

Abstract
Since it has been established that the injection of plasmid DNA can lead to an efficient expression of a specific protein in vivo, nonviral gene therapy approaches have been considerably improved, allowing clinical trials. However, the use of antibiotic resistance genes as selection markers for plasmid production raises safety concerns which are often pointed out by the regulatory authorities. Indeed, a horizontal gene transfer to patient's bacteria cannot be excluded, and residual antibiotic in the final product could provoke allergic reactions in sensitive individuals. A new generation of plasmid backbones devoid of antibiotic resistance marker has emerged to increase the safety profile of nonviral gene therapy trials. This article reviews the existing strategies for plasmid maintenance and, in particular, those that do not require the use of antibiotic resistance genes. They are based either on the complementation of auxotrophic strain, toxin–antitoxin systems, operator–repressor titration, RNA markers, or on the overexpression of a growth essential gene. Minicircles that allow removing of the antibiotic resistance gene from the initial vector will also be discussed. Furthermore, reported use of antibiotic-free plasmids in preclinical or clinical studies will be listed to provide a comprehensive view of these innovative technologies.
Introduction
Gene therapy approaches are of increasing interest, and >1,700 clinical trials have already been approved worldwide.1 Since the discovery that plasmid DNA injection can be an efficient way to express a specific protein in vivo,2 the so-called “nonviral” gene therapy field has been growing. The production of plasmid DNA requires specific markers able to select plasmid-containing strain after bacterial transformation and during the amplification process. When plasmids are passed on to the daughter cells, the system is segregationally stable only if each daughter cell contains at least one copy of the plasmids.3 Plasmid stability should be correlated with the number of copies of the plasmid in the parent bacteria: high-copy plasmids such as the pUC vectors which are maintained at several hundred copies per cell should be highly stable because the occurrence of plasmid-free daughter cells is statistically improbable. However such plasmids are lost at an alarming high rate under nonselective conditions.4 Indeed, plasmid imposes an additional metabolic load for the bacteria which strongly influence its growth rate and plasmid-free cells that enjoy a nontrivial advantage and can overgrow the whole population (for review,3,4). Therefore, it is essential to impose a selection pressure favorable to the selection of bacteria containing the plasmid of interest. Genes conferring resistance to antibiotics are often used as plasmid selection markers and require growing the bacteria in an antibiotic-containing culture medium. However, this approach raises several safety concerns that should be carefully considered.
These considerations have not escaped the attention of regulatory authorities. The European Pharmacopoeia 7.0 states, “Unless otherwise justified and authorised, antibiotic-resistance genes used as selectable genetic markers, particularly for clinically useful antibiotics, are not included in the vector construct. Other selection techniques for the recombinant plasmid are preferred”.5 World Health Organization (WHO) guidelines demand that “the possibility of expression of such gene sequences in mammalian cells or in micro-organisms which are potentially pathogenic, and the possible clinical consequences of such expression, should be considered”.6 In the same way, the European Agency for the Evaluation of Medicinal Products (EMA) specifies that “consideration should be given to avoiding their use, where feasible”. It states also that “special attention should be given to the nature of the selection marker. The use of certain selection markers, such as resistance to antibiotics, which may adversely impact on other clinical therapies in the target population, should be avoided” and that “it is undesirable to use in production, agents which are known to provoke sensitivity in certain individuals, for example, β-lactam antibiotics”.7 Finally, US Food and Drug Administration recommends that “penicillin and other β-lactam antibiotics be avoided during production, due to the risk of serious hypersensitivity reactions in patients. If antibiotic selection is used during production, it is preferable not to use selection markers which confer resistance to antibiotics in significant clinical use, in order to avoid unnecessary risk of spread of antibiotic resistance traits to environmental microbes. Also, residual antibiotic in the final product should be quantitated when possible, and the potential for allergy considered”.8 These concerns are of special importance when considering vaccination, where a very large population is treated in a preventive manner, and plasmid-based genetic vaccination represents the actual major development axis for plasmids as a therapeutic agent.
It is timely to consider innovative approaches concerning bacteria selection during plasmid production. This article reviews the existing strategies for plasmid maintenance and, in particular, those that do not require the use of antibiotic resistance genes. Minicircle strategy that involves removing of the antibiotic resistance gene from the initial vector will also be discussed. Furthermore, reported use of antibiotic-free plasmids for preclinical or clinical studies will be discussed. These antibiotic-free approaches will certainly know a growing success in the next few years because of their improved safety profiles.
Antibiotic Resistance Genes
Over the last century, antibiotics became a main treatment to fight bacterial infections. However, bugs have developed their own shield, and antibiotic resistances compromise the efficacy of current therapies. Emergence and spreading of bacteria resistant to high doses of antibiotics have been favored by their overuse and misuse, and there is now evidence of increasing number of multiple-drug resistant organisms (for review9,10). Bacteria can acquire resistance to antibiotics by either genetic mutation or by horizontal gene transfer (accepting resistance genes from other bacteria).
Several antibiotic resistance genes have been widely used as selectable markers in routine biotechnology. The bla gene encodes a β-lactamase that confers resistance to ampicillin which is commonly known as a broad-spectrum antibiotic. The use of this resistance gene as a selection marker is not currently acceptable for clinical trial because of the risk of spreading in the environment and because of potential horizontal gene transfers which could provide pathogenic bacteria with resistance to antibiotics that are used for patient treatment. The nptII gene appears to be a more appropriate selection marker as it confers resistance to kanamycin that is not so commonly used to treat human infections because of its numerous side effects.
Another concern about the use of antibiotic resistance genes for selection is that bacterial cultures require a large amount of antibiotics which are expensive compounds. It is difficult to avoid their degradation or inactivation, and antibiotics may not be completely effective, in particular in continuous culture conditions limiting the scale-up. Moreover, residues of antibiotics could contaminate the final product even after purification. This could be particularly problematic for patients with hypersensitivity which is relatively common for β-lactam antibiotics.11
It is also important to note that the synthesis of antibiotic resistance proteins recruits part of the available metabolic resources in bacteria. Moreover, markers are usually produced at levels far exceeding those that are necessary for plasmid maintenance and selection. The metabolic energy consumed to synthesize these enzymatic markers cannot be devoted to plasmid production, and this metabolic burden has thus an impact on culture performance and on plasmid yield.12,13
Also, antibiotic resistance genes are large prokaryotic genes, which induce two drawbacks: (i) they increase plasmid size, which induces a decrease in transfection efficiency, and (ii) they bear unmethylated CpG sequences specific to prokaryotic backbones, which induce the innate immune system. The effect of plasmid size on gene transfer efficiency has been studied in vitro and in vivo. To achieve transfection, a plasmid has to find its way to the nucleus through the relatively viscous cytosolic environment. The nuclear pore by itself is also considered as a strong barrier for plasmid diffusion to the nucleus.14,15 For both these reasons, it might be assumed that smaller size plasmids might reach more efficiently the cell nucleus during any transfection procedures, whether mediated by a chemical vector or through a physical delivery method such as electrotransfer. Indeed, it has been shown that smaller plasmids bearing the same expression cassette as larger ones were more efficiently expressed after in vivo gene transfer and that the increase in gene transfer efficiency was inversely correlated to plasmid size both in vitro and in vivo.16,17,18 Antibiotic-free plasmid systems would be equally beneficial in the context of gene or protein delivery using bacteria,19,20 since such a system is required for plasmid maintenance within the vector. In that case, a particular attention should be paid to the choice of the selection strategy and any modification of the bacterial vector should be carefully considered. Also, activation of the innate immunity by prokaryotic plasmid backbone must be taken into account. Double stranded prokaryotic DNA is a potent activator of immune system through recognition of unmethylated CpG sequences by the Toll-like receptor 9.21 Other DNA-recognizing proteins trigger innate immunity, such as the DNA-dependent activator of interferon-regulatory factors.22 If innate immunity activation might represent a potential adjuvant advantage for DNA-based vaccination, on the other side, it represents an unwanted negative effect for gene therapy strategies implying a sustained, long-lasting expression of the therapeutic transgene. In this latter case, plasmids devoid of prokaryotic antibiotic resistance gene and bearing a shorter prokaryotic backbone may display a decisive advantage.
New Selection Markers for Plasmid Production
Complementation of auxotrophic bacterial strains
Auxotrophy is defined as the inability of an organism to synthesize a particular organic compound required for its growth. To produce antibiotic-free plasmid by using this strategy, the bacterial strain is first modified by introducing a deletion or a nonsense point mutation into an essential or conditionally essential chromosomal gene resulting in auxotrophy. Bacterial growth is restored upon introduction into those strains of a plasmid either carrying the deleted gene or coding a suppressor tRNA which allows a complete translation of the truncated protein (Figure 1). Several target genes have been studied in the last few years and the developed strategies showed promising results (Table 1).
Figure 1.

Auxotrophy of bacterial strain and prototrophic growth restoration by plasmid DNA.
Table 1. Auxotrophic complementation approaches for plasmid production.

Glycine auxotrophy was obtained after chromosomal disruption of the glyA gene that encodes serine hydroxymethyl transferase which is involved in the main glycine biosynthesis pathway in Escherichia coli E. coli.23 The resulting mutant strain can grow fast only if glycine is added to the culture medium. A complementation plasmid with a functional copy of the glyA gene was constructed providing a serine hydroxymethyl transferase source and thus allowing growth of the auxotrophic bacterial strain. Plasmid constructs based on this backbone could therefore be selected and maintained in culture without addition of antibiotics. The capability of this system for recombinant overproduction of rhamnulose 1-phosphate aldolase was evaluated, obtaining high cell density cultures and productivity levels comparable to those obtained with a conventional system.23 QAPRTase, an enzyme implied in de novo nicotinamide adenine dinucleotide biosynthesis, appeared recently as another attractive target for auxotrophy-based system.24 The fact that the QAPRTase gene is ubiquitous in bacteria and mammals addresses a key point in term of biosafety reducing side effects of a potential horizontal gene transfer. Bacteria deficient in QAPRTase are able to survive in common rich media such as lysogeny broth medium (which contains abundant nicotinamide adenine dinucleotide precursors) but under conditions of nutritional deficiency the de novo nicotinamide adenine dinucleotide biosynthesis pathway becomes necessary for bacterial growth. The growth status of the transformed strains was better than that of the prototrophic reference strain as the overproduction of QAPRTase did not lead to metabolic-burden effect. Similarly, translation initiation factor 1 auxotrophic strain was developed by chromosomal deletion of the infA gene.25 The main advantage of infA targeting is that this protein is made of only 71 amino acids, and plasmids carrying this selectable marker can therefore be maintained small. Growth rates of the control and the plasmid-harbouring strains are indistinguishable from each other.
Two other groups studied an alternative way to get and bypass auxotrophy. Auxotrophic strains were obtained by introducing an amber nonsense mutation into the target gene. The plasmids contained an amber suppressor tRNA (around 100 bp) which inserts an amino acid in response to UAG thus allowing a complete translation of the truncated protein. The first plasmid developed, called pCOR,26,27 aimed to overcome an arginine auxotrophy. Interestingly, it also contains a conditional origin of replication that requires the trans-acting π initiator protein encoded by the pir gene integrated into the E. coli host chromosome for its propagation, thus limiting the risk of plasmid dissemination to other bacterial strains. High yields of supercoiled pCOR were obtained by high cell-density fermentation with a yield of 100 mg/l. Furthermore, a particular combination of mutations of the π initiator protein has lead to a threefold- to fivefold increase in supercoiled monomer pCOR plasmid per biomass unit.27
In another approach, a thymidine auxotrophic strain was isolated for the production of pFAR plasmids. This antibiotic-free system offers the possibility of using a commercially available thymidine-free medium. At similar optical density or after overnight growth, the amount of purified pFAR plasmids was equivalent to that of a pVAX2 derivative prepared from DH5α grown in lysogenic broth medium supplemented with kanamycin.28 The small size pFAR plasmid led to high transgene expression in several tissues.
Toxin–antitoxin-based systems
Toxin–antitoxin systems are made of two key elements: a biologically active protein molecule and the corresponding inhibitor. Such mechanisms have been developed by bacteria as metabolism regulators ensuring an adequate response to environmental changes.29 Some well-described toxin–antitoxin systems, also called postsegregational killing systems, have emerged as alternatives to the use of antibiotic for plasmid selection.
Killer loci were inserted into the plasmid as selection marker. The selection is based on the fact that toxin and antitoxin elements have different half-life times, the poison protein being more stable. After the loss of the plasmid, expression of both of them stops but the toxin remains in the cell for a longer period of time and thus induces the killing of the bacteria (Figure 2). This method presents several advantages: (i) no changes must be made on the host's chromosome although chromosomal integration of the “poison” protein might confer some advantage (such as in the following example), (ii) killer loci are generally small, and (iii) media or reactor configuration changes do not impact the selection efficiency.30
Figure 2.

Toxin–antitoxin-based systems.
The small CcdB protein is an inhibitor of the essential gyrase and is toxic for enterobacteriaceae as E. coli31 but not for eukaryotic cells. In the same operon, the ccdA gene encodes the antidote which interacts with the toxin and the resulting complex strongly represses the ccd promoter. Separation of the components of this ccd operon has led to a more efficient system for bacterial selection.32 The ccdB poison gene was integrated into the bacteria chromosome downstream to the ccd promoter, and the ccdA gene was inserted into an expression plasmid. In case of the absence or loss of the plasmid, the toxin is produced and induces cell death (Figure 2). The separation of both components ensured efficient killing of plasmid-free cells but also an increase of plasmid-containing bacteria and of plasmid production. The same approach was used to produce proteins as vaccines or therapeutical proteins. It was shown that this method enables to reach higher yields of protein of interest compared to standard method based on antibiotic resistance selection.33 However, as for the auxotrophy complementation approaches, this strategy requires modification of the host bacterial genome.
Operator–repressor titration
Operator–repressor systems can be used to reversibly control the expression of a gene. Several studies aimed at introducing such regulatory sequences upstream from a specific gene in the chromosome of E. coli. Plasmids containing one or several operator sequences were engineered and, when introduced into bacteria, were able to competitively titrate the repressor allowing thus expression of the gene (Figure 3).
Figure 3.

Operator–repressor titration.
To demonstrate applicability of this system, the Kanr gene was placed under the control of the lac operator/promoter and inserted into the bacterial chromosome. The resulting strain was unable to grow in the presence of kanamycin because of repression of the kanamycin resistance gene, but after transformation with a high copy number plasmid (it was critical that the plasmid copy number per cell was sufficient to achieve repressor titration) containing the lac operator, kan expression was derepressed thus allowing selection.34 This selection method was adapted to obtain a totally antibiotic-free procedure. A lac operator/promoter was inserted upstream of the essential chromosomal dapD gene, and a new pORT plasmid able to titrate the repressor was constructed.35 The DapD protein is involved in the lysine/diaminopimelate pathway, and the dapD repression is lethal when bacteria are grown in lysine/diaminopimelate-free media. The pORT vector titrates the repressor, thus allowing the selection of plasmid-containing bacteria. As in the case of other methods described above, it is necessary to modify the chromosomal genome.
RNA-based selection markers
Other selection markers are based on antisense RNA regulators. An RNA IN-sacB sequence was chromosomally integrated and constitutively expressed acting as a selection marker since cells containing sacB encoding levansucrase are killed in the presence of sucrose36 (Figure 4). A rescuing plasmid expressing a 150-bp RNA-OUT antisense RNA was constructed. The antisense RNA-OUT hybridized with the RNA-IN sequence and silenced sacB expression thus allowing growth in sucrose-complemented media. Replacement of kanamycin resistance marker with RNA-OUT was not detrimental to plasmid production yield and quality.36 This vector was recently improved by incorporation of transient expression enhancers which appeared more potent alternatives to improve transgene expression for gene therapy or vaccination.37
Figure 4.

RNA-based selection markers.
A similar approach was developed using the origin of replication (ori)-encoded RNAI which is common in ColE1-based plasmids (Figure 4). Similarly to the operator–repressor titration strategy, an essential gene of E. coli was placed under the control of an operator–repressor system but, in that case, the repressor was fused to an RNAII sequence. When plasmids are present in the cells, RNAI derived from the origin of replication hybridizes to the RNAII sequence, inhibiting the repressor translation.38 Using this strategy, a twofold increase in the overall plasmid yield was obtained compared to the conventional approach using a kanamycin marker gene probably because of a combination of several effects: a decrease in metabolic load, a decrease in the plasmid size, and an increase in replication rate. The main advantage of this approach is that it does not require additional sequence on the plasmid backbone because the selection marker is part of the replication origin.
Overexpression of a growth essential gene
It has been shown that overexpression of certain growth essential genes (fabI or murA) in E. coli reduced their susceptibility to antimicrobial compounds.39 The fabI gene encodes an enoyl ACP reductase that catalyzes fatty acid elongation. Its overexpression resulted in reduced susceptibility to triclosan, an antibacterial agent acting as a chemical inhibitor of enoyl ACP reductase. This characteristic allowed the development of a new cloning vector pFab40 which codes for fabI (860 bp). If fabI overexpression and triclosan were toxic when used alone, their combination resulted in enhanced growth of bacteria and in plasmid production (Figure 5). An advantage over the other antibiotic-free systems is that this approach does not require mutant host strain and is thus easier to develop. On the other side, triclosan has to be present in the bacterial cell culture and has to be carefully eliminated from purified plasmid DNA. Moreover, this method requires gene overexpression which could represent a burden of energy.
Figure 5.

Overexpression of a growth essential gene.
Minicircles
In the minicircle strategy, an antibiotic resistance gene is first included into the constructs as a selection marker and then eliminated by site-specific recombination in E. coli (for review,41,42). Minicircles were first obtained by att site-specific recombination mediated by the phage Λ integrase.16,43,44 Another approach consisted in the Cre-mediated and Cre-directed excision of the bacterial vector sequences to create minicircles.45 After purification, the resulting minicircles are small supercoiled DNA molecules containing almost exclusively the gene of interest and its regulating sequence motifs and are devoid of antibiotic resistance gene. Previously, purification was obtained by two time-consuming and labour-intensive steps: the bacterial DNA backbone was digested by a restriction enzyme, and the miniplasmid was then purified by ultracentrifugation on cesium chloride. Two strategies have been developed to facilitate minicircle purification. Inclusion of the endonuclease I-SceI together with its recognition site in the plasmid backbone allowed linearization and degradation of the backbone and production of purified minicircles.46,47 Beside, a new affinity-based chromatographic purification approach allowed isolation of highly pure minicircles.48
If antibiotic are still required for minicircle production, the removing of the antibiotic resistance gene combined with a careful elimination of antibiotic residues in the product make this approach highly promising as it offers small, CpG-free, supercoiled products suitable for gene therapy treatments.
Reported Uses of Antibiotic-Free Plasmids for Gene Therapy
Till now, only some studies have reported the use of antibiotic-free plasmids in preclinical and clinical models. This is probably mainly because of the fact that most of these approaches are relatively recent and that the kanamycin selection marker remains allowed for gene therapy clinical trials.5,6,7,8 However, several studies have been performed using antibiotic-free plasmids with the objective of increasing biosafety (Table 2).
Table 2. Preclinical and clinical studies using nonantibiotic plasmids.
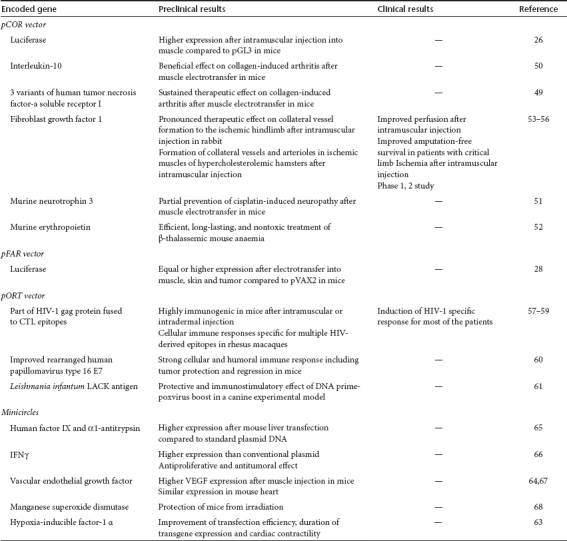
All studies that aimed at comparing the efficiency of these plasmids with common vectors containing antibiotic resistance gene reported equal or higher efficacy of the antibiotic-free plasmids in term of luciferase expression.26,28 The pCOR plasmid developed at the end of the nineties has been used in several preclinical models such as arthritis,49,50 neuropathy,51 β-thalassemia,52 and ischemia,53,54 and no vector-related toxicity has been reported. The promising results obtained in rabbit and hamster after intramuscular injection of pCOR encoding fibroblast growth factor 1 led to human phase 1 and 2 clinical studies which have suggested an improvement of perfusion and an increase in amputation-free survival in patients with critical limb ischemia.55,56 The more recent pFAR plasmid was compared to a vector containing Kanr and equal or higher expression of luciferase was obtained with the pFAR when electrotransfered into muscle, skin, or tumor.28 The pORT vector was chosen to develop a HIV-1 DNA vaccine, and its delivery induced a specific immune response in both animals and patients.57,58,59 The pORT plasmid encoding an artificial HPV-16 E7-gene was also able to generate an immune response after intramuscular injection into mice.60 Finally, the same vector expressing Leishmania infantum LACK antigen was shown protective in a canine experimental model.61 The pORT plasmid AMEP coding for the recombinant disintegrin domain of ADAM 1562 is presently being evaluated in a phase 1 clinical trial to treat advanced or metastatic melanoma by using electrotransfer (BioAlliance Pharma, personal communication, 20 November 2010). The Staby system based on the use of toxin–antitoxin system was tested in preclinical trials and compared to a conventional KanR plasmid. It was shown that equal or higher expression and immunogenicity are obtained in mice using the antibiotic-free plasmid (Delphi Genetics, personal communication, 30 May 2011). Minicircles have been used in several preclinical mice models and have demonstrated equal or higher expression in heart,63,64 liver,65 tumor,66 or muscle64,67 and are therefore another attractive alternative for gene therapy of various diseases.
It is important to underline that toxicity or vector-related side effects have never been reported and that, when compared to classical plasmids, antibiotic-free systems revealed equivalent or even higher efficiency. These first data ranked therefore antibiotic-free plasmids among the very promising vectors for future gene therapy clinical trials.
Discussion and Future Directions
During the last decades, gene therapy has known a growing interest and has met many preclinical successes both for treatment and prevention of a wide variety of diseases. Today, it is critical to be aware of the potential safety concerns of the gene therapy approaches and to offer therapies that meet the highest expectations of effectiveness and safety. The use of antibiotic resistance genes as selection markers raises concerns which are regularly pointed out by the authorities.5,6,7,8 We can reasonably anticipate that, in the next few years, antibiotic resistance genes as selection markers will become more restricted or even forbidden for a clinical use because of the risk of horizontal gene transfer and because of the allergic potential of the compounds used for plasmid maintenance. In this paper, we reviewed the new generation of plasmid backbones devoid of antibiotic resistance marker which has recently emerged. An ideal selection marker should have all the following properties: (i) if a horizontal gene transfer occurs, it should not impact the recipient cell. In particular, the plasmid should not provide the bacteria with any benefit as it is the case for antibiotic resistance genes. (ii) The selection marker should have a limited impact on the plasmid size and should not induce immune activation. (iii) The marker cannot be toxic for eukaryotic cells. (iv) Plasmid maintenance should not require the presence of potentially harmful compounds or very sensitive detection methods should be applied to guarantee their complete removal. (v) Plasmid yields should be high even in large culture, and the culture media required should be easily available and inexpensive to allow scaling-up. As available data about purity and yields using the different approaches is very heterogeneous, it would be interesting to perform a systematic comparative study.
Several of the strategies described in this review meet many of the required characteristics, and when more preclinical and clinical data will be provided proving both efficiency and safety, these vectors will certainly play a major role in the development of the future gene therapies.
REFERENCES
- The Journal of Gene Medicine Clinical Trial site 2011 ). < www.wiley.com/legacy/wileychi/genmed/clinical/ >
- Wolff JA, Malone RW, Williams P, Chong W, Acsadi G, Jani A.et al. (1990Direct gene transfer into mouse muscle in vivo Science 2474949 Pt 11465–1468. [DOI] [PubMed] [Google Scholar]
- Friehs K. Plasmid copy number and plasmid stability. Adv Biochem Eng Biotechnol. 2004;86:47–82. doi: 10.1007/b12440. [DOI] [PubMed] [Google Scholar]
- Summers D. Timing, self-control and a sense of direction are the secrets of multicopy plasmid stability. Mol Microbiol. 1998;29:1137–1145. doi: 10.1046/j.1365-2958.1998.01012.x. [DOI] [PubMed] [Google Scholar]
- European Pharmacopeia 7.0 (2011), 5.14. Gene transfer medical products for human use: 648.
- World Health Organization. Guidelines for assuring the quality and nonclinical safety evaluation of DNA vaccines 2005 ). < http://www.who.int/biological/publications/ trs/areas/vaccines/dna/Annex%201_DNA%20vaccines.pdf >
- The European Agency for the Evaluation of Medicinal Products. Note for Guidance on the Quality, Preclinical and Clinical Aspects of Gene Transfer Medicinal Products < < http://www.ema.europa.eu/docs/en_GB/ document_library/Scientific_guideline/2009/10/WC500003977.pdf > ( 2001
- US Food and Drug Administration. Guidance for human somatic cell therapy and gene therapy (1998 ). < http://www.fda.gov/downloads/ BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/ CellularandGeneTherapy/ucm081670.pdf >
- Nicolau DP. Current challenges in the management of the infected patient. Curr Opin Infect Dis. 2011;24 Suppl 1:S1–10. doi: 10.1097/01.qco.0000393483.10270.ff. [DOI] [PubMed] [Google Scholar]
- Alanis AJ. Resistance to antibiotics: are we in the post-antibiotic era. Arch Med Res. 2005;36:697–705. doi: 10.1016/j.arcmed.2005.06.009. [DOI] [PubMed] [Google Scholar]
- Solensky R. Hypersensitivity reactions to beta-lactam antibiotics. Clin Rev Allergy Immunol. 2003;24:201–220. doi: 10.1385/CRIAI:24:3:201. [DOI] [PubMed] [Google Scholar]
- Rozkov A, Avignone-Rossa CA, Ertl PF, Jones P, O'Kennedy RD, Smith JJ.et al. (2004Characterization of the metabolic burden on Escherichia coli DH1 cells imposed by the presence of a plasmid containing a gene therapy sequence Biotechnol Bioeng 88909–915. [DOI] [PubMed] [Google Scholar]
- Cunningham DS, Koepsel RR, Ataai MM., and, Domach MM. Factors affecting plasmid production in Escherichia coli from a resource allocation standpoint. Microb Cell Fact. 2009;8:27. doi: 10.1186/1475-2859-8-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escriou V, Ciolina C, Helbling-Leclerc A, Wils P., and, Scherman D. Cationic lipid-mediated gene transfer: analysis of cellular uptake and nuclear import of plasmid DNA. Cell Biol Toxicol. 1998;14:95–104. doi: 10.1023/a:1007425803756. [DOI] [PubMed] [Google Scholar]
- Escriou V, Carrière M, Bussone F, Wils P., and, Scherman D. Critical assessment of the nuclear import of plasmid during cationic lipid-mediated gene transfer. J Gene Med. 2001;3:179–187. doi: 10.1002/jgm.174. [DOI] [PubMed] [Google Scholar]
- Kreiss P, Cameron B, Darquet AM, Scherman D., and, Crouzet J. Production of a new DNA vehicle for gene transfer using site-specific recombination. Appl Microbiol Biotechnol. 1998;49:560–567. doi: 10.1007/s002530051213. [DOI] [PubMed] [Google Scholar]
- Kreiss P, Cameron B, Rangara R, Mailhe P, Aguerre-Charriol O, Airiau M.et al. (1999Plasmid DNA size does not affect the physicochemical properties of lipoplexes but modulates gene transfer efficiency Nucleic Acids Res 273792–3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloquel C, Fabre E, Bureau MF., and, Scherman D. Plasmid DNA electrotransfer for intracellular and secreted proteins expression: new methodological developments and applications. J Gene Med. 2004;6 Suppl 1:S11–S23. doi: 10.1002/jgm.508. [DOI] [PubMed] [Google Scholar]
- Gardlik R, Behuliak M, Palffy R, Celec P., and, Li CJ. Gene therapy for cancer: bacteria-mediated anti-angiogenesis therapy. Gene Ther. 2011;18:425–431. doi: 10.1038/gt.2010.176. [DOI] [PubMed] [Google Scholar]
- Pálffy R, Gardlík R, Hodosy J, Behuliak M, Resko P, Radvánský J.et al. (2006Bacteria in gene therapy: bactofection versus alternative gene therapy Gene Ther 13101–105. [DOI] [PubMed] [Google Scholar]
- Krieg AM. CpG motifs in bacterial DNA and their immune effects. Annu Rev Immunol. 2002;20:709–760. doi: 10.1146/annurev.immunol.20.100301.064842. [DOI] [PubMed] [Google Scholar]
- Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T.et al. (2007DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response Nature 448501–505. [DOI] [PubMed] [Google Scholar]
- Vidal L, Pinsach J, Striedner G, Caminal G., and, Ferrer P. Development of an antibiotic-free plasmid selection system based on glycine auxotrophy for recombinant protein overproduction in Escherichia coli. J Biotechnol. 2008;134:127–136. doi: 10.1016/j.jbiotec.2008.01.011. [DOI] [PubMed] [Google Scholar]
- Dong WR, Xiang LX., and, Shao JZ. Novel antibiotic-free plasmid selection system based on complementation of host auxotrophy in the NAD de novo synthesis pathway. Appl Environ Microbiol. 2010;76:2295–2303. doi: 10.1128/AEM.02462-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hägg P, de Pohl JW, Abdulkarim F., and, Isaksson LA. A host/plasmid system that is not dependent on antibiotics and antibiotic resistance genes for stable plasmid maintenance in Escherichia coli. J Biotechnol. 2004;111:17–30. doi: 10.1016/j.jbiotec.2004.03.010. [DOI] [PubMed] [Google Scholar]
- Soubrier F, Cameron B, Manse B, Somarriba S, Dubertret C, Jaslin G.et al. (1999pCOR: a new design of plasmid vectors for nonviral gene therapy Gene Ther 61482–1488. [DOI] [PubMed] [Google Scholar]
- Soubrier F, Laborderie B., and, Cameron B. Improvement of pCOR plasmid copy number for pharmaceutical applications. Appl Microbiol Biotechnol. 2005;66:683–688. doi: 10.1007/s00253-004-1729-9. [DOI] [PubMed] [Google Scholar]
- Marie C, Vandermeulen G, Quiviger M, Richard M, Préat V., and, Scherman D. pFARs, plasmids free of antibiotic resistance markers, display high-level transgene expression in muscle, skin and tumour cells. J Gene Med. 2010;12:323–332. doi: 10.1002/jgm.1441. [DOI] [PubMed] [Google Scholar]
- Bukowski M, Rojowska A., and, Wladyka B. Prokaryotic toxin-antitoxin systems–the role in bacterial physiology and application in molecular biology. Acta Biochim Pol. 2011;58:1–9. [PubMed] [Google Scholar]
- Pecota DC, Kim CS, Wu K, Gerdes K., and, Wood TK. Combining the hok/sok, parDE, and pnd postsegregational killer loci to enhance plasmid stability. Appl Environ Microbiol. 1997;63:1917–1924. doi: 10.1128/aem.63.5.1917-1924.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Melderen L. Molecular interactions of the CcdB poison with its bacterial target, the DNA gyrase. Int J Med Microbiol. 2002;291:537–544. doi: 10.1078/1438-4221-00164. [DOI] [PubMed] [Google Scholar]
- Szpirer CY., and, Milinkovitch MC. Separate-component-stabilization system for protein and DNA production without the use of antibiotics. BioTechniques. 2005;38:775–781. doi: 10.2144/05385RR02. [DOI] [PubMed] [Google Scholar]
- Peubez I, Chaudet N, Mignon C, Hild G, Husson S, Courtois V.et al. (2010Antibiotic-free selection in E. coli: new considerations for optimal design and improved production Microb Cell Fact 965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SG, Cranenburgh RM, Weiss AM, Wrighton CJ, Sherratt DJ., and, Hanak JA. Repressor titration: a novel system for selection and stable maintenance of recombinant plasmids. Nucleic Acids Res. 1998;26:2120–2124. doi: 10.1093/nar/26.9.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cranenburgh RM, Hanak JA, Williams SG., and, Sherratt DJ. Escherichia coli strains that allow antibiotic-free plasmid selection and maintenance by repressor titration. Nucleic Acids Res. 2001;29:E26. doi: 10.1093/nar/29.5.e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luke J, Carnes AE, Hodgson CP., and, Williams JA. Improved antibiotic-free DNA vaccine vectors utilizing a novel RNA based plasmid selection system. Vaccine. 2009;27:6454–6459. doi: 10.1016/j.vaccine.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luke JM, Vincent JM, Du SX, Gerdemann U, Leen AM, Whalen RG.et al. (2011Improved antibiotic-free plasmid vector design by incorporation of transient expression enhancers Gene Ther 18334–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mairhofer J, Cserjan-Puschmann M, Striedner G, Nöbauer K, Razzazi-Fazeli E., and, Grabherr R. Marker-free plasmids for gene therapeutic applications–lack of antibiotic resistance gene substantially improves the manufacturing process. J Biotechnol. 2010;146:130–137. doi: 10.1016/j.jbiotec.2010.01.025. [DOI] [PubMed] [Google Scholar]
- Xu HH, Real L., and, Bailey MW. An array of Escherichia coli clones over-expressing essential proteins: a new strategy of identifying cellular targets of potent antibacterial compounds. Biochem Biophys Res Commun. 2006;349:1250–1257. doi: 10.1016/j.bbrc.2006.08.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh S., and, Good L. Plasmid selection in Escherichia coli using an endogenous essential gene marker. BMC Biotechnol. 2008;8:61. doi: 10.1186/1472-6750-8-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayrhofer P, Schleef M., and, Jechlinger W. Use of minicircle plasmids for gene therapy. Methods Mol Biol. 2009;542:87–104. doi: 10.1007/978-1-59745-561-9_4. [DOI] [PubMed] [Google Scholar]
- Gill DR, Pringle IA., and, Hyde SC. Progress and prospects: the design and production of plasmid vectors. Gene Ther. 2009;16:165–171. doi: 10.1038/gt.2008.183. [DOI] [PubMed] [Google Scholar]
- Darquet AM, Cameron B, Wils P, Scherman D., and, Crouzet J. A new DNA vehicle for nonviral gene delivery: supercoiled minicircle. Gene Ther. 1997;4:1341–1349. doi: 10.1038/sj.gt.3300540. [DOI] [PubMed] [Google Scholar]
- Darquet AM, Rangara R, Kreiss P, Schwartz B, Naimi S, Delaère P.et al. (1999Minicircle: an improved DNA molecule for in vitro and in vivo gene transfer Gene Ther 6209–218. [DOI] [PubMed] [Google Scholar]
- Bigger BW, Tolmachov O, Collombet JM, Fragkos M, Palaszewski I., and, Coutelle C. An araC-controlled bacterial cre expression system to produce DNA minicircle vectors for nuclear and mitochondrial gene therapy. J Biol Chem. 2001;276:23018–23027. doi: 10.1074/jbc.M010873200. [DOI] [PubMed] [Google Scholar]
- Chen ZY, He CY., and, Kay MA. Improved production and purification of minicircle DNA vector free of plasmid bacterial sequences and capable of persistent transgene expression in vivo. Hum Gene Ther. 2005;16:126–131. doi: 10.1089/hum.2005.16.126. [DOI] [PubMed] [Google Scholar]
- Kay MA, He CY., and, Chen ZY. A robust system for production of minicircle DNA vectors. Nat Biotechnol. 2010;28:1287–1289. doi: 10.1038/nbt.1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayrhofer P, Blaesen M, Schleef M., and, Jechlinger W. Minicircle-DNA production by site specific recombination and protein-DNA interaction chromatography. J Gene Med. 2008;10:1253–1269. doi: 10.1002/jgm.1243. [DOI] [PubMed] [Google Scholar]
- Bloquel C, Bessis N, Boissier MC, Scherman D., and, Bigey P. Gene therapy of collagen-induced arthritis by electrotransfer of human tumor necrosis factor-alpha soluble receptor I variants. Hum Gene Ther. 2004;15:189–201. doi: 10.1089/104303404772679995. [DOI] [PubMed] [Google Scholar]
- Saidenberg-Kermanac'h N, Bessis N, Deleuze V, Bloquel C, Bureau M, Scherman D.et al. (2003Efficacy of interleukin-10 gene electrotransfer into skeletal muscle in mice with collagen-induced arthritis J Gene Med 5164–171. [DOI] [PubMed] [Google Scholar]
- Pradat PF, Finiels F, Kennel P, Naimi S, Orsini C, Delaere P.et al. (2001Partial prevention of cisplatin-induced neuropathy by electroporation-mediated nonviral gene transfer Hum Gene Ther 12367–375. [DOI] [PubMed] [Google Scholar]
- Fabre EE, Bigey P, Beuzard Y, Scherman D., and, Payen E. Careful adjustment of Epo non-viral gene therapy for beta-thalassemic anaemia treatment. Genet Vaccines Ther. 2008;6:10. doi: 10.1186/1479-0556-6-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witzenbichler B, Mahfoudi A, Soubrier F, Le Roux A, Branellec D, Schultheiss HP.et al. (2006Intramuscular gene transfer of fibroblast growth factor-1 using improved pCOR plasmid design stimulates collateral formation in a rabbit ischemic hindlimb model J Mol Med 84491–502. [DOI] [PubMed] [Google Scholar]
- Caron A, Michelet S, Caron A, Sordello S, Ivanov MA, Delaère P.et al. (2004Human FGF-1 gene transfer promotes the formation of collateral vessels and arterioles in ischemic muscles of hypercholesterolemic hamsters J Gene Med 61033–1045. [DOI] [PubMed] [Google Scholar]
- Comerota AJ, Throm RC, Miller KA, Henry T, Chronos N, Laird J.et al. (2002Naked plasmid DNA encoding fibroblast growth factor type 1 for the treatment of end-stage unreconstructible lower extremity ischemia: preliminary results of a phase I trial J Vasc Surg 35930–936. [DOI] [PubMed] [Google Scholar]
- Nikol S, Baumgartner I, Van Belle E, Diehm C, Visoná A, Capogrossi MC, TALISMAN 201 investigators et al. Therapeutic angiogenesis with intramuscular NV1FGF improves amputation-free survival in patients with critical limb ischemia. Mol Ther. 2008;16:972–978. doi: 10.1038/mt.2008.33. [DOI] [PubMed] [Google Scholar]
- Mwau M, Cebere I, Sutton J, Chikoti P, Winstone N, Wee EG.et al. (2004A human immunodeficiency virus 1 (HIV-1) clade A vaccine in clinical trials: stimulation of HIV-specific T-cell responses by DNA and recombinant modified vaccinia virus Ankara (MVA) vaccines in humans J Gen Virol 85Pt 4911–919. [DOI] [PubMed] [Google Scholar]
- Hanke T., and, McMichael AJ. Design and construction of an experimental HIV-1 vaccine for a year-2000 clinical trial in Kenya. Nat Med. 2000;6:951–955. doi: 10.1038/79626. [DOI] [PubMed] [Google Scholar]
- Wee EG, Patel S, McMichael AJ., and, Hanke T. A DNA/MVA-based candidate human immunodeficiency virus vaccine for Kenya induces multi-specific T cell responses in rhesus macaques. J Gen Virol. 2002;83 Pt 1:75–80. doi: 10.1099/0022-1317-83-1-75. [DOI] [PubMed] [Google Scholar]
- Ohlschläger P, Pes M, Osen W, Dürst M, Schneider A, Gissmann L.et al. (2006An improved rearranged Human Papillomavirus Type 16 E7 DNA vaccine candidate (HPV-16 E7SH) induces an E7 wildtype-specific T cell response Vaccine 242880–2893. [DOI] [PubMed] [Google Scholar]
- Ramos I, Alonso A, Peris A, Marcen JM, Abengozar MA, Alcolea PJ.et al. (2009Antibiotic resistance free plasmid DNA expressing LACK protein leads towards a protective Th1 response against Leishmania infantum infection Vaccine 276695–6703. [DOI] [PubMed] [Google Scholar]
- Daugimont L, Vandermeulen G, Defresne F, Bouzin C, Mir LM, Bouquet C.et al. (2011Antitumoral and antimetastatic effect of antiangiogenic plasmids in B16 melanoma: Higher efficiency of the recombinant disintegrin domain of ADAM 15 Eur J Pharm Biopharm 78314–319. [DOI] [PubMed] [Google Scholar]
- Huang M, Chen Z, Hu S, Jia F, Li Z, Hoyt G.et al. (2009Novel minicircle vector for gene therapy in murine myocardial infarction Circulation 12011 SupplS230–S237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenler S, Andersson A, Simonson OE, Lundin KE, Chen ZY, Kay MA.et al. (2009Gene transfer to mouse heart and skeletal muscles using a minicircle expressing human vascular endothelial growth factor J Cardiovasc Pharmacol 5318–23. [DOI] [PubMed] [Google Scholar]
- Chen ZY, He CY, Ehrhardt A., and, Kay MA. Minicircle DNA vectors devoid of bacterial DNA result in persistent and high-level transgene expression in vivo. Mol Ther. 2003;8:495–500. doi: 10.1016/s1525-0016(03)00168-0. [DOI] [PubMed] [Google Scholar]
- Wu J, Xiao X, Zhao P, Xue G, Zhu Y, Zhu X.et al. (2006Minicircle-IFNgamma induces antiproliferative and antitumoral effects in human nasopharyngeal carcinoma Clin Cancer Res 124702–4713. [DOI] [PubMed] [Google Scholar]
- Chang CW, Christensen LV, Lee M., and, Kim SW. Efficient expression of vascular endothelial growth factor using minicircle DNA for angiogenic gene therapy. J Control Release. 2008;125:155–163. doi: 10.1016/j.jconrel.2007.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Epperly MW, Kay MA, Chen ZY, Dixon T, Franicola D.et al. (2008Radioprotection in vitro and in vivo by minicircle plasmid carrying the human manganese superoxide dismutase transgene Hum Gene Ther 19820–826. [DOI] [PMC free article] [PubMed] [Google Scholar]