

Abstract
Gene-directed enzyme prodrug therapy (GDEPT) has been investigated as a means of cancer treatment without affecting normal tissues. This system is based on the delivery of a suicide gene, a gene encoding an enzyme which is able to convert its substrate from non-toxic prodrug to cytotoxin. In this experiment, we have developed a targeted suicide gene therapeutic system that is completely contained within tumor-tropic cells and have tested this system for melanoma therapy in a preclinical model. First, we established double stable RAW264.7 monocyte/macrophage-like cells (Mo/Ma) containing a Tet-On® Advanced system for intracellular carboxylesterase (InCE) expression. Second, we loaded a prodrug into the delivery cells, double stable Mo/Ma. Third, we activated the enzyme system to convert the prodrug, irinotecan, to the cytotoxin, SN-38. Our double stable Mo/Ma homed to the lung melanomas after 1 day and successfully delivered the prodrug-activating enzyme/prodrug package to the tumors. We observed that our system significantly reduced tumor weights and numbers as targeted tumor therapy after activation of the InCE. Therefore, we propose that this system may be a useful targeted melanoma therapy system for pulmonary metastatic tumors with minimal side effects, particularly if it is combined with other treatments.
Keywords: B16-F10, Mouse lung melanoma, Mouse monocytes, Targeted cell delivery, Suicide therapy
Introduction
A major objective of anticancer therapy is to target the treatment to the tumors without affecting normal tissues. Strategies such as gene-directed enzyme/prodrug therapy (GDEPT) [1, 2] and antibody-directed enzyme/prodrug therapy (ADEPT) [3] have been developed for this purpose with varying degrees of success [4]. Nanoparticle delivery enhanced by attaching tumor cell- or tumor vascular-specific ligands to the nanoparticles [5] has achieved some success. Targeted cytotherapy utilizing ‘delivery’ cells such as stem cells [6–11], or other cells [12, 13] represents another strategy aimed at more focused cancer therapy.
Irinotecan (CPT-11) is a prodrug that is converted by carboxylesterase in the liver to a potent topoisomerase I inhibitor, SN-38. Irinotecan is approved clinically for colorectal carcinoma therapy [14]. Irinotecan was shown to strongly inhibit metastasis in a B16-F10 mouse lung melanoma model [15], and has been used alone or in combination to treat other solid cancers, especially those involving the gastrointestinal tract [16]. However, the use of this prodrug is limited because only 2–5% of the injected dose is converted to active SN-38, and the drug has undesirable side effects such as diarrhea and neutropenia [17]. Moreover, SN-38 has poor solubility [14]. Carboxylesterase (CE) produced by rabbit liver is much more efficient than human CE in the conversion of irinotecan to SN-38. A secreted form of rabbit CE delivered by neural stem cells has been shown to be effective after systemic administration of irinotecan in several preclinical models [18–20]. However, despite its advantages, expression of rabbit CE in a human host could result in immune consequences [21]. This could result in its inactivation or other undesirable outcomes.
We hypothesized that it might be possible to sequester both a prodrug and the prodrug-activating enzyme within the same cell for targeting B16-F10 lung melanomas in mice. Bioengineered, tumor-homing cells could be given in large numbers intravenously, thereby producing a self-contained ‘stealth’ prodrug/activator delivery system to circumvent both immune complications and the side effects inherent with irinotecan given systemically. Accordingly, we engineered mouse monocyte/macrophage cells (Mo/Ma; RAW 264.7, ATCC TIB-78) for TetOn regulated production of intracellular rabbit carboxylase InCE (InCE), pre-loaded them overnight with irinotecan, inoculated them systemically to mice with B16-F10luc2 lung melanomas, and administered tetracycline (doxycycline) when we estimated that the engineered Mo/Ma had reached the tumors. Here we show that we could attenuate the lung melanomas using this targeted cytotherapy approach.
Materials and methods
Reagents and cells
RAW 264.7 mouse monocyte/macrophage (Mo/Ma) cells were purchased from ATCC (Manassas, VA). Irinotecan and doxycycline were from Sigma (St. Louis, MO) and Clontech (Mountain View, CA); the TetOn system was from Clontech. Hygromycin and G418 were from Invivogen (San Diego, CA) and Invitrogen (Carlsbad, CA). B16-F10-luc2 cells were obtained from Caliper Life Sciences (Hopkinton, MA).
Cell culture
RAW264.7 cells were cultured in RMPI containing 10% fetal bovine serum (Sigma) and 1× penicillin–streptomycin in a 37°C humidified incubator with 5% CO2. Irinotecan was added in fresh medium overnight and removed the following morning. Double-stable Mo/Ma were maintained in the RPMI medium described above, with 100 μg/ml G418 and 100 μg/ml hygromycin added to preserve stable transfection. B16-F10-luc2 cells were cultured in RMPI containing 10% fetal bovine serum (Sigma) in a 37°C humidified incubator with 5% CO2.
Generation of double-stable cells for inducible expression of InCE
We cloned the rabbit carboxylesterase (InCE) gene by PCR from a rabbit liver cDNA library from Biochain (Hayward, CA). Primers used were as follows: forward primer, 5′-GCTTGAATTCCGCCACCATGTGGCTCTG-3′ and reverse primer, 5′-CGTGCTAGCTCACAGCTCAATGT-3′. Both primers contained restriction sites for EcoR1 and Nhe1 to facilitate subcloning. The PCR product was run on a 1% agarose gel to confirm proper amplicon size, then excised from the gel and ligated into the TOPO TA cloning vector pCR 2.1 (Invitrogen) for 20 min. Then we transformed into Escherichia coli TOP10 competent cells by heat shock (42°C) and streaked LB agar plates containing ampicillin (mp) and x-gal. Positive transformants were selected using x-gal and amp and grown up in LB agar containing amp. Then, the gene, which expresses an intracellular form of rabbit carboxylesterase (InCE) from this vector, was excised with EcoR1 and Nhe1and ligated into the pTRE-Tight plasmid (Clontech). Again, positive transformants were selected using amp and colony PCR. Plasmid containing the InCE gene was isolated from these bacteria using an Endo-Free Maxi kit (Qiagen). We then created a double-stable Mo/Ma line. First, we transfected RAW 264.7 cells with the pTet-On Advanced plasmid (Clontech) followed by selection in G418 media (500 μg/ml). We then transfected the selected cells with the InCE-recombinant pTRE-Tight plasmid (InCE) and selected stable transformants with hygromycin (500 μg/ml).
Homing study
To evaluate that double stable Mo/Ma cells home to the B16-F10-luc2, magnetic nanoparticles were synthesized as described by Zhang et al. [22], and then ligand exchanged and porphyrin-labeled as described by Balivada et al. [23].
Irinotecan loading of Mo/Ma and evaluation of irinotecan and SN-38 content
To evaluate irinotecan loading, 10 μM of the prodrug was added to medium, incubated with double stable Mo/Ma cells overnight, and removed by thorough washing with PBS. Loading was verified by examining the cells’ UV fluorescence on an epifluorescent microscope (Carl Zeiss, New York) using an ultraviolet filter (Carl Zeiss, New York) immediately after washing and 24 h later. Cells were lifted immediately after washing, counted using a hemocytometer, and evaluated for viability with trypan blue staining. Cells were then lysed using a sonicator at a No. 6 setting (Fisher Scientific, Waltham, MA). Irinotecan loading was quantified by measuring fluorescence (excitation: 377 nm, emission, 427 nm) of the cells on a fluorimeter (ISA FluoroMax 2 Jobin-Yvon-Spex, Edison, New Jersey) and comparing it to the fluorescence of a standard curve of irinotecan in solution. To evaluate irinotecan conversion to SN-38 by the InCE, double stable Mo/Ma monocytes were first loaded with irinotecan at 10 μM overnight and washed as described above. Then, 1 μg/ml of doxycycline was added to the medium for 24 h. After washing, cells were lifted, counted, and viability assessed by trypan blue exclusion. Cells were lysed and fluorescence (excitation: 377 nm, emission: 549 nm) was determined and compared to an SN-38 standard curve to estimate the amount of SN-38 per cell. The amount of SN-38 in the medium after the 24 h doxycycline incubation was also evaluated at this time.
Co-cultures with B16-F10-luc2
To test the cytoxic effect of irinotecan-loaded double stable Mo/Ma on B16-F10-luc2 melanoma cell lines, the cells were co-cultured. The irinotecan-loaded Mo/Ma and B16-F10-luc2 melanoma cells were plated in a ratio of 1 (600 cells) to 5 (2400 cells) into wells of a 96 well plate and allowed to adhere overnight in medium that consisted of a 1:1 ratio of double stable Mo/Ma and B16-F10-luc2 media; we found that both cells could thrive in this hybrid media. The MTT assay (Roche, Mannheim, Germany) was carried out to assess cell proliferation. To perform this assay, 10 μl of MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, a tetrazole) solution was added to each well of the 96 well plate and incubated for 4 h at 37°C. One hundred microliter of solubilization solution (10% sodium dodecyl sulfate in 0.01 M HCl) was then added to each well and incubated overnight. Absorbance was read at 590 nm with a reference filter of 690 nm.
In vivo experiments
Eight week old C57/BL6 female mice (Charles River, Wilmington, MA) were given 2.5 × 105 B16-F10-luc2 mouse melanoma cells intravenously. In preparation for injection 4 days later, double-stable Mo/Ma cells were incubated with 10 μM irinotecan overnight followed by washing. Two million of the irinotecan-loaded double stable Mo/Ma cells were given intravenously 5 days after tumor cell inoculation. One day later, 2 mg/kg of doxcycline was administered intraperitoneally (I.P.). This protocol was repeated two more times with irinotecan-loaded double stable Mo/Ma cells given at 10 and 15 days, respectively, after tumor cell inoculation.
Estimate of tumor burden
Mice were euthanized on day 18 after tumor cell inoculation. To determine the effect of irinotecan-loaded double stable Mo/Ma therapy, we measured lung weights and counted total tumor numbers of superficial tumors on lungs per mouse after photographing both dorsal and ventral lung surfaces. Tumor counts were manually done from these photographs. Other tissues that were collected included spleen, liver, and kidney.
Results
Construction of plasmid and Tet-On double stable Mo/Ma cell lines
The InCE PCR product from a rabbit liver cDNA library was amplified by PCR. As shown in Fig. 1a, we observed the 1683 bp PCR product of InCE. Then, we ligated this sequence with the TOPO TA-vector and transformed E. coli TOP10. Transformed cells were plated on Luria agar (LA) plates containing amp and x-gal. We selected positive transformants (white colonies) to confirm inserts by enzyme digestion. To express InCE in a regulated expression vector, we digested both the pTRE-Tight vector and the TOPO TA vector containing the InCE sequence with EcoR1 and Nhe1. We ligated the InCE insert with the pTre-Tight expression vector from the Tet-On system (Fig. 1b). We confirmed the presence of the InCE insert by electrophoresis (Fig. 1b, c).
Fig. 1.

In vitro experiments with InCE. a PCR was performed and the PCR product detected. The arrow indicates the InCE PCR product. b Enzyme digestion of pTRE-InCE plasmid. The upper arrow indicates the pTRE-tight vector and the lower arrow indicates the InCE insert. c Construction of the pTRE-InCE plasmid. d Expression level of InCE from double stable Mo/Ma cells with and without doxycycline. We estimated the level of InCE by O-nitrophenyl acetate (O-NPA). Then we spectrophotometrically measured absorbance at A415 nm. Results are presented as the mean ± standard errors of mean (SEM), *P>0.0016, compared with the level of control
After construction of the double stable Mo/Ma cells as described in the “Material and methods” section above, we estimated expression of InCE by the O-nitrophenyl acetate (O-NPA) method [19]. Results are shown in Fig. 1d.
Homing study for double stable Mo/Ma cells
To verify that double stable Mo/Ma cells homed to tumors, we loaded double stable Mo/Ma with magnetic nanoparticles containing 15 μg/ml of iron. Then we injected 1 million cells I.V. into mice. After 1 day, mice were sacrificed and various organs (lung, kidney, liver, spleen) were harvested, fixed, snap frozen, and sectioned. We stained lung using Prussian blue and a nuclear fast red counterstain (Fig. 2). The turquoise color and arrows in Fig. 2 show iron-containing Mo/Ma cells stained with Prussian blue.
Fig. 2.

Homing of Mo/Ma to the B16-F10-luc2 melanoma tumors 1 day after 1 × 106 double stable Mo/Ma administration by I.V. We loaded Mo/Ma with magnetic nanoparticles containing 15 μg/ml of iron. Nuclei are visible as red color inside the turquoise color. The tumor section was viewed and photographed at ×20 magnification. The boxed area is enlarged and monocytes are circled and marked with black arrows for better visualization, while melanoma granules are marked with white arrows
Evaluation of cell content of irinotecan and SN-38
We compared irinotecan levels in cells to a standard curve and found that 0.45 nmol of irinotecan had loaded in the 2 × 106 Mo/Ma (data not shown). 24 h after loading, Mo/Ma still contained most of the irinotecan loaded (Supplemental Fig. 1). Also, we estimated the amount of SN-38 produced in irinotecan-loaded Mo/Ma in the presence of doxycycline (1 μg/ml). According to the standard curve, 2 × 106 Mo/Ma cells contained 0.15 nmol of SN-38 24 h after doxycycline addition (data not shown).
Irinotecan-loaded Mo/Ma cell lines inhibit B16-F10 melanoma tumors in vitro
To validate prodrug efficiency for B16-F10-luc2 melanoma cell lines in vitro, we co-cultured them with double stable Mo/Ma cells as described in Methods above. After cells were plated, we added doxycycline to the co-cultures to a final concentration of 1 μg/ml. As shown in Fig. 3, addition of doxycycline to co-cultures of InCE-expressing double stable Mo/Ma with B16-F10-luc2 melanoma cells decreased total cell proliferation measured by MTT assay, as compared to co-cultures without doxycycline.
Fig. 3.

Direct co-culture of irinotecan-loaded double stable Mo/Ma attenuated the growth of B16-F10-luc2 when doxycycline was added to the medium. The MTT assay was carried out after 1, 2 and 3 days. Blue bars represent cultures without doxycycline and red bars represent cultures with doxycycline. Irinotecan-loaded double stable Mo/Ma were co-cultured with 2.4 × 103 B16-F10-luc2 (Irinotecan-loaded double stable Mo/Ma:B16-F10-luc2 ratio = 1:5). Results are presented as the mean ± SEM; *P>0.01, compared with the level of control
Weight and number of tumors after treatments
Melanoma-bearing mice were intravenously inoculated with irinotecan-loaded double stable Mo/Ma cells; 1 day later, mice were given doxycycline (2 mg/kg) to induce intracellular InCE expression. We repeated these treatments three times during 18 days. After 18 days, mice were anesthetized by 100 mg/kg of ketamine and 15 mg/kg of xylazine. Then 150 mg/kg luciferin (Caliper Life Sciences) was given I.P. followed by a 5 min wait. Mice were euthanized and lungs removed from them; lungs were placed in weigh boats in an IVIS Lumina imager (Caliper Life Sciences). Luciferase bioluminescence, which reflects numbers of B16-F10-luc2 cells present, was assessed (Fig. 4). As shown, we observed a reduced luciferase signal in lungs receiving both irinotecan-loaded double stable Mo/Ma and doxycycline. We also measured tumor weights and compared them to controls (Fig. 5). In this experiment, lung tumor weights were significantly reduced in the treatment group receiving irinotecan-loaded Mo/Ma with doxycycline (P value<0.1) compared to the group receiving only irinotecan-loaded Mo/Ma. The average number of tumors in this treatment group was significantly decreased relative to the group receiving irinotecan-loaded Mo/Ma alone (P value<0.05) (Fig. 6a). In terms of average tumor size, most tumors observed in all three groups were quite small (smaller than<1 mm). Treatment did not reduce numbers of medium (1–2 mm) or large tumors (greater than>2 mm) (Fig. 6b). Tumors were not noted in other organs, except one tumor was noted in perirenal adipose tissue in a mouse in the tumor alone group.
Fig. 4.
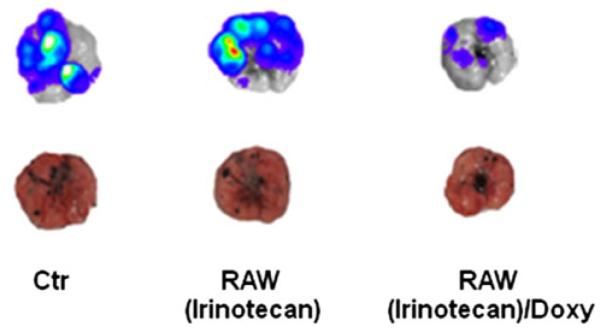
Effect of various treatments on B16-F10-luc2 tumor-bearing mice. Bioluminescent and photographic images of excised mouse lungs with tumors 3 weeks after injection of B16-F10-luc2 melanoma cells. RAW(Irinotecan): double stable Mo/Ma after loading with irinotecan in the absence of doxycycline; RAW(Irinotecan)/Doxy: double stable Mo/Ma after loading with irinotecan in the presence of doxycycline
Fig. 5.

In vivo assays. a Lung weights in mice after different treatments. b Representative images of excised lungs from different treatments. RAW(Irinotecan): double stable Mo/Ma after loaded irinotecan in the absence of doxycycline; RAW(Irinotecan)/Doxy: double stable Mo/Ma after loaded irinotecan in the presence of doxycycline. *Statistically significant (P value>0.1)
Fig. 6.

The average number and average size of tumors in the mouse lungs after treatments. RAW(Irinotecan): double stable Mo/Ma after loading with irinotecan in the absence of doxycycline; RAW(Irinotecan)/Doxy: double stable Mo/Ma after loading with irinotecan in the presence of doxycycline. *Statistically significant (P value>0.1)
Discussion
Here we have shown for the first time that a prodrug/prodrug activating enzyme system self-contained within tumor-homing cells can attenuate preclinical metastatic melanoma. The delivery cells, in this case mouse monocyte/macrophage RAW 264.7 (Mo/Ma) cells, were engineered to synthesize an intracellular form of InCE in a tightly regulated fashion; the prodrug irinotecan was carried within the targeting cells themselves. This paradigm circumvented the necessity of systemic administration of irinotecan. Hence, multiple levels of control were built into the system to preclude damage to normal tissues while delivering a potent chemotherapy in localized fashion to the tumor.
Irinotecan is the water-soluble form of camptothecin [24–26] and is used clinically or in clinical trials to treat several types of human cancer [1, 27]. It has been shown to markedly inhibit B16-F10 lung tumors in mice [15] In contrast to some prodrugs, irinotecan itself has side effects such as severe diarrhea and neutropenia [28] when it is converted by CE in the liver to SN-38, a potent topoisomerase I inhibitor [29]. Rabbit carboxylesterase (InCE) has been shown to be 100–1000-fold more efficient than human carboxylesterase in converting irinotecan to SN-38 [19]. To date, stem cell-directed CE has involved a secreted form of the prodrug-activating enzyme [18–20]. However, rabbit CE secreted extracellularly could lead to an immunological response in human tissue, inactivating the enzyme or causing otherwise unwanted effects [30]. Thus, an advantage of the self-contained system described here is that the InCE is expressed only intracellulary, taking advantage of the much greater efficiency of the rabbit InCE compared to human CE while minimizing exposure to the immune system.
In a clinical situation, autologous human peripheral monocytes isolated by pheresis would be used instead of mouse cells. Double-stable transfectants can require an unreasonable amount of time to generate. However, we have evidence that by fine-tuning plasmid ratios, it is possible to introduce the regulated system by transient transfection (unpublished data). Systems such as the sleeping beauty transposon system or AAV vectors could also be used. Thus, there are transfection systems which would allow the use of human delivery cells, making such a construct potentially feasible.
Here, we used rabbit CE to maximize the chances of showing proof of concept. In a clinical situation, human carboxylesterase could be the gene used. Even if rabbit CE were used, it should largely remain within the stealth delivery cells, and the total amount of expression would be relatively low. Therefore, we feel that the possibility of a deleterious immune reaction would be slim. The stealth delivery cells should primarily be at the tumor site, where activation of the innate immune response may be beneficial. Alternatively, tumor sites are known for being immunoimpaired tissues, which could minimize any immune damping of the enzyme activity.
We initially used a nanoparticle formulation containing irinotecan to load the targeting cells. However, we observed that control cells incubated with free irinotecan were loaded with comparable efficiency. Since a nanoparticle system is not necessary, the approach could be more readily adapted to laboratories without nanoparticle expertise. The CPT-11 prodrug causes minimal effect within monocytes (data not shown).
In the studies here, some leakage from monocytes was observed (data not shown). This leakage could potentially produce undesired side effects, but none were seen. The lack of toxicity here can be explained by the low levels of irinotecan administered. We administered 0.45 nmol irinotecan in two million loaded Mo/Ma. This corresponds to a maximum possible released amount of 8.8 μg/kg. Experimental mouse studies of irinotecan commonly use dosages ranging from 30 to 100 mg/kg [31–34]. The ED50 of SN-38 is in the range of 1–2.5 mg/kg [17]. Clearly, little or no toxicity should arise from drug dosages on the order of 1,000 times less than the ED50. Given the evidence that the loaded Mo/Ma home to the tumor (Fig. 2), intratumoral concentrations of drug could be orders of magnitude higher than systemic levels, thus explaining the efficacy of the treatment system seen here.
An advantage of the system described here would be realized if the active drug were more lipophilic than the parent compound. This is indeed the case. SN-38, in contrast to irinotecan, is very membrane-permeable [34] and thus can move out of the cell to result in a desirable bystander effect on cancer cells. There are other examples where the cytotoxic product moves easily through the membrane but the prodrug does not. This is exemplified by the conversion of the prodrug CB1954 by nitroreductase to its active form [2].
The present system could be improved in several ways. The dose of doxycycline that was used in these pilot studies was quite low (2 mg/kg). This dose could be increased to as much as 50 mg/kg [35]. Thus, more intracellular InCE could have been synthesized, resulting in more SN-38. This approach could also be combined with other treatments. For example, it has recently been shown that Moxifloxacin [36] enhances the anti-tumor and anti-angiogenic activity of irinotecan in human xenograft tumors.
We found that the engineered mouse monocytes homed effectively to tumors. Monocytes and/or macrophages are often found as tumor-associated cells [37]. Rat monocytes were shown to efficiently invade rat glioma spheroids in vitro, and peritoneal macrophages specifically migrated to rat gliomas after intravenous or intracarotid administration [38]. Tie2-expressing monocytes were used successfully for targeted delivery of alpha interferon to xenotransplanted U87 gliomas with reduction of growth and metastasis [13]. Valable et al. administered P388-D1 monocytes IV to rats with C6 gliomas and observed that they accumulated in the gliomas but also were found in liver and spleen [39]. The liver/spleen cells were thought to indicate that some of these cells home towards the reticuloendothelial system on their way to elimination from the body [39–42].
The RAW264.7 cell line employed here is a monocyte-macrophage line widely used as a model for mouse monocyte-macrophage studies. These cells are leukemic cells that might home more efficiently to tumors than normal mouse monocytes, and it would be valuable to compare this ability to peripheral blood-derived monocytes. However, here our objective was to show proof of concept for this cell-based delivery system, so we chose a cell line that would be more amenable to the necessary genetic engineering. The double stable mouse monocytes loaded with irinotecan but not subjected to doxycycline-induced release of InCE did not reduce lung melanoma growth; in fact, they slightly stimulated growth (Fig. 5b). Other work has shown slight stimulation of growth when unmodified delivery cells are given, which is not surprising since these cells are often recruited into tumors as accessory cells. In the suicide gene scenario described here, the delivery cells are killed along with cancer cells, removing potential undesirable consequences of their continued coexistence in the tumor microenvironment.
Many other potent suicide gene therapy systems are available for which it would be advantageous to avoid toxic effects of the prodrug itself give systemically. In some cases, this is due to the fact that potential activating enzymes such as aldehyde oxidase, cytochrome P450 reductase, and glutathione S-transferase have such wide tissue distribution that targeting is difficult [2]. Other examples exist where a potential prodrug has unfortunate side effects. For example, a mutant form of nitroreductase can convert gemcitabine to a more toxic form. The present delivery system could potentially be used as a strategy to reduce required doses of other chemotherapeutic agents such as dacarbazine. It could also be used with other CE-activated prodrugs. For example, a CE-cleavable doxorubricin prodrug has recently been described [43]. Thus, the system we describe here holds potential for further development as a stealth enzyme activating prodrug system for targeted therapy of melanoma and other types of cancer [5].
Supplementary Material
Acknowledgements
This work was supported by NIH 1R21CA135599, the Terry C. Johnson Center for Basic Cancer Research at Kansas State University, Kansas State University Targeted Excellence, Kansas State Legislative Appropriation, and the Kansas Agricultural Experiment Station.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s11033-011-0720-7) contains supplementary material, which is available to authorized users.
Contributor Information
Gwi-Moon Seo, Department of Anatomy and Physiology, College of Veterinary Medicine, Kansas State University, 1800 Denison Avenue, Manhattan, KS 66506, USA.
Raja Shekar Rachakatla, Department of Anatomy and Physiology, College of Veterinary Medicine, Kansas State University, 1800 Denison Avenue, Manhattan, KS 66506, USA.
Sivasai Balivada, Department of Anatomy and Physiology, College of Veterinary Medicine, Kansas State University, 1800 Denison Avenue, Manhattan, KS 66506, USA.
Marla Pyle, Department of Anatomy and Physiology, College of Veterinary Medicine, Kansas State University, 1800 Denison Avenue, Manhattan, KS 66506, USA.
Tej B. Shrestha, Department of Anatomy and Physiology, College of Veterinary Medicine, Kansas State University, 1800 Denison Avenue, Manhattan, KS 66506, USA
Matthew T. Basel, Department of Anatomy and Physiology, College of Veterinary Medicine, Kansas State University, 1800 Denison Avenue, Manhattan, KS 66506, USA
Carl Myers, Department of Diagnostic Medicine/Pathobiology, College of Veterinary Medicine, Kansas State University, Manhattan, KS 66506, USA.
Hongwang Wang, Department of Chemistry, Kansas State University, Manhattan, KS 66506, USA.
Masaaki Tamura, Department of Anatomy and Physiology, College of Veterinary Medicine, Kansas State University, 1800 Denison Avenue, Manhattan, KS 66506, USA.
Stefan H. Bossmann, Department of Chemistry, Kansas State University, Manhattan, KS 66506, USA
Deryl L. Troyer, Department of Anatomy and Physiology, College of Veterinary Medicine, Kansas State University, 1800 Denison Avenue, Manhattan, KS 66506, USA
References
- 1.Azrak RG, Cao S, Slocum HK, Toth K, Durrani FA, Yin MB, Pendyala L, Zhang W, McLeod HL, Rustum YM. Therapeutic synergy between irinotecan and 5-fluorouracil against human tumor xenografts. Clin Cancer Res. 2004;10:1121–1129. doi: 10.1158/1078-0432.ccr-0913-3. [DOI] [PubMed] [Google Scholar]
- 2.Rooseboom M, Commandeur JN, Vermeulen NP. Enzyme-catalyzed activation of anticancer prodrugs. Pharmacol Rev. 2004;56:53–102. doi: 10.1124/pr.56.1.3. [DOI] [PubMed] [Google Scholar]
- 3.Friedlos F, Denny WA, Palmer BD, Springer CJ. Mustard prodrugs for activation by Escherichia coli nitroreductase in gene-directed enzyme prodrug therapy. J Med Chem. 1997;40:1270–1275. doi: 10.1021/jm960794l. [DOI] [PubMed] [Google Scholar]
- 4.Greco O, Dachs GU. Gene directed enzyme/prodrug therapy of cancer: historical appraisal and future prospectives. J Cell Physiol. 2001;187:22–36. doi: 10.1002/1097-4652(2001)9999:9999<::AID-JCP1060>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 5.Liu Y, Miyoshi H, Nakamura M. Nanomedicine for drug delivery and imaging: a promising avenue for cancer therapy and diagnosis using targeted functional nanoparticles. Int J Cancer. 2007;120:2527–2537. doi: 10.1002/ijc.22709. [DOI] [PubMed] [Google Scholar]
- 6.Aboody KS, Brown A, Rainov NG, Bower KA, Liu S, Yang W, Small JE, Herrlinger U, Ourednik V, Black PM, Breakefield XO, Snyder EY. Neural stem cells display extensive tropism for pathology in adult brain: evidence from intracranial gliomas. Proc Natl Acad Sci USA. 2000;97:12846–12851. doi: 10.1073/pnas.97.23.12846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Studeny M, Marini FC, Champlin RE, Zompetta C, Fidler IJ, Andreeff M. Bone marrow-derived mesenchymal stem cells as vehicles for interferon-beta delivery into tumors. Cancer Res. 2002;62:3603–3608. [PubMed] [Google Scholar]
- 8.Studeny M, Marini FC, Dembinski JL, Zompetta C, Cabreira-Hansen M, Bekele BN, Champlin RE, Andreeff M. Mesenchymal stem cells: potential precursors for tumor stroma and targeted-delivery vehicles for anticancer agents. J Natl Cancer Inst. 2004;96:1593–1603. doi: 10.1093/jnci/djh299. [DOI] [PubMed] [Google Scholar]
- 9.Rachakatla RS, Marini F, Weiss ML, Tamura M, Troyer D. Development of human umbilical cord matrix stem cell-based gene therapy for experimental lung tumors. Cancer Gene Ther. 2007;14:828–835. doi: 10.1038/sj.cgt.7701077. [DOI] [PubMed] [Google Scholar]
- 10.Rachakatla RS, Pyle MM, Ayuzawa R, Edwards SM, Marini FC, Weiss ML, Tamura M, Troyer D. Combination treatment of human umbilical cord matrix stem cell-based interferon-beta gene therapy and 5-fluorouracil significantly reduces growth of metastatic human breast cancer in SCID mouse lungs. Cancer Invest. 2008;26:662–670. doi: 10.1080/07357900701871134. [DOI] [PubMed] [Google Scholar]
- 11.Ganta C, Chiyo D, Ayuzawa R, Rachakatla R, Pyle M, Andrews G, Weiss M, Tamura M, Troyer D. Rat umbilical cord stem cells completely abolish rat mammary carcinomas with no evidence of metastasis or recurrence 100 days post-tumor cell inoculation. Cancer Res. 2009;69:1815–1820. doi: 10.1158/0008-5472.CAN-08-2750. [DOI] [PubMed] [Google Scholar]
- 12.Arbab AS, Pandit SD, Anderson SA, Yocum GT, Bur M, Frenkel V, Khuu HM, Read EJ, Frank JA. Magnetic resonance imaging and confocal microscopy studies of magnetically labeled endothelial progenitor cells trafficking to sites of tumor angiogenesis. Stem Cells. 2006;24:671–678. doi: 10.1634/stemcells.2005-0017. [DOI] [PubMed] [Google Scholar]
- 13.De Palma M, Mazzieri R, Politi LS, Pucci F, Zonari E, Sitia G, Mazzoleni S, Moi D, Venneri MA, Indraccolo S, Falini A, Guidotti LG, Galli R, Naldini L. Tumor-targeted interferon-alpha delivery by Tie2-expressing monocytes inhibits tumor growth and metastasis. Cancer Cell. 2008;14:299–311. doi: 10.1016/j.ccr.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 14.Huang B, Desai A, Tang S, Thomas TP, Baker JR., Jr The synthesis of a c(RGDyK) targeted SN38 prodrug with an indolequinone structure for bioreductive drug release. Org Lett. 2010;12:1384–1387. doi: 10.1021/ol1002626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matsuzaki T, Yokokura T, Mutai M, Tsuruo T. Inhibition of spontaneous and experimental metastasis by a new derivative of camptothecin, CPT-11, in mice. Cancer Chemother Pharmacol. 1988;21:308–312. doi: 10.1007/BF00264196. [DOI] [PubMed] [Google Scholar]
- 16.Arimori K, Kuroki N, Kumamoto A, Tanoue N, Nakano M, Kumazawa E, Tohgo A, Kikuchi M. Excretion into gastrointestinal tract of irinotecan lactone and carboxylate forms and their pharmacodynamics in rodents. Pharm Res. 2001;18:814–822. doi: 10.1023/a:1011040529881. [DOI] [PubMed] [Google Scholar]
- 17.O’Reilly S, Rowinsky EK. The clinical status of irinotecan (CPT-11), a novel water soluble camptothecin analogue: 1996. Crit Rev Oncol Hematol. 1996;24:47–70. doi: 10.1016/1040-8428(96)00211-9. [DOI] [PubMed] [Google Scholar]
- 18.Aboody KS, Bush RA, Garcia E, Metz MZ, Najbauer J, Justus KA, Phelps DA, Remack JS, Yoon KJ, Gillespie S, Kim SU, Glackin CA, Potter PM, Danks MK. Development of a tumor-selective approach to treat metastatic cancer. PLoS One. 2006;1:e23. doi: 10.1371/journal.pone.0000023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Danks MK, Morton CL, Krull EJ, Cheshire PJ, Richmond LB, Naeve CW, Pawlik CA, Houghton PJ, Potter PM. Comparison of activation of CPT-11 by rabbit and human carboxylesterases for use in enzyme/prodrug therapy. Clin Cancer Res. 1999;5:917–924. [PubMed] [Google Scholar]
- 20.Danks MK, Yoon KJ, Bush RA, Remack JS, Wierdl M, Tsurkan L, Kim SU, Garcia E, Metz MZ, Najbauer J, Potter PM, Aboody KS. Tumor-targeted enzyme/prodrug therapy mediates long-term disease-free survival of mice bearing disseminated neuroblastoma. Cancer Res. 2007;67:22–25. doi: 10.1158/0008-5472.CAN-06-3607. [DOI] [PubMed] [Google Scholar]
- 21.Humerickhouse R, Lohrbach K, Li L, Bosron WF, Dolan ME. Characterization of CPT-11 hydrolysis by human liver carboxylesterase isoforms hCE-1 and hCE-2. Cancer Res. 2000;60:1189–1192. [PubMed] [Google Scholar]
- 22.Zhang G, Liao Y, Baker I. Surface engineering of core/ shell iron/iron oxide nanoparticles from microemulsions for hyperthermia. Mater Sci Eng C. 2010;30:92–97. doi: 10.1016/j.msec.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Balivada S, Rachakatla RS, Wang H, Samarakoon TN, Dani RK, Pyle M, Kroh FO, Walker B, Leaym X, Koper OB, Tamura M, Chikan V, Bossmann SH, Troyer DL. A/C magnetic hyperthermia of melanoma mediated by iron(0)/iron oxide core/shell magnetic nanoparticles: a mouse study. BMC Cancer. 2010;10:119. doi: 10.1186/1471-2407-10-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sawada S, Okajima S, Aiyama R, Nokata K, Furuta T, Yokokura T, Sugino E, Yamaguchi K, Miyasaka T. Synthesis and antitumor activity of 20(S)-camptothecin derivatives: carbamate-linked, water-soluble derivatives of 7-ethyl-10-hydroxycamptothecin. Chem Pharm Bull (Tokyo) 1991;39:1446–1450. doi: 10.1248/cpb.39.1446. [DOI] [PubMed] [Google Scholar]
- 25.Sawada S, Yokokura T, Miyasaka T. Synthesis and antitumor activity of A-ring or E-lactone modified water-soluble prodrugs of 20(S)-camptothecin, including development of irinotecan hydrochloride trihydrate (CPT-11) Curr Pharm Design. 1995;1:113–132. [Google Scholar]
- 26.Miyasaka T, Sawada S, Nokata K, Sugino E, Mutai M. Camptothecin derivatives and process for preparing same. #4,604,463 US Patent. 1986
- 27.Wu MH, Yan B, Humerickhouse R, Dolan ME. Irinotecan activation by human carboxylesterases in colorectal adenocarcinoma cells. Clin Cancer Res. 2002;8:2696–2700. [PubMed] [Google Scholar]
- 28.Innocenti F, Undevia SD, Iyer L, Chen PX, Das S, Kocherginsky M, Karrison T, Janisch L, Ramirez J, Rudin CM, Vokes EE, Ratain MJ. Genetic variants in the UDP-glucuronosyl-transferase 1A1 gene predict the risk of severe neutropenia of irinotecan. J Clin Oncol. 2004;22:1382–1388. doi: 10.1200/JCO.2004.07.173. [DOI] [PubMed] [Google Scholar]
- 29.van Ark-Otte J, Kedde MA, van der Vijgh WJ, Dingemans AM, Jansen WJ, Pinedo HM, Boven E, Giaccone G. Determinants of CPT-11 and SN-38 activities in human lung cancer cells. Br J Cancer. 1998;77:2171–2176. doi: 10.1038/bjc.1998.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oosterhoff D, Pinedo HM, van der Meulen IH, de Graaf M, Sone T, Kruyt FA, van Beusechem VW, Haisma HJ, Gerritsen WR. Secreted and tumour targeted human carboxylesterase for activation of irinotecan. Br J Cancer. 2002;87:659–664. doi: 10.1038/sj.bjc.6600519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bissery MC, Vrignaud P, Lavelle F, Chabot GG. Experimental antitumor activity and pharmacokinetics of the camptothecin analog irinotecan (CPT-11) in mice. Anticancer Drugs. 1996;7:437–460. doi: 10.1097/00001813-199606000-00010. [DOI] [PubMed] [Google Scholar]
- 32.Bissery MC, Vrignaud P, Lavelle F, Chabot GG. Preclinical antitumor activity and pharmacokinetics of irinotecan (CPT-11) in tumor-bearing mice. Ann N Y Acad Sci. 1996;803:173–180. doi: 10.1111/j.1749-6632.1996.tb26386.x. [DOI] [PubMed] [Google Scholar]
- 33.Kaneda N, Nagata H, Furuta T, Yokokura T. Metabolism and pharmacokinetics of the camptothecin analogue CPT-11 in the mouse. Cancer Res. 1990;50:1715–1720. [PubMed] [Google Scholar]
- 34.Tardi PG, Dos Santos N, Harasym TO, Johnstone SA, Zisman N, Tsang AW, Bermudes DG, Mayer LD. Drug ratio-dependent antitumor activity of irinotecan and cisplatin combinations in vitro and in vivo. Mol Cancer Ther. 2009;8:2266–2275. doi: 10.1158/1535-7163.MCT-09-0243. [DOI] [PubMed] [Google Scholar]
- 35.Zabala M, Wang L, Hernandez-Alcoceba R, Hillen W, Qian C, Prieto J, Kramer MG. Optimization of the Tet-on system to regulate interleukin 12 expression in the liver for the treatment of hepatic tumors. Cancer Res. 2004;64:2799–2804. doi: 10.1158/0008-5472.can-03-3061. [DOI] [PubMed] [Google Scholar]
- 36.Reuveni D, Halperin D, Fabian I, Tsarfaty G, Askenasy N, Shalit I. Moxifloxacin increases anti-tumor and anti-angiogenic activity of irinotecan in human xenograft tumors. Biochem Pharmacol. 2010;79:1100–1107. doi: 10.1016/j.bcp.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 37.Solinas G, Germano G, Mantovani A, Allavena P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J Leukoc Biol. 2009;86:1065–1073. doi: 10.1189/jlb.0609385. [DOI] [PubMed] [Google Scholar]
- 38.Strik HM, Hulper P, Erdlenbruch B, Meier J, Kowalewski A, Hemmerlein B, Gold R, Bahr M. Models of monocytic invasion into glioma cell aggregates. Anticancer Res. 2006;26:865–871. [PubMed] [Google Scholar]
- 39.Valable S, Barbier EL, Bernaudin M, Roussel S, Segebarth C, Petit E, Remy C. In vivo MRI tracking of exogenous monocytes/macrophages targeting brain tumors in a rat model of glioma. Neuroimage. 2008;40:973–983. doi: 10.1016/j.neuroimage.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 40.Burke B, Sumner S, Maitland N, Lewis CE. Macrophages in gene therapy: cellular delivery vehicles and in vivo targets. J Leukoc Biol. 2002;72:417–428. [PubMed] [Google Scholar]
- 41.Burke B. Macrophages as novel cellular vehicles for gene therapy. Expert Opin Biol Ther. 2003;3:919–924. doi: 10.1517/14712598.3.6.919. [DOI] [PubMed] [Google Scholar]
- 42.Burke B, Giannoudis A, Corke KP, Gill D, Wells M, Ziegler-Heitbrock L, Lewis CE. Hypoxia-induced gene expression in human macrophages: implications for ischemic tissues and hypoxia-regulated gene therapy. Am J Pathol. 2003;163:1233–1243. doi: 10.1016/S0002-9440(10)63483-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barthel BL, Zhang Z, Rudnicki DL, Coldren CD, Polinkovsky M, Sun H, Koch GG, Chan DC, Koch TH. Preclinical efficacy of a carboxylesterase 2-activated prodrug of doxazolidine. J Med Chem. 2009;52:7678–7688. doi: 10.1021/jm900694z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.