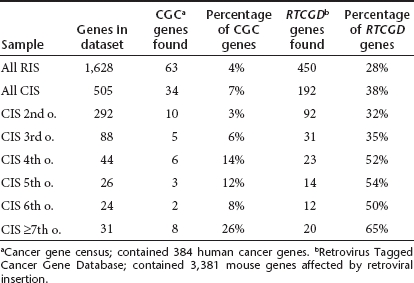
Table 6. Common insertion sites (CIS) of order seven or higher of 454 X-SCID samples and comparison with Sanger CIS.
Discussion
Because of the limited number of subjects in individual clinical phase I trials and the small animal hosts of preclinical gene therapy models, single insertion studies can only allow limited statistical evaluation on how intensively the insertion site preferences of retrovirus vectors influence functional integrity and fate of the targeted cells in vivo. Differences in annotations of the mammalian genomes used for mapping and differences in bioinformatical and statistical approaches to analyze RIS sequences have hampered comprehensive comparative integrome studies. We reasoned that a meta-analysis comparing and pooling data from numerous RIS datasets of clinical and preclinical gene therapy trials should allow to substantially extend the insights in insertion preferences of gammaretroviral vectors. We performed a comparative bioinformatical analysis that overcomes technical limitations by realigning the raw sequence data of distinct preclinical and clinical RIS datasets resulting from transduction of primitive hematopoietic stem and progenitor cells to an identical build of the species' genome. This reanalysis found largely identical results as previously obtained in the individual studies. However, we noticed also differences due to varying analysis parameters compared to the original studies (e.g., genome annotation, determination of the gene(s) next to an integration site, criteria for CIS, statistical analysis). These changes demonstrate clearly the necessity of standardized genome annotations and bioinformatical/mathematical analyses for meta-analyses as presented here.
The combined view of the resulting information set encompassed data from 24 individual patients, 1 normal donor, 142 mice, 23 primates, and 1 in vitro set of transduced cells with a combined content of 3,863 insertions. Retrovirus vector insertions were not at all randomly distributed, and had far more influence on the biological fate of engrafted cells than had initially been anticipated. Across all datasets, the affinity of the viral insertion repertoire to genes was quite surprising. When analyzing both human and nonhuman primate datasets obtained from post-transplant samples, we found that ~73% of RIS were located in or within 10 kb of RefSeq genes. A similar frequency of integrants located in and/or near RefSeq genes was found in pretransplant samples, reflecting a very high affinity of gammaretroviruses for these genomic regions.
CIS affecting the same genomic region in different cells of the same or different individuals provided a simple but highly effective statistical tool to demonstrate nonrandomness in the distribution of insertions. In addition, CIS allow for an estimate of potential genomic sites of functional deregulation in a background of sites without such effects.31,32,33 The likelihood of detecting CIS increases with the number of RIS retrieved. However, even after statistical sample-size correction, the frequency of CIS was much higher than can be anticipated based on the gene preference of murine leukemia virus-derived vectors alone. Our analysis of the pooled datasets indicated that >40% of gammaretroviral RIS in engrafted hematopoietic cells were clustered in genomic regions. CIS regions were predominantly identical between independent studies, even when conducted in different species, with their overall target area representing a very small fraction of the entire genome. The majority of frequently affected (“higher order”) CIS genes are known to promote cellular transformation (as listed in RTCGD and CGC cancer gene databases29,30) and have been found activated by gammaretroviral insertion in preclinical18,34,35 and clinical studies.3,8,10
In pretransplantation samples, the proportion of 2nd order CIS was >10-fold higher than the expected random value calculated. The number of pretransplantation CIS may point to a preference for specific gene regions already at the time of transduction, which could be due to differences in the accessibility of genes to vector insertion or due to the presence of transcription factors that facilitate insertion into these specific genomic areas.25 In lentiviruses it has been shown that regulatory viral LTR elements present in the preintegration complex can bind a variety of transcription factors.36 For retroviruses only a correlation between transcription factor-binding sites and retroviral insertion has been described.37,38 At the moment, there is no direct evidence for tethering leading to integration for gammaretroviral vectors.39
Whether highly overrepresented insertion loci might be related to the underlying clinical condition, vector design or transgene effects has been ground for speculation.3,6,17,20,21 Our data do not fully support this hypothesis. We found that prominent CIS (i.e., of 5th to 7th orders) were targeted by gammaretroviral insertion in many data sets, indicating that the mechanism of selection is not restricted to the particular constellations in single trials. However, some of these studies might share characteristics relevant for the interpretation of this finding. These include a selective advantage resulting from transgene expression at different levels, different types of immunodeficiency that influence bone marrow and immune function, and specific vector elements.
If CIS are a statistical indicator of clonal in vivo selection, we hypothesized that ranking the most frequently encountered CIS should predict which loci most likely produce clinically symptomatic manifestations of insertional mutagenesis.30 Strikingly, the top five most frequent CIS were exactly those loci associated with clinically relevant adverse insertional side effects that have occurred in clinical retrovirus gene therapy: LMO2, MDS1/EVI1, PRDM16, SETBP1, and CCND2. Moreover, MDS1/EVI1 was the most preferred insertion region in mice and nonhuman primates. Unfortunately, the available transduced human CD34+ cells cannot strictly be used as a “starting cell population” to measure the degree of clone selection upon engraftment of transduced cells in vivo: The pretransplantation CD34+ cells are highly heterogeneous and only a very small fraction of the cells (<1%) has long-term repopulation capacity. Hence, this cell subset is not available for integration analysis and direct comparison of the integration pattern of the transduced heterogeneous CD34+ cells with the pattern observed in engrafted cells to assess potential signs of clone selection may lead to interpretation errors. Thus, in the meta-analysis presented here, we focused on the description of the presence of CIS rather than draw conclusions on frequency and extent of clonal selection.
While the development of a leukemia depends on secondary events,16,35 our data demonstrate that functional effects of mutagenesis in the CIS gene regions as a first event is not related to single insertions into particular codons. Insertion locations in a particular CIS often differ by tens of thousands of base pairs. Therefore, we can hypothesize that their biological effects are not likely caused by direct mutagenic effects on the primary sequence, but rather result from e.g., changing the resident gene's activity by enhancer interference, trans-splicing or incapacitation of the original gene's transcript.
For a stratification of CIS according to gene function, involved gene loci were assessed by GO analysis and IPA. GO analysis revealed significantly overrepresented gene categories in post-transplant samples. These genes were involved in the regulation of cellular processes or cell death. The proteins are likely to have kinase- or transferase activity. Ingenuity analysis revealed that vectors were found in or near genes that are involved in physiological functions of the hematopoietic system, a preference already evident before transplantation.
Our comparative analysis elucidates a surprisingly high degree of retroviral insertion preferences in our in vitro and in vivo data sets. Depending on the function of the activated genes, one can hypothesize that in vivo selection of clones following engraftment may positively influence the therapeutic success of gene therapy by favoring vector-containing clones over nontransduced cells, but may reach an increased incidence of progression to abnormal clonality, overt myelodysplasia or leukemia. Quantitative determination of CIS is a valuable tool for the identification of the genomic context in which adverse events are more likely to occur.34 Prior to the clinical utilization of new vectors, it appears both necessary and feasible to assess safety with a large-scale RIS analysis from preclinical in vitro and in vivo models using high throughput screening of transduced cells. An acceleration of the insertion retrieval procedure could in future result in (i) a faster monitoring of the genomic RIS distribution which might substantially help to detect insertional side effects very early and to ameliorate the treatment of the patient and (ii) a prescreening of clones with potentially safe integrations if only a limited number of transduced clones are assumed to be used for clinical gene transfer (e.g., induced pluripotent stem cells).40 Analyzing CIS might also help to define genes whose temporary expression can foster engraftment and expansion of stem cells in a clinical setting.
Materials and Methods
Sequencing and insertion site analyses. LAM-PCR, LM-PCR, bacterial cloning and Sanger sequencing were performed as previously described.41,42 The RIS datasets included were from a CML gene marking study,19 two SCID-X1 clinical trials,20,21 a CGD clinical trial3 an ADA-SCID clinical trial,24 a pretransplantation dataset from human CD34+ cells,25 a subset of murine studies22 and a rhesus macaque study.23
To verify data obtained using shotgun cloning of LAM-PCR products in bacteria, direct high throughput pyrosequencing of a single clinical trial was performed and compared to the results of the conventional cloning method. Comparisons were conducted using different cell fractions from all patients of the X-SCID, London, UK trial at variable time points. The RIS were determined by direct 454 sequencing (Roche) of LAM amplification products.43 To analyze different samples in parallel in one sequencing run, a fusion primer containing one of 24 distinct barcodes was used to amplify the conventional LAM-PCR product.44,45,46 The LTR-fusion primers and linker fusion primers (MWG Biotech, Ebersberg, Germany) were designed as recommended by Roche for amplicon sequencing and used at concentrations of 5.6 pmol. 30–100 ng of purified LAM-PCR products were used as template for fusion primer PCR. PCR was performed by initial denaturation 2 minutes at 95 °C followed by 11 cycles of 95 °C for 45 seconds, 60 °C for 45 seconds and 72 °C for 1 minute, terminated by a final extension of 5 minutes at 72 °C. 5 µl of PCR product were visualized on a 2% agarose gel, and DNA concentrations of each PCR product were quantified (Nanodrop Technologies, Wilmington, DE). The barcoded PCR products were pooled and sequenced with the 454 GS FLX platform (Roche).
In the context of the European 6th framework project (CONSERT), we have developed an insertion site database (LAM-PCR database) that can store, reanalyze and update the location and associated features of retroviral vector integrants. The software trims, aligns, locates and annotates the genomic human or murine RIS. The sequences obtained were analyzed by uploading the reads as a FastA format to the LAM-PCR database (https://consert.gatc-biotech.com/lampcr/index.html). We have used NCBI BLAST for the alignment of the sequences on human (Build 35) or mouse (Build 37) genome assembly. The RIS of the primate study have only been aligned to the human genome. For the analysis of the SCID sequences obtained by 454 (Roche) high throughput sequencing we used the LAM-PCR Database with NCBI BLAST Build 36.2. The sequences are available in the database. User name and password should be requested from the corresponding author. The blast results for each sequence are given in Supplementary Table S3.
Definition of CIS. The determination of CIS has been performed manually. In brief, we have measured the distance between individual integrants independently of being located in or outside of gene-coding regions. Two RIS form a CIS if they fell within a 30 kb window. We call such an insertion region CIS of 2nd order. A CIS of 3rd order bears three RIS in a 50 kb window and a CIS of 4th order four RIS in a 100 kb window. For CIS of 5th order or higher we assumed a window of 200 kb. These definitions imply that a CIS of higher order always contains at least one CIS of lower order.
GO analysis and IPA. To classify vector-targeted genes according to GO terms and/or their interactions, for each RIS we classified the closest RefSeq genes interrupted by the vector or within 10 kb of the RIS. GO analysis was performed using the publicly available NIH-DAVID Bioinformatics resources (http://david.niaid.nih.gov/david/ease.htm)47 and IPA analysis using a license for the IPA bioinformatics application, version 7.5 (www.ingenuity.com). In these analyses, each gene was scored only once, even if multiple RIS were located within the gene or within 10 kb of the gene.
Biostatistics. Computer simulations were used to derive datasets of 10,000 synthetic random insertion events for each analysis. Comparisons between the observed and expected properties of experimental versus random RIS were made. To assess the randomness in the occurrence of CIS, we applied mathematical models of CIS formation accounting for the number of RIS, size of the genome, known insertion preferences and other parameters as previously described.31
The comparison of the number of CIS before transplant and after transplant was analyzed using a modified Monte Carlo approach which adjusts for the differences in the number of insertion sites in the two datasets. In brief, let npre and npost be the numbers of RIS before and after transplantation, and let RISpre and RISpost be the set of the exact positions of these RIS, respectively. In our analysis it turned out that npre < npost. Random samples of size npre were drawn repeatedly (10,000 simulation runs) from RISpost, and for each of these samples the numbers of CIS of order 2,…,8 were counted. This yielded simulated distributions (n = 10,000) of the number of CIS (of each order), with which the observed numbers of CIS in RISpre were then compared to obtain P values. This comparison was not biased by the differences in the sample sizes npre, npost because the random samples drawn from RISpost contained the same numbers of RIS as RISpre.
The overrepresentation of gene categories affected by an insertion was examined using the output of the NIH-DAVID Bioinformatic resources software. Overrepresented gene categories were determined by Fisher's exact test, which compares the proportion of genes having at least one RIS and belonging to a particular category, with the proportion of all genes that fall into this category. The results were adjusted for multiple testing using the Bonferroni correction of the P values provided by the software. Note that this analysis presupposes that under the null hypothesis all genes have the same probability of being affected by a RIS. This is only an approximation, given that even in the special case of uniform distribution of the RIS this probability depends on the length of the genes.
Statement. Human and animal studies have been approved by the authors institutional review boards. For the human studies the Declaration of Helsinki protocols were followed and patients gave their written informed consent.
SUPPLEMENTARY MATERIAL Table S1. Identical insertion regions between mouse and human/nonhuman primate dataset. Table S2. Table of insertion sites (RIS) of 454 sequenced SCID UK samples. Table S3. Table of insertion sites (RIS) of Sanger sequenced samples.
Acknowledgments
Funding was provided by NIH R01 CA 112470-01 (http://www.nih.gov), by the Deutsche Forschungsgemeinschaft DFG (http://www.dfg.de), grant Ka976/5-3, SCHM 2134/1-1 and SFB738-C3 by the Bundesministerium für Bildung und Forschung BMBF (www.bmbf.de), grant 01GU0601 (TreatID) and 01GU0809 (iGene), the Netherlands Health Research Organization ZonMw (www.zonmw.nl), Translational Gene Therapy Program, project 43100016, and by the European Commission's 5th and 6th Framework Programs (http://ec.europa.eu/research), Contracts QLK3-CT-2001-00427-INHERINET, LSHB-CT-2004-005242-CONSERT, LSHB-CT-2006-018933 and LSHB-CT-2006-19038. The funders had no role in study design, data collection, and analysis, decision to publish, or preparation of the manuscript.
Supplementary Material
Identical insertion regions between mouse and human/nonhuman primate dataset.
Table of insertion sites (RIS) of 454 sequenced SCID UK samples.
Table of insertion sites (RIS) of Sanger sequenced samples.
REFERENCES
- Cavazzana-Calvo M, Hacein-Bey S, de Saint Basile G, Gross F, Yvon E, Nusbaum P.et al. (2000Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease Science 288669–672. [DOI] [PubMed] [Google Scholar]
- Gaspar HB, Parsley KL, Howe S, King D, Gilmour KC, Sinclair J.et al. (2004Gene therapy of X-linked severe combined immunodeficiency by use of a pseudotyped gammaretroviral vector Lancet 3642181–2187. [DOI] [PubMed] [Google Scholar]
- Ott MG, Schmidt M, Schwarzwaelder K, Stein S, Siler U, Koehl U.et al. (2006Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1 Nat Med 12401–409. [DOI] [PubMed] [Google Scholar]
- Seger R, Siler U, Reichenbach J, Notheis G, Wintergerst U, Belohradsky B.et al. (2008Immediate clinical benefit, but variable long-term correction of X-linked CGD by gene therapy in children Human Gene Therapy 191097 [Google Scholar]
- Aiuti A, Cattaneo F, Galimberti S, Benninghoff U, Cassani B, Callegaro L.et al. (2009Gene therapy for immunodeficiency due to adenosine deaminase deficiency N Engl J Med 360447–458. [DOI] [PubMed] [Google Scholar]
- Boztug K, Schmidt M, Schwarzer A, Banerjee PP, Díez IA, Dewey RA.et al. (2010Stem-cell gene therapy for the Wiskott-Aldrich syndrome N Engl J Med 3631918–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S, Hauer J, Lim A, Picard C, Wang GP, Berry CC.et al. (2010Efficacy of gene therapy for X-linked severe combined immunodeficiency N Engl J Med 363355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S, von Kalle C, Schmidt M, Le Deist F, Wulffraat N, McIntyre E.et al. (2003A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency N Engl J Med 348255–256. [DOI] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P.et al. (2003LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1 Science 302415–419. [DOI] [PubMed] [Google Scholar]
- Howe SJ, Mansour MR, Schwarzwaelder K, Bartholomae C, Hubank M, Kempski H.et al. (2008Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients J Clin Invest 1183143–3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein S, Ott MG, Schultze-Strasser S, Jauch A, Burwinkel B, Kinner A.et al. (2010Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease Nat Med 16198–204. [DOI] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S, Garrigue A, Wang GP, Soulier J, Lim A, Morillon E.et al. (2008Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1 J Clin Invest 1183132–3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Jenkins NA., and, Copeland NG. Insertional mutagenesis identifies genes that promote the immortalization of primary bone marrow progenitor cells. Blood. 2005;106:3932–3939. doi: 10.1182/blood-2005-03-1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calmels B, Ferguson C, Laukkanen MO, Adler R, Faulhaber M, Kim HJ.et al. (2005Recurrent retroviral vector integration at the Mds1/Evi1 locus in nonhuman primate hematopoietic cells Blood 1062530–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kustikova O, Fehse B, Modlich U, Yang M, Düllmann J, Kamino K.et al. (2005Clonal dominance of hematopoietic stem cells triggered by retroviral gene marking Science 3081171–1174. [DOI] [PubMed] [Google Scholar]
- Li Z, Düllmann J, Schiedlmeier B, Schmidt M, von Kalle C, Meyer J.et al. (2002Murine leukemia induced by retroviral gene marking Science 296497. [DOI] [PubMed] [Google Scholar]
- Woods NB, Bottero V, Schmidt M, von Kalle C., and, Verma IM. Gene therapy: therapeutic gene causing lymphoma. Nature. 2006;440:1123. doi: 10.1038/4401123a. [DOI] [PubMed] [Google Scholar]
- Seggewiss R, Pittaluga S, Adler RL, Guenaga FJ, Ferguson C, Pilz IH.et al. (2006Acute myeloid leukemia is associated with retroviral gene transfer to hematopoietic progenitor cells in a rhesus macaque Blood 1073865–3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glimm H, Schmidt M, Fischer M, Schwarzwaelder K, Wissler M, Klingenberg S.et al. (2005Efficient marking of human cells with rapid but transient repopulating activity in autografted recipients Blood 106893–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deichmann A, Hacein-Bey-Abina S, Schmidt M, Garrigue A, Brugman MH, Hu J.et al. (2007Vector integration is nonrandom and clustered and influences the fate of lymphopoiesis in SCID-X1 gene therapy J Clin Invest 1172225–2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzwaelder K, Howe SJ, Schmidt M, Brugman MH, Deichmann A, Glimm H.et al. (2007Gammaretrovirus-mediated correction of SCID-X1 is associated with skewed vector integration site distribution in vivo J Clin Invest 1172241–2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kustikova OS, Geiger H, Li Z, Brugman MH, Chambers SM, Shaw CA.et al. (2007Retroviral vector insertion sites associated with dominant hematopoietic clones mark “stemness” pathways Blood 1091897–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hematti P, Hong BK, Ferguson C, Adler R, Hanawa H, Sellers S.et al. (2004Distinct genomic integration of MLV and SIV vectors in primate hematopoietic stem and progenitor cells PLoS Biol 2e423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiuti A, Cassani B, Andolfi G, Mirolo M, Biasco L, Recchia A.et al. (2007Multilineage hematopoietic reconstitution without clonal selection in ADA-SCID patients treated with stem cell gene therapy J Clin Invest 1172233–2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattoglio C, Facchini G, Sartori D, Antonelli A, Miccio A, Cassani B.et al. (2007Hot spots of retroviral integration in human CD34+ hematopoietic cells Blood 1101770–1778. [DOI] [PubMed] [Google Scholar]
- Wu X, Luke BT., and, Burgess SM. Redefining the common insertion site. Virology. 2006;344:292–295. doi: 10.1016/j.virol.2005.08.047. [DOI] [PubMed] [Google Scholar]
- Bussey KJ, Kane D, Sunshine M, Narasimhan S, Nishizuka S, Reinhold WC.et al. (2003MatchMiner: a tool for batch navigation among gene and gene product identifiers Genome Biol 4R27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alibés A, Yankilevich P, Cañada A., and, Díaz-Uriarte R. IDconverter and IDClight: conversion and annotation of gene and protein IDs. BMC Bioinformatics. 2007;8:9. doi: 10.1186/1471-2105-8-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R.et al. (2004A census of human cancer genes Nat Rev Cancer 4177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akagi K, Suzuki T, Stephens RM, Jenkins NA., and, Copeland NG. RTCGD: retroviral tagged cancer gene database. Nucleic Acids Res. 2004;32 Database issue:D523–D527. doi: 10.1093/nar/gkh013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abel U, Deichmann A, Bartholomae C, Schwarzwaelder K, Glimm H, Howe S.et al. (2007Real-time definition of non-randomness in the distribution of genomic events PLoS ONE 2e570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Ridder J, Uren A, Kool J, Reinders M., and, Wessels L. Detecting statistically significant common insertion sites in retroviral insertional mutagenesis screens. PLoS Comput Biol. 2006;2:e166. doi: 10.1371/journal.pcbi.0020166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehse B., and, Roeder I. Insertional mutagenesis and clonal dominance: biological and statistical considerations. Gene Ther. 2008;15:143–153. doi: 10.1038/sj.gt.3303052. [DOI] [PubMed] [Google Scholar]
- Modlich U, Kustikova OS, Schmidt M, Rudolph C, Meyer J, Li Z.et al. (2005Leukemias following retroviral transfer of multidrug resistance 1 (MDR1) are driven by combinatorial insertional mutagenesis Blood 1054235–4246. [DOI] [PubMed] [Google Scholar]
- Baum C, Düllmann J, Li Z, Fehse B, Meyer J, Williams DA.et al. (2003Side effects of retroviral gene transfer into hematopoietic stem cells Blood 1012099–2114. [DOI] [PubMed] [Google Scholar]
- Ariumi Y, Serhan F, Turelli P, Telenti A., and, Trono D. The integrase interactor 1 (INI1) proteins facilitate Tat-mediated human immunodeficiency virus type 1 transcription. Retrovirology. 2006;3:47. doi: 10.1186/1742-4690-3-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frith MC, Fu Y, Yu L, Chen JF, Hansen U., and, Weng Z. Detection of functional DNA motifs via statistical over-representation. Nucleic Acids Res. 2004;32:1372–1381. doi: 10.1093/nar/gkh299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felice B, Cattoglio C, Cittaro D, Testa A, Miccio A, Ferrari G.et al. (2009Transcription factor binding sites are genetic determinants of retroviral integration in the human genome PLoS ONE 4e4571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattoglio C, Pellin D, Rizzi E, Maruggi G, Corti G, Miselli F.et al. (2010High-definition mapping of retroviral integration sites identifies active regulatory elements in human multipotent hematopoietic progenitors Blood 1165507–5517. [DOI] [PubMed] [Google Scholar]
- Papapetrou EP, Lee G, Malani N, Setty M, Riviere I, Tirunagari LM.et al. (2011Genomic safe harbors permit high ß-globin transgene expression in thalassemia induced pluripotent stem cells Nat Biotechnol 2973–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kustikova OS, Baum C., and, Fehse B. Retroviral integration site analysis in hematopoietic stem cells. Methods Mol Biol. 2008;430:255–267. doi: 10.1007/978-1-59745-182-6_18. [DOI] [PubMed] [Google Scholar]
- Schmidt M, Schwarzwaelder K, Bartholomae C, Zaoui K, Ball C, Pilz I.et al. (2007High-resolution insertion-site analysis by linear amplification-mediated PCR (LAM-PCR) Nat Methods 41051–1057. [DOI] [PubMed] [Google Scholar]
- Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA.et al. (2005Genome sequencing in microfabricated high-density picolitre reactors Nature 437376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parameswaran P, Jalili R, Tao L, Shokralla S, Gharizadeh B, Ronaghi M.et al. (2007A pyrosequencing-tailored nucleotide barcode design unveils opportunities for large-scale sample multiplexing Nucleic Acids Res 35e130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer M, Stenzel U., and, Hofreiter M. Parallel tagged sequencing on the 454 platform. Nat Protoc. 2008;3:267–278. doi: 10.1038/nprot.2007.520. [DOI] [PubMed] [Google Scholar]
- Bushman FD. Retroviral integration and human gene therapy. J Clin Invest. 2007;117:2083–2086. doi: 10.1172/JCI32949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC.et al. (2003DAVID: Database for Annotation, Visualization, and Integrated Discovery Genome Biol 4P3. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Identical insertion regions between mouse and human/nonhuman primate dataset.
Table of insertion sites (RIS) of 454 sequenced SCID UK samples.
Table of insertion sites (RIS) of Sanger sequenced samples.