

Abstract
Integrin receptors bind extracellular matrix proteins, and this link between the cell membrane and the surrounding matrix may translate skeletal loading to biologic activity in osteoprogenitor cells. The interaction between integrin and growth factor receptors allows for mechanically induced regulation of growth factor signaling. Skeletal unloading leads to decreased bone formation and osteoblast proliferation that can be explained in part by a failure of insulin-like growth factor-I (IGF-I) to activate its signaling pathways in unloaded bone. The aim of this study is to determine whether unloading induced resistance is specific for IGF-I or common to other skeletal growth factors, and to examine the regulatory role of integrins in IGF-I signaling. Bone marrow osteoprogenitor (BMOp) cells were isolated from control or hindlimb suspended rats. Unloaded BMOp cells treated with IGF-I failed to respond with increased proliferation, receptor phosporylation, or signaling activation in the setting of intact ligand binding; whereas the platelet-derived growth factor (PDGF) response was fully intact. Pre-treatment of control BMOp cells with an integrin inhibitor, echistatin, failed to disrupt PDGF signaling, but blocked IGF-I signaling. Recovery of IGF-I signaling in unloaded BMOp cells followed the recovery of marked reduction in integrin expression induced by skeletal unloading. Selective targeting of integrin subunits with siRNA oligonucleotides revealed that integrin β1 and β3 are required for normal IGF-I receptor phosphorylation. We conclude that integrins, in particular integrin β3, are regulators of IGF-I, but not PDGF, signaling in osteoblasts, suggesting that PDGF could be considered for investigation in prevention and/or treatment of bone loss during immobilization and other forms of skeletal unloading.
Keywords: skeletal unloading, insulin-like growth factor, platelet-derived growth factor, osteoblast, integrin
Introduction
Skeletal unloading by hindlimb elevation results in decreased bone formation, osteoblast number and osteoblast maturation in rats.(1–8) Bone marrow osteoprogenitor (BMOp) cells from unloaded bone are impaired with respect to proliferation and differentiation in vitro.(9–11) Although the phenomenon of decreased bone formation during skeletal unloading is well known, the underlying mechanism remains unclear.
IGF-I is required for the proliferation of most cell types and promotes cell survival by inhibiting apoptosis. In bone, IGF-I is one of the most abundant growth factors and plays an important role in regulating bone formation.(12,13) IGF-I stimulates osteoblast proliferation and promotes bone formation.(14–17) The anti-apoptotic effects of IGF-I may regulate bone formation by increasing the life span of osteoblasts. Binding of IGF-I to its receptor stimulates autophosphorylation of specific tyrosine residues, which creates multiple docking sites for various intracellular signaling substrates and in turn activates two distinct pathways: Ras/Raf/MEK/MAPK, which promotes proliferation, and PI3K/Akt that inhibits apoptosis.(18–21)
We have demonstrated that skeletal unloading induces resistance to IGF-I that is specific to unloaded bones.(5,6) IGF-I administered concurrently with skeletal unloading fails to stimulate bone formation in unloaded bones but acts normally in the weight bearing forelimbs.(5) Additionally, IGF-I fails to maintain osteoprogenitor number and proliferation suppressed by unloading. The resistance to IGF-I persists in BMOp cells isolated from unloaded bones in that proliferation does not increase in response to IGF-I treatment as is found in those isolated from normally loaded bone. The BMOp cells from unloaded bone demonstrate a failure to activate the IGF-I receptor and downstream signaling cascades when treated with IGF-I.
Integrin receptors may be a mechanical load sensitive regulator of IGF-I signaling in osteoblasts. Integrins are membrane bound heterodimeric receptors that bind to their own limited set of extra-cellular matrix and/or cell surface proteins. Ligand binding to integrins induces cytoskeletal reorganization and recruitment of various signaling proteins to the cell membrane. Integrin receptor regulation of growth factor receptors has been described in assorted tissues, and the role of integrin αVβ3 in the regulation of IGF-I receptor function has been described in detail in vascular smooth muscle cells.(22,23) In the non-loaded state, the unstimulated integrin αVβ3 receptor causes an alteration in the recruitment of the SHP-2 phosphatase that augments activity and subsequently deactivates the IGF-I receptor. The role of integrin αVβ3 in the regulation of the IGF-I receptor in osteoblasts is suggested by the impairment of IGF-I receptor activation in BMOp cells by echistatin, an integrin antagonist specific for α5β1 and αVβ3 integrins,(24) and the reduction in the expression of integrin αVβ3 in BMOp cells isolated from unloaded bone, which fail to respond to IGF-I.(6) Evaluation of IGF-I signaling in BMOp cells from unloaded bone revealed a failure of initial receptor phosphorylation that is independent of phosphatase activity in that IGF-I resistance persists in the setting of treatment with a phosphatase inhibitor, suggesting a different mechanism of integrin regulation of IGF-I receptor in osteoblasts than in vascular smooth muscle cells.(6)
This evidence led us to question whether skeletal unloading induced resistance is specific to IGF-I or affects other skeletal growth factors, and whether it is directly influenced by alterations of integrins. We compared the receptor activation, downstream signaling, and the proliferative response of BMOp cells isolated from loaded and unloaded bones to administration of IGF-I or platelet-derived growth factor (PDGF). Receptors for PDGF and IGF-I are both members of the cell surface tyrosine kinase family of receptors, and PDGF is an important mediator of bone formation and fracture healing. Studies have shown that PDGF enhances primary rat osteoblastic cell proliferation,(25) collagen synthesis in rat osteoblast cultures,(26) bone formation in subcutaneously implanted matrix,(27) and is expressed in healing mandibular fractures in rats.(28) We compared the effects of echistatin and the pattern of integrin expression over time in culture on growth factor receptor activation. Finally, specific integrin subunits were targeted for knockdown to determine their regulatory role of IGF-I signaling in osteoblasts. Our data indicate that the resistance to IGF-I induced by skeletal unloading is not shared by PDGF suggesting distinct mechanisms, in particular the selective role of integrins, by which these growth factor receptors are regulated in BMOp cells.
Material and Methods
Animal protocols
Male Sprague-Dawley (SD) rats at 11 weeks of age were purchased (Simonsen, Gilroy, CA), fed standard laboratory rat chow (22/5 Rodent Diet (W); Harlan Teklad) containing 1.13% calcium and 0.94% phosphorus, maintained on a 12:12-hours light-dark cycle, and allowed to acclimate to individual housing for one week. At 12 weeks of age the rats were randomly divided into two groups, skeletal unloaded using the tail suspension hindlimb elevation model as previously described(2–5) or loaded via normal ambulation. The unloaded group was hindlimb elevated for 7 days; the other group served as the normally loaded pair-fed controls. These studies were approved by the Institutional Animal Care and Use Committee of the San Francisco Veterans Affairs Medical Center, where the studies were performed.
BMOp cell culture
The tibial and femoral BMOp cells were harvested using techniques previously described.(5,6) Briefly, the marrow from the tibiae and femora was collected in primary culture medium (α-MEM containing L-glutamine and nucleosides; Mediatech), supplemented with 10% FBS (Atlanta Biologicals), 100U/mL penicillin/streptomycin (Mediatech), and 0.25μg/mL amphotericin B (Life Technologies). A single-cell suspension from each rat was obtained by repeated passage through an 18-gauge needle and counted with a hemacytometer. The cells were plated at a cell density of 3×105 cells/cm2. Nonadherent cells were removed by aspiration, and the primary medium was replenished on day 5 after plating. Medium was changed to differentiation medium, primary culture medium supplemented with 50μg/mL ascorbic acid and 3mM β-glycerophosphate, at day 7 after plating. Differentiation medium was subsequently refreshed twice weekly.
siRNA Knockdown
BMOp cells were isolated from the long bones of control rats as detailed above and plated at a cell density of 6×105 cells/cm2. At day 4 in culture, cells were treated (siLentfect, Biorad) with a 4 duplex pool of siRNA oligonucleotides targeting the β1 or β3 integrin subunit (siβ1 or siβ3, 25nM, Dharmacon) or a non-targeting oligonucleotide pool (siCont, 25nM, Dharmacon). The medium was refreshed after an overnight incubation and then serum deprived at day 7 in culture in preparation for IGF-I stimulation.
Proliferation Assay
BMOp cells were harvested as described above and plated in 6-well plates. The cultures were switched to primary culture medium containing 1% FBS on day 7 after plating. After 24 hrs, the cells were rinsed once with PBS and switched to FBS-free primary culture medium supplemented with 0–100ng/mL recombinant human IGF-I (gracious gift from Chiron) or 0–10ng/mL PDGF-BB (Sigma) for 24 hrs at 37°C. During the last 4 hrs of the treatment, the cultures were labeled with 10μM bromodeoxyuridine (BrdU, Xymed). BrdU incorporation during DNA synthesis was measured by means of a Cell Proliferation ELISA BrdU-Kit (Roche). The absorbance (at 370nm) of the BrdU incorporation in the culture was normalized to cell number by the absorbance of the crystal violet stain of an additional parallel culture that was treated in the same manner.
Signaling cascade activation
BMOp cell cultures were established as detailed above. At a designated day in culture, cells were rinsed with PBS, and the medium was changed to FBS-free primary culture medium. After overnight incubation at 37°C, the BMOp cells were treated with IGF-I (10ng/mL), PDGF (1ng/mL), or vehicle for a 10-minute incubation. Total cell lysates were collected immediately. BMOp cells from loaded bones were pre-treated with echistatin (100nM, Sigma) or vehicle 12 hours prior to IGF-I or PDGF treatment.
Immunoprecipitation and Western blotting
Treated BMOp cells were washed twice with ice-cold PBS that contained 0.1% NaF and Na3VO4 and solubilized in lysis buffer consisting of 1% Triton X-100, 0.1% SDS, 1% sodium deoxycholate, 1mM EDTA, 50mM HEPES, 0.1% NaF, 0.1% Na3VO4, 150mM NaCl, 100μg/mL phenylmethanesulfonyl fluoride (PMSF), and protease inhibitor cocktail (Complete Mini; Roche). The protein concentration in the cell lysates was measured using a BCA protein assay kit (Pierce Biotechnology). For the phosphorylated IGF-I receptor assessment, equivalent protein samples (300–500μg) of the cell lysates were immunoprecipitated using rabbit antibody to the IGF-I receptor β chain (C-20, Santa Cruz Biotechnology) and Ultralink Protein A/G agarose beads (Pierce). Immunoprecipitated material was solubilized in SDS sample buffer (2% SDS, 10mM DTT, 10% glycerol, 10mM Tris-HCl, 0.01% bromphenol blue), boiled at 95°C for 5 minutes, and separated by SDS-PAGE. Separated peptides were transferred onto polyvinylidene difluoride (PVDF) membranes. After blocking with 5% nonfat milk TBS/Tween buffer, the PVDF membranes were incubated with mouse antibody to phospho-tyrosine (PY99, 1:500 dilution, Santa Cruz) at 4°C overnight, followed by incubation with horseradish peroxidase (HRP)-conjugated anti-mouse IgG antibody (1:6667 dilution, Amersham) at room temperature for one hour. The blots were developed using a chemiluminescent substrate (SuperSignal West Dura; Pierce) according to the manufacturer’s instructions and exposure to radiographic film. The phosphorylated IGF-I receptor bands were digitally scanned and quantification of all blots was performed on a Macintosh computer using Multi-Analysit software (Bio-Rad Laboratories) or Kodak 1D Image Analysis software v3.5. The blots were stripped of the phospho-specific antibodies by incubating for one hour in stripping buffer (2% SDS, 100mM 2-mercaptoethanol, 62.5mM Tris-HCl [pH 6.7]) at room temperature, and reprobed with primary antibody directed at the IGF-I receptor β chain (C-20, 1:1000 dilution, Santa Cruz) at 4°C overnight. Total IGF-I receptor amount was detected and quantified as detailed above.
To measure the activation of the PDGF receptor and downstream components of the IGF-I and PDGF signaling pathways, equivalent protein samples (50–100μg) of the cell lysates in SDS sample buffer were separated by SDS-PAGE and transferred as described above. After blocking with 5% bovine serum albumin TBS/Tween buffer, the PVDF membranes were incubated with rabbit antibodies to phospho-PDGF receptor (1:1000 dilution, Cell Signaling) or phospho-p44/42 MAPK (1:2000 dilution, phospho-ERK1/2, Thr202/Tyr204; Cell Signaling) at 4°C overnight, followed by incubation with HRP-conjugated anti-rabbit IgG at room temperature for one hour. The blots were developed, and phospho-specific proteins quantified as described above. The blots were stripped of the phospho-specific antibodies as detailed above and incubated with primary antibody directed at the PDGF receptor (1:1000 dilution, Cell Signaling), p44/42 MAPK (1:1000 dilution, ERK1/2, Cell Signaling), integrin β1 (1:1000, Chemicon), or actin, used as a loading control (1:2000 dilution, Sigma) at 4°C overnight. Protein amount was quantified as detailed above.
IGF-I Receptor Binding assay
Des(1–3)IGF-I, an analog with normal affinity for the IGF-I receptor but decreased affinity for IGF binding proteins was obtained from GroPep Pty. Ltd. (Adelaide, Australia). Cultures of BMOp cells were established as described above and plated at 1 X 106 cells per well in 12-well plates. The cultures were rinsed twice with PBS and switched to FBS-free medium on day 7. After 24 hrs, the cultures were exposed to 25pM 125I-IGF-I (specific activity > 74TBq/mmol; Amersham Pharmacia Biotech) and increasing doses (0, 0.01, 0.1, 1, 10, or 100nM) of unlabeled IGF-I or des IGF-I for 10 minutes at 37°C. At the end of the incubation, the cultures were put on ice, rinsed three times with ice-cold PBS, and solubilized in 0.5M NaOH. The receptor-bound 125I-IGF-Ilevels were determined by detection in a γ-counter.
Quantitative Real-Time PCR
Total RNA was harvested from cultured cells using the RNeasy Mini extraction kit (Qiagen). For each sample, 2μg of total RNA was reverse-transcribed in 100μL of a reaction mixture that contained 10mM Tris-HCl (pH 8.3), 50mM KCl, 7.5mM MgCl2, 1mM each dNTP, 5μM random primers (GIBCO BRL), 0.4U/μL RNase inhibitor (Roche), and 2.5U/μL Moloney murine leukemia virus reverse transcriptase (M-MLV) (GIBCO BRL) at 25°C for 10 minutes, 48°C for 40 minutes, 95°C for 5 minutes, and 4°C (stored). The sequences of the qRTPCR primers used for measurement of the integrin subunits of rats are listed in Table 1 or published.(29) These primers were designed using Primer Express Software (Applied Biosystems). Primers were synthesized by the Biomolecular Resource Center (University of California, San Francisco, CA). The control mitochondrial L19 primer set was obtained commercially (Applied Biosystems). qRTPCR was carried out in triplicate with 20-μL reaction volumes of 10μL SYBR Green PCR Master Mix (Applied Biosystems), 500nM of each primer, and 1μL cDNA template. The fluorescence intensity is directly proportional to the accumulation of PCR product and can be detected with an ABI Prism 7900HT (Applied Biosystems). After the final cycle of PCR amplification, the dissociation curve analysis was performed to confirm the presence of a single PCR product and lack of primer dimers. Analysis was carried out using the sequence detection software supplied with the ABI Prism 7900HT. The number of PCR cycles (threshold cycles [Ct]) required for the fluorescent intensities to exceed a threshold just above background was calculated for the test reactions.
Table 1.
Primers for Real-Time PCR
| Integrin | Primer | Sequence (5′-3′) | PCR product bp (sequence) | GeneBank Accession No. |
|---|---|---|---|---|
| α1 | Forward Reverse |
CGTTAGCCTCACCGTCAAAAC GCATCTCCTCAATTCCCGAAA |
101 (2936–3036) | X52140 |
| α2 | Forward Reverse |
ACACACAGCCTCACCCGTATC TCAGCCAGCAGGAGACGTTA |
101 (520–620) | AB067445 |
| αV | Forward Reverse |
CGTGAAGGCGCAGAATCA CTTCGTTGTTCCGGACAACC |
101 (72–172) | S58528 |
| β1 | Forward Reverse |
AGGGTGAAGCTCACGTGCAT AATAAGAACAATTCCGGCAACC |
101 (2176–2276) | U12309 |
| β3 | Forward Reverse |
TCGTCGGAGTCCAACATCT CAGAGACTCATCCGAGCAGGA |
101 (79–179) | AJ440952 |
| β5 | Forward Reverse |
CGATGCTGTCTACCCACGTG AGACTGTGTGGAATGCTCGCT |
101 (1492–1592) | NM147139 |
The Ct values were determined for three test reactions in each sample and averaged. The ΔCt values were obtained by subtracting the L19 (as endogenous control) Ct values from the target gene Ct values of the same samples. The relative quantification of the target genes was given by 2−ΔCt.
Statistical analysis
Data are presented as mean ± SD or +/− SEM. All data were analyzed using ANOVA followed by a posthoc Fisher’s protected least significant difference (PLSD) test with a StatView 5.0 program (SAS Institute). Student’s t-test was also used for analyzing the effects of IGF-I and PDGF treatment in vitro on BMOp cells. Statistical significance was stated for p < 0.05.
Results
Skeletal unloading impairs IGF-I but not PDGF induced proliferation
We examined the direct effects of IGF-I and PDGF on proliferation of BMOp cells isolated from loaded and unloaded bone. IGF-I treatment increased BrdU incorporation dose dependently in BMOp cells from normally loaded bones (Fig 1A). In BMOp cells from unloaded bones basal BrdU incorporation was reduced, and IGF-I failed to stimulate proliferation in these cells (Fig 1A). In contrast, PDGF stimulated BrdU incorporation in the BMOp cells from both loaded and unloaded bones (Fig 1B). In fact, PDGF increased BrdU incorporation in the BMOp cells from unloaded bones to the level observed in the BMOp cells from loaded bones. To confirm that the changes in proliferation response to growth factors reflected changes in the signaling cascade activation we looked at ERK1/2 phosphorylation.
Figure 1. Skeletal unloading affects proliferation of BMOp cells in response to IGF-I and PDGF in vitro.
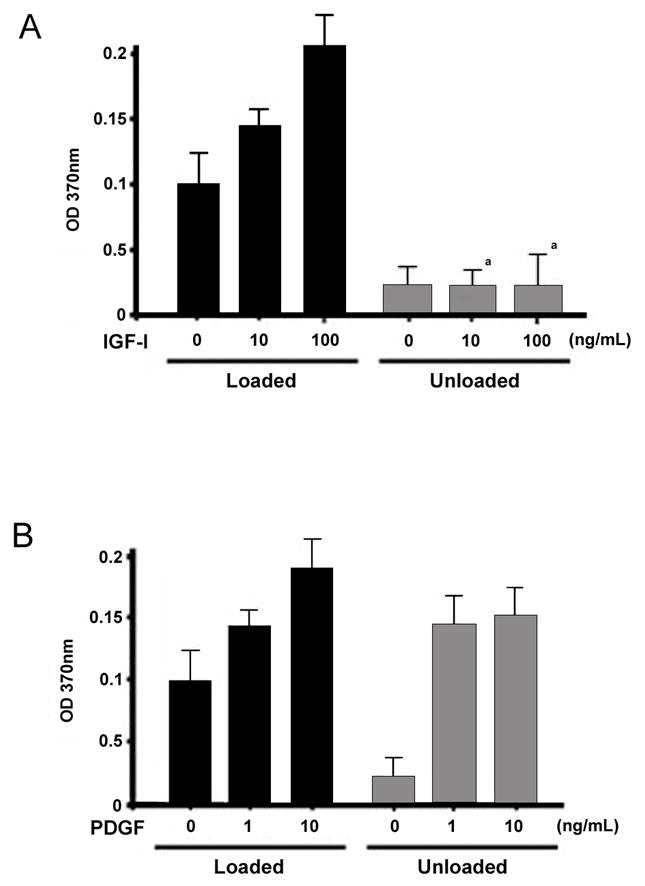
BMOp cells from loaded and unloaded bones were incubated with IGF-I (A) or PDGF (B) for 24 hrs at day 7 in culture. During the last 4 hours, the cultures were labeled with BrdU and absorbance quantifies the BrdU incorporation and proliferation. Means ± SD, n = 3. a p < 0.05 vs. Loaded + IGF-I.
As in our previous investigations, we found that skeletal unloading blocked the ability of IGF-I to activate the MAPK pathway. The brisk phosphorylation of ERK1/2 following treatment of BMOp cells from loaded bone with IGF-I is abolished in BMOp cells from unloaded bone (Fig 2A). This blunting of the phosphorylation of ERK1/2 is not due to skeletal loading or unloading induced changes in total ERK1/2 levels. To determine whether this pathway remained intact when activated by PDGF, we examined the ability of PDGF to stimulate ERK1/2 phosphorylation in BMOp cells from loaded and unloaded bone. PDGF stimulated brisk phosphorylation of ERK1/2 in BMOp cells from both loaded and unloaded bone, and the magnitude of response was equal (Fig 2B). Thus the impairment of the proliferative response of BMOp cells by skeletal unloading does not affect the signaling pathways of all receptor tyrosine kinases, as PDGF signaling remains intact.
Figure 2. Effect of skeletal unloading on growth factor stimulated phosphorylation of ERK1/2.
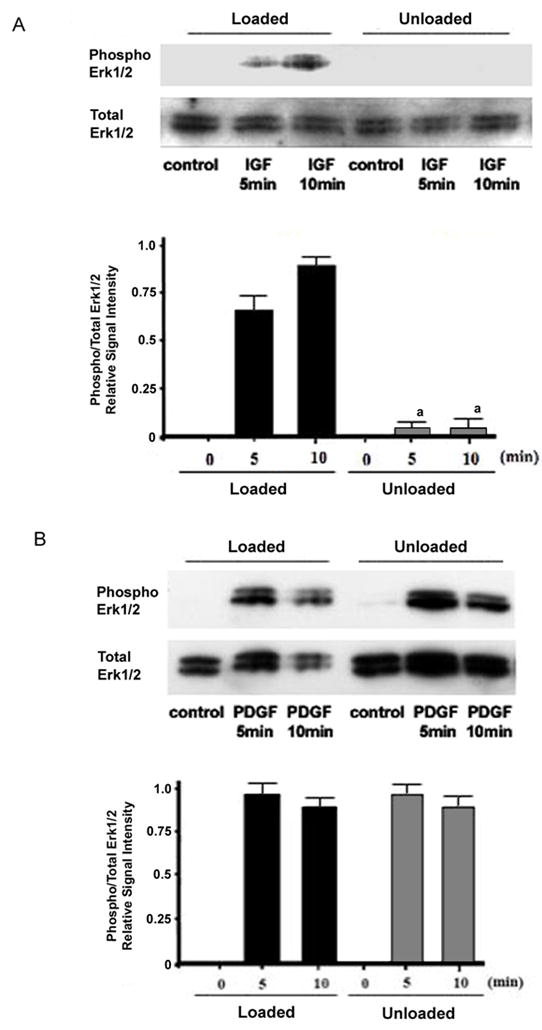
BMOp cells from loaded and unloaded bones were serum deprived at day 7 in culture, and then treated with IGF-I (A) or PDGF (B). Representative immunoblots illustrate skeletal unloading induced impairment of IGF-I stimulated ERK1/2 phosphorylation. Relative signal intensities of the ratio of phosphorylated to total ERK1/2 were evaluated. Means ± SD, n = 3. a p < 0.05 vs. Loaded + IGF-I.
Skeletal unloading specifically disrupts IGF-I receptor activation but not ligand binding
To identify the signaling step at which skeletal unloading differentially impacts the proliferative response to IGF-I and PDGF we looked at IGF-I and PDGF receptor activation. Skeletal unloading significantly blunted ligand induced IGF-I receptor activation without altering the total receptor levels (Fig 3A). In contrast to IGF-I, PDGF stimulated equally PDGF receptor phosphorylation in BMOp cells whether isolated from loaded or unloaded bone (Fig 3B). As with the IGF-I receptor, immunoblots of total cell lysate revealed no differences in PDGF receptor expression due to changes in the skeletal loading status (not shown).
Figure 3. Effect of skeletal unloading on the activation of PDGF and IGF-I receptors.
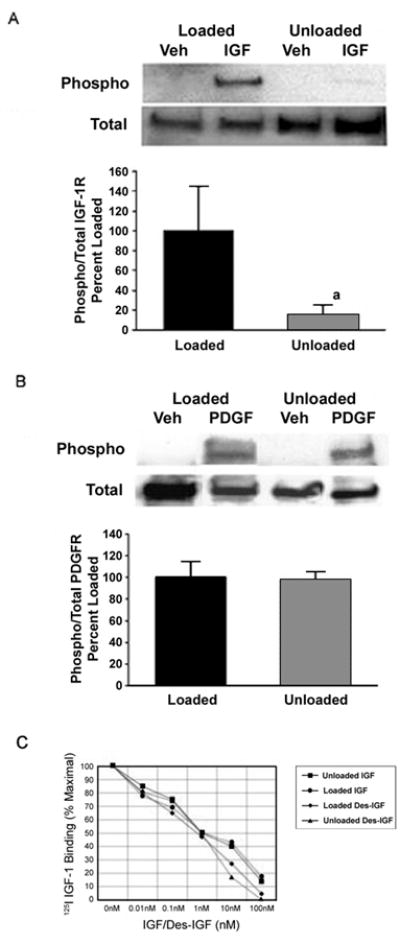
BMOp cells from loaded and unloaded bones were serum deprived at day 7 in culture, and then treated with IGF-I (A) or PDGF (B). Representative immunoblots illustrate skeletal unloading induced impairment of IGF-I receptor phosphorylation. Relative signal intensities of the ratio of phosphorylated to total IGF-I or PDGF receptors were evaluated. Means ± SD, n = 3. a p < 0.05 vs. Loaded. (C) BMOp cells from loaded and unloaded bones were serum deprived at day 7 in culture, and then exposed to 25pM 125I-IGF and various doses of unlabeled IGF-I or des-IGF-I for 10 minutes. Data are means of triplicates. Error bars are not shown to simplify presentation, but in all cases enclosed an SD of less than 10% of the mean. These values are expressed as a percentage of maximal specific 125I-IGF binding that was comparable for BMOp cells isolated from loaded and unloaded bones.
We next examined receptor binding using both IGF-I and its des 1–3 analog to determine whether the failure of IGF-I to activate its receptor in BMOp cells isolated from unloaded bone was due to an inability to bind to its receptor or competitive binding with IGF binding proteins. Des-IGF-I has an affinity for the IGF-I receptor that is comparable to that of IGF-I but has much weaker affinity for IGF-I binding proteins.(30) Neither IGF-I nor des-IGF-I was impaired in its binding to BMOp cells from unloaded or loaded bones (Fig 3C). Additionally, there is no evidence that skeletal unloading induced an increase in IGF binding protein sequestration of IGF-I, as IGF-I and des-IGF-I binding curves were the same. These findings indicate that the disruption in IGF-I signaling in BMOp cells from unloaded bone, manifesting as the failure of IGF-I to activate its receptor, is downstream of ligand binding.
Integrin regulation of the IGF-I receptor
Integrins are known to modulate the response to growth factors in a number of different cell types,(31–33) and their impact on both IGF-I and PDGF signaling has been observed.(22,23) Pre-treatment of BMOp cells from loaded bone with the integrin antagonist echistatin recreated the IGF-I resistance phenomenon of BMOp cells from unloaded bone described above. Echistatin dramatically impaired IGF-I induced cognate receptor phosphorylation without changing IGF-I receptor expression (Fig 4A). This impairment of ligand induced receptor phosphorylation by echistatin was not observed in the PDGF treated BMOp cells (Fig 4B). This observation suggests a regulatory role of integrins for IGF-I, but not PDGF, signaling in bone.
Figure 4. Effect of echistatin on the activation of the PDGF and IGF-I receptors.
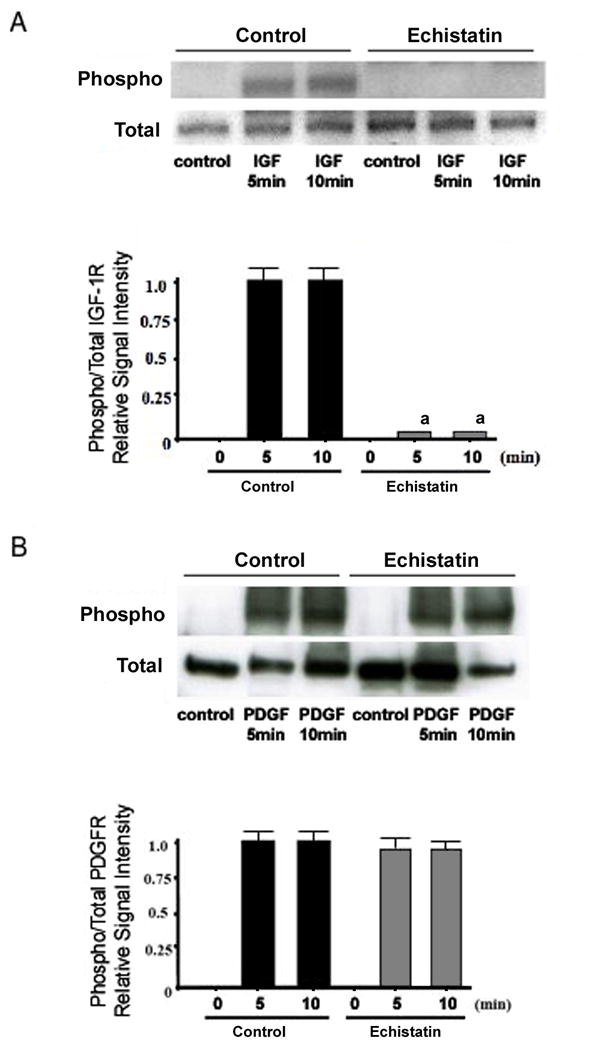
BMOp cells from loaded bones were serum deprived at day 7 in culture, and then treated with IGF-I (A) or PDGF (B). Twelve hours prior, echistatin (100nM) or vehicle was added. Representative immunoblots illustrate echistatin-induced impairment of IGF-I receptor phosphorylation. Relative signal intensities of the ratio of phosphorylated to total IGF-I and PDGF receptors were evaluated. Means ± SD, n = 3. a p < 0.05 vs. Control.
The expression of integrin subunits and markers of mesenchymal differentiation relative to a control gene, L19, in the BMOp cells from normally loaded bone at day 7 in culture is shown in Table 2. These unloaded progenitor cells at day 7 show a 3 fold increase in PPARγ2 expression (p < 0.05), however, there is a non-significant increase (120%) in the expression of Runx2. Characterization of the BMOp cells from unloaded bones at days 5 and 7 in culture revealed a reduction in the expression of a number of integrin subunits involved in the cellular response to growth factors, namely α1, α2, αV, β1, β3, and β5 (Fig 5A). This reduction in integrin subunit mRNA expression temporally coincides with the observed impairment of IGF-I receptor activation. Measurement of the dominant integrin, β1, confirmed the reduction in protein expression of this integrin subunit in BMOp cells isolated from unloaded bone at day 7 in culture (Fig 5B). The recovery of integrin subunit expression in BMOp cells isolated from unloaded bone occurred over time in culture. By day 14 in culture, the level of expression of each integrin subunit in BMOp cells from both loaded and unloaded bone had equalized, and the equal expression persisted at day 21 in culture. Immunoblotting for the β1 integrin subunit confirmed this recovery over time in culture. Actin expression showed modest increases as function of time in culture likely reflecting increased cytoskeleton and BMOp cell differentiation over time and more importantly did not change in response to differences in loading conditions in vivo.
Table 2.
Gene Expressiona
| Gene | Expression | Gene | Expression |
|---|---|---|---|
| α1 | 0.190 (0.020) b | β1 | 4.934 (2.308) b |
| α2 | 0.008 (0.003) b | β3 | 0.197 (0.070) b |
| αV | 0.589 (0.105) b | β5 | 1.691 (0.233) b |
|
| |||
| PPARγ | 0.022 (0.004) c | Runx2 | 2.938 (0.593) c |
Fold expression relative to L19 in BMOp cells isolated from loaded animals at day 7 in culture
Mean (SD), n = 8
Mean (SD), n = 3
Figure 5. Recovery of skeletal unloading induced reduction in expression of integrin subunits.
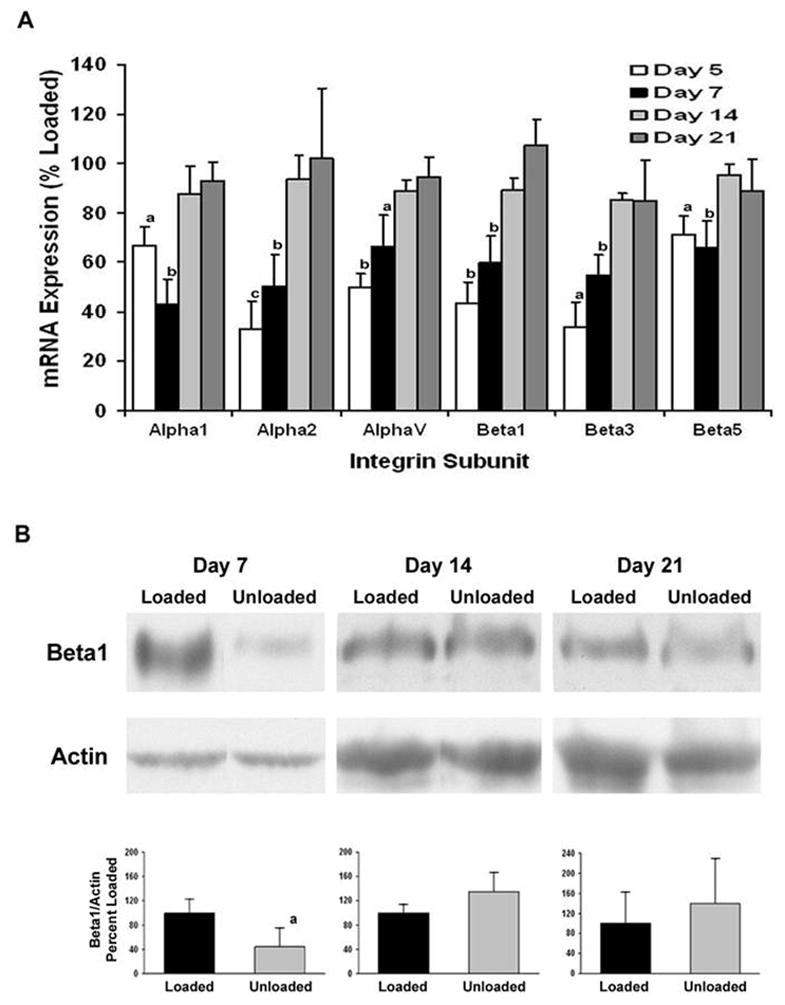
The BMOp cells from loaded and unloaded bones were grown in culture. Total RNA was harvested at days 5, 7, 14, and 21 in culture and cell lysates collected at days 7, 14, and 21 in culture. A) Relative mRNA expression of the integrin subunits normalized to L19 of the unloaded BMOp cells as a percentage of that of loaded BMOp are displayed. Means ± SEM, of four independent BMOp cell pools at day 5, thirteen at day 7, six at day 14 and four at day 21. a p < 0.05, b p < 0.01, c p = 0.055. B) A representative immunoblot illustrates that expression of the β1 integrin subunit was significantly less than that in loaded BMOp cells after 7 days but not 14 or 21 days in culture. Relative signal intensities of the ratio of β1 integrin to actin were evaluated. Means ± SD, n = 3. a p < 0.05 vs. Loaded.
The effect of the recovery of integrin expression over time in culture in BMOp cells from unloaded bone on IGF-I signaling was assessed. Relative to the blunted IGF-I signaling at day 7 in culture, BMOp cells from unloaded bones at days 14 and 21 demonstrated a recovery of ligand induced IGF-I receptor phosphorylation as the ratio of phosphorylated to total signal intensities was equivalent to that of BMOp cells from loaded bone (Fig 6A). Additionally, normalization of the brisk ERK1/2 phosphorylation in response to IGF-I confirmed recovery of the unloading induced impairment of MAPK signaling pathway over time in culture (Fig 6B). Overall, loss of skeletal unloading induced IGF-I resistance was associated with the recovery of integrin subunit expression in the BMOp cells.
Figure 6. Recovery of IGF-I signaling in BMOp cells from unloaded bone followed longitudinally.

BMOp cells from loaded and unloaded bones were grown in culture over 21 days. At days 14 and 21, cultures were serum deprived and then treated with IGF-I. Representative immunoblots illustrate recovery of ligand induced IGF-I receptor (A) and ERK1/2 (B) phosphorylation at 14 and 21 days in culture. Relative signal intensities of the ratio of phosphorylated to total IGF-I receptor and ERK1/2 were evaluated. Means ± SD, n = 4. There were no significant differences.
We then targeted integrin subunits for knockdown using siRNA technology to assess the role of specific integrin subunits in the regulation of IGF-I signaling in BMOp cells. Treatment of BMOp cells with siRNA oligonucleotide pools, siβ1 or siβ3, significantly reduced the expression of the β1 or β3 integrin subunit relative to that of the non-targeting siRNA treated, siCont, by the same magnitude observed in BMOp cells isolated from unloaded bones at day 7 in culture relative to those from loaded bones (Fig 7A). There was no significant off-target reduction; however, a mild reduction in β3 subunit expression was observed in the siβ1 treated cells (Fig 7A). Treatment of BMOp cells with non-targeting siRNA oligonucleotides did not disrupt the expression of integrin subunits relative to the expression in non-treated cells (not shown). Immunoblotting confirmed the efficiency and specificity of siRNA knockdown (Fig 7B).
Figure 7. Effects of targeted integrin subunit knockdown on IGF-I signaling.

(A) BMOp cells from loaded bones were treated with specific (siβ1 or siβ3) or non-targeting control (siCont) siRNA oligonucleotides on day 4 in culture. At day 7, cultures were serum deprived and then treated with IGF-I. RNA expression of β1 or β3 integrin subunits following targeted siRNA treatment was specifically and significantly reduced. Means ± SEM, n = 4, a p < 0.05 vs. siCont. (B) A representative immunoblot confirms the diminished expression of β1 and β3 subunit following siRNA treatment. (C) IGF-IR activation is impaired by β1 or β3 integrin subunit knockdown. (D) Relative signal intensities of the ratio of phosphorylated to total IGF-I receptor were evaluated. Means ± SEM, n = 3, a p < 0.05 vs. siCont.
These siRNA treated cells were then exposed to IGF-I to assess the role of the β1 and β3 integrin subunits in the integrity of IGF-I signaling in BMOp cells. Phosphorylation of the IGF-IR was not stimulated by knockdown of either the β1 or β3 integrin subunit in the basal state nor were the levels of total IGF-I receptor affected (Fig 7C). Ligand induced phosphorylation of the IGF-IR was blunted by knockdown of either the β1 or β3 integrin subunit. The magnitude of impairment of IGF-IR activation was significant and of similar magnitude as in unloaded BMOp cells when β3 subunit was targeted, whereas the degree of impairment with β1 knockdown was less potent and did not reach significance (Fig 7D). The effect of knockdown of both β1 and β3 integrin subunits by combined siRNA treatment, siβ1 and siβ3, on IGF-IR activation was equivalent to the effect β3 knockdown alone (not shown). The siRNA knockdown caused stimulation of basal pERK levels, which compromised the assessment of signaling downstream of IGF-IR as it was not possible to differentiate the effect of IGF-I treatment.
Discussion
The hindlimb suspension model is used as an animal model to investigate the mechanisms of bone loss caused by unloading from disuse or space flight. With this model, we and others have demonstrated that skeletal unloading causes suppression of osteoblast numbers, proliferation, and bone formation. We have demonstrated that glucocorticoids have a minimal effect on osteoblast function as we have found plasma corticosterone levels during hindlimb unloading are the same as those found in control animals, and adrenalectomy does not alter the unloading effect on bones.(34) The bone marrow stromal cells isolated from unloaded bones express a higher level of the adipocyte differentiation marker, PPARγ2, suggesting a lower osteogenic potential. However, the level of Runx2 expression in these adherent cells is not decreased, suggesting there is a pool of available osteoprogenitor cells, but they are resistant to bone formation signals. The cross-talk and stimulation of osteoclasts may also be impaired as there is lower propensity to form osteoclasts; the number of TRAP+ osteoclasts is 63% of controls, p < 0.05 (not shown).
Thus, we considered the possibility that the response of osteoblasts and osteoblast precursors to receptor tyrosine kinase growth factors would be impaired by skeletal unloading and not to non-receptor tyrosine kinase factors like TGFβ, that have been shown to blunt unloading induced bone loss.(29) In this study, we found that this expectation was only realized, in that not all receptor tyrosine kinase growth factors were impacted. In previous studies(5,6,35,36) we have shown that resistance to IGF-I plays an important role in the impaired osteoblast function and decrease in bone formation during skeletal unloading. The IGF-I resistance in bones caused by skeletal unloading persists in the BMOp cells isolated from unloaded bones. Our present results demonstrate that the resistance to IGF-I caused by skeletal unloading is not shared by PDGF. PDGF, unlike IGF-I, signaling was fully intact and capable of stimulating BMOp cells from unloaded bones.
Unloading induced resistance of IGF-I is characterized by a failure of IGF-I to stimulate cognate receptor phosphorylation and to activate proliferative and anti-apoptotic signaling cascades. We show that this is not a function of decreased receptor quantity or impaired IGF-I receptor affinity. Unloading does not induce changes in IGF binding proteins as ligand binding affinity curves are unchanged with des-IGF-I, an analogue with minimal binding protein affinity. PDGF signaling, on the other hand, is unaffected by skeletal unloading as demonstrated by completely intact PDGF receptor and downstream signaling phosphorylation in response to PDGF.
The difference in integrity of the PDGF and IGF-I signaling cascades in response to skeletal unloading is due to selective integrin receptor regulation of IGF-I but not PDGF signaling in BMOp cells. Previously, we demonstrated that αVβ3 expression in BMOp cells was reduced by skeletal unloading, and echistatin, a disintegrin selective for αVβ3 or α5β1, blocked IGF-IR phosphorylation.(6) Echistatin causes an IGF-I resistance phenomenon similar to that induced by unloading, but it fails to block ligand induced PDGF receptor activation. Skeletal unloading induces a dramatic reduction of numerous integrin subunits, and despite this reduction skeletal unloading fails to block PDGF signaling.
The marked reduction of integrin subunits in unloaded BMOp cells provides a mechanism of the “memory” BMOp cells have of their previous loaded state. BMOp cells demonstrate unloading induced resistance to IGF-I both in vivo and in vitro. The memory of the unloaded state is a transient phenomenon in vitro. In recently isolated unloaded BMOp cells integrin expression is reduced, which causes IGF-I resistance and contributes to the reduction in cell colonies formed.(5) However, as these cultures grow these cells recover integrin expression, as well as IGF-I signaling and receptor phosphorylation. This recovery of unloaded cells is contemporary with BMOp differentiation and maturation, with normal alkaline phosphatase and mineralization activity per cell noted.(5) Integrin subunits are markers of functional osteoblasts.
Integrin regulation of IGF-I signaling has been described in various tissues. Impaired IGF-I receptor activation by disruption of ligand occupancy of integrins with echistatin,(6,37) and ligand independent activation of IGF-I receptor by mechanical stimulation of osteosarcoma cells with fluid flow(37) demonstrates a role of integrins in IGF-I signaling. However, a specific integrin regulator of IGF-I signaling in osteoblasts and osteoblast precursors is unknown. Studies from Clemmons and co-workers(22,23) have detailed a proposed mechanism of interaction between the integrin and IGF-I signaling pathways. In porcine vascular smooth muscle cells, integrin β3 phosphorylation recruits the phosphatase SHP-2 to the membrane and enables the transfer of SHP-2 to SHPS-1 away from the IGF-I receptor, enhancing IGF-I receptor phosphorylation and signaling. Blocking integrin β3 activation prevents SHP-2 sequestering and terminates IGF-I signaling.
The SHP-2 oriented mechanism incompletely describes the nature of integrin regulation of the IGF-I receptor in osteoblasts. We have shown that unloading induced impairment of IGF-I receptor activation is not recovered by pretreatment with the phosphatase inhibitor orthovanadate,(6) thus IGF-I resistance is not a function of increased phosphatase activity but rather a loss of kinase activity. Mechanical stimulation of osteosarcoma cells in a serum-free environment phosphorylated the IGF-I receptor, which was blocked by the disintegrin, echistatin.(37) These observations suggest activity of an integrin mediated non-receptor kinase is required for IGF-I receptor activation. Recently, IGF-I has been shown to directly bind integrin β3 and to stimulate a complex formation of the IGF-I receptor and αVβ3 integrin. IGF-1 mutated to disrupt integrin β3 binding only retained IGF-I receptor binding but failed to phosphorylate the IGF-I receptor, activate downstream AKT and ERK signaling pathways, stimulate proliferation, and stimulate integrin/IGF-I receptor complex formation.(38) This complex would allow cross talk between the two signaling pathways and recruitment of non-receptor kinases to directly phosphorylate the IGF-I receptor. Focal adhesion kinases, components of the integrin signaling cascade, are required for growth factor response, regulating response with intrinsic kinase activity or providing binding sites for other non-receptor kinases.(39,40) Specifically, FAK and PYK2 have been shown to directly interact with the insulin and IGF-I receptors.(41,42)
We used siRNA knockdown to target integrin β1 and β3 specifically to determine whether either integrin subunit regulates IGF-I signaling in BMOp cells. We did not assess the effect on PDGF signaling as unloading induced reductions in integrin expression did not affect PDGFR activation. Integrin β1 was included as a candidate subunit because it is predominately expressed in bone and, like β3, has been shown to modulate IGF-I signaling(43,44) in other cell types. Our results show that a reduction in β3 expression significantly reduced IGF-I receptor phosphorylation whereas a reduction in β1 expression had a less consistent effect. This may indicate that β3 is more critical for IGF-I signaling than β1 or that because of the high level of expression of β1 in these cells sufficient β1 levels remain after siβ1 treatment to maintain some level of IGF-I responsiveness. In either case the siRNA knockdown studies demonstrate that integrin β3, and possibly β1, function as regulators of the IGF-I receptor in rat BMOp cells that may be responsive to mechanical stimulation. The siRNA knockdown model was not equivalent to the skeletal unloading model as it was not sufficient to assess the effect of targeted integrin knockdown on signaling downstream of IGF-IR. Although IGF-IR phosphorylation was specifically manipulated by siRNA treatment, basal pERK levels were uniformly increased in response to siRNA treatment, and this lowered the sensitivity to detects changes in response to IGF-I exposure.
In conclusion, skeletal unloading causes bone loss that is associated with impaired IGF-I but not PDGF signaling. Our results indicate that the mechanism by which skeletal unloading leads to IGF-I resistance has little impact on the proliferative response to PDGF. IGF-I signaling in osteoblast precursors is more sensitive to the unloading induced reductions in integrins, especially β3, than PDGF. The integrity of PDGF signaling can be utilized as PDGF is known to stimulate osteoblast proliferation and differentiation.(45) Localized use of PDGF therapy to repair periodontal defects is a FDA approved indication, and systemic use in an osteoporotic animal model demonstrated increased osteoblast proliferation, bone formation, and BMD.(46) Potentially, PDGF may be an option to treat bone loss or prevent disuse osteoporosis in conditions of skeletal unloading like prolonged bedrest, space flight, or neurologic injury.
Acknowledgments
This work was supported by NIH grants RO1 DK54792 (DB), T32 DK07161-33 and DK07418-27 (RL), NASA grant NNA04CC67G (DB), National Space Biomedical Research Institute grant through NASA NCC 9-58 (RL), and Veterans Administration Research Enhancement Award Program (DB). We thank Scott J Munson for technical assistance.
Footnotes
Disclosures:
All authors state that they have no conflicts of interest.
Authors’ role: Study design: RL, SN, TS, BH, and DB. Study conduct: RL, SN, TK, YW, and HE. Data collection: RL, SN, TK, and TS. Data analysis: RL, SN, TK, and TS. Data interpretation: RL, SN, TS, BH, and DB. Drafting manuscript: RL and SN. Revising manuscript content: RL, SN, BH, and DB. Approving final version of manuscript: RL, SN, TK, YW, TS, HE, BH, and DB. RL takes responsibility for the integrity of the data analysis.
Contributor Information
Roger K Long, Email: roger.long@ucdmc.ucdavis.edu.
Shigeki Nishida, Email: snishida@ca2.so-net.jp.
Takuo Kubota, Email: takuo.kubota@ucsf.edu.
Yongmei Wang, Email: yongmei.wang@ucsf.edu.
Takeshi Sakata, Email: sakata@reimeikai.com.
Hashem Z Elalieh, Email: hashem.elalieh@ucsf.edu.
Bernard P Halloran, Email: Bernard.Halloran@ucsf.edu.
Daniel D Bikle, Email: Daniel.Bikle@ucsf.edu.
References
- 1.Bikle DD, Halloran BP. The response of bone to unloading. J Bone Miner Metab. 1999;17(4):233–44. doi: 10.1007/s007740050090. [DOI] [PubMed] [Google Scholar]
- 2.Globus RK, Bikle DD, Morey-Holton E. The temporal response of bone to unloading. Endocrinology. 1986;118(2):733–42. doi: 10.1210/endo-118-2-733. [DOI] [PubMed] [Google Scholar]
- 3.Halloran BP, Bikle DD, Harris J, Foskett HC, Morey-Holton E. Skeletal unloading decreases production of 1,25-dihydroxyvitamin D. Am J Physiol. 1993;264(5 Pt 1):E712–6. doi: 10.1152/ajpendo.1993.264.5.E712. [DOI] [PubMed] [Google Scholar]
- 4.Halloran BP, Bikle DD, Harris J, Tanner S, Curren T, Morey-Holton E. Regional responsiveness of the tibia to intermittent administration of parathyroid hormone as affected by skeletal unloading. J Bone Miner Res. 1997;12(7):1068–74. doi: 10.1359/jbmr.1997.12.7.1068. [DOI] [PubMed] [Google Scholar]
- 5.Sakata T, Halloran BP, Elalieh HZ, Munson SJ, Rudner L, Venton L, Ginzinger D, Rosen CJ, Bikle DD. Skeletal unloading induces resistance to insulin-like growth factor I on bone formation. Bone. 2003;32(6):669–80. doi: 10.1016/s8756-3282(03)00088-7. [DOI] [PubMed] [Google Scholar]
- 6.Sakata T, Wang Y, Halloran BP, Elalieh HZ, Cao J, Bikle DD. Skeletal unloading induces resistance to insulin-like growth factor-I (IGF-I) by inhibiting activation of the IGF-I signaling pathways. J Bone Miner Res. 2004;19(3):436–46. doi: 10.1359/JBMR.0301241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matsumoto T, Nakayama K, Kodama Y, Fuse H, Nakamura T, Fukumoto S. Effect of mechanical unloading and reloading on periosteal bone formation and gene expression in tail-suspended rapidly growing rats. Bone. 1998;22:89S–93S. doi: 10.1016/s8756-3282(98)00018-0. [DOI] [PubMed] [Google Scholar]
- 8.Dehority W, Halloran BP, Bikle DD, Curren T, Kostenuik PJ, Wronski TJ, Shen Y, Rabkin B, Bouraoui A, Morey-Holton E. Bone and hormonal changes induced by skeletal unloading in the mature male rat. Am J Physiol. 1999;276:E62–E69. doi: 10.1152/ajpendo.1999.276.1.e62. [DOI] [PubMed] [Google Scholar]
- 9.Machwate M, Zerath E, Holy X, Hott M, Modrowski D, Malouvier A, Marie PJ. Skeletal unloading in rat decreases proliferation of rat bone and marrow-derived osteoblastic cells. Am J Physiol. 199(264):E790–E799. doi: 10.1152/ajpendo.1993.264.5.E790. [DOI] [PubMed] [Google Scholar]
- 10.Zhang R, Supowit SC, Klein GL, Lu Z, Christensen MD, Lozano R, Simmons DJ. Rat tail suspension reduces messenger RNA level for growth factors and osteopontin and decreases the osteoblastic differentiation of bone marrow stromal cells. J Bone Miner Res. 1995;10(3):415–423. doi: 10.1002/jbmr.5650100312. [DOI] [PubMed] [Google Scholar]
- 11.Kostenuik PJ, Halloran BP, Morey-Holton ER, Bikle DD. Skeletal unloading inhibits the in vitro proliferation and differentiation of rat osteoprogenitor cells. Am J Physiol. 1997;273:E1133–E1139. doi: 10.1152/ajpendo.1997.273.6.e1133. [DOI] [PubMed] [Google Scholar]
- 12.Van Wyk JJ, Smith EP. Insulin-like growth factors and skeletal growth: Possibilities for therapeutic interventions. J Clin Endocrinol Metab. 1999;84:4349–4354. doi: 10.1210/jcem.84.12.6201. [DOI] [PubMed] [Google Scholar]
- 13.LeRoith D. Insulin-like growth factor I receptor signaling-overlapping or redundant pathways? Endocrinology. 2000;141:1287–1288. doi: 10.1210/endo.141.4.7475. [DOI] [PubMed] [Google Scholar]
- 14.Canalis E. Effect of insulin like growth factor I on DNA and protein synthesis in cultured rat calvaria. J Clin Invest. 1980;66:709–719. doi: 10.1172/JCI109908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hock JM, Centrella M, Canalis E. Insulin-like growth factor I has independent effects on bone matrix formation and cell replication. Endocrinology. 1988;122:254–260. doi: 10.1210/endo-122-1-254. [DOI] [PubMed] [Google Scholar]
- 16.Canalis E, McCarthy T, Centrella M. Isolation and characterization of insulin-like growth factor I (somatomedin-C) from cultures of fetal rat calvariae. Endocrinology. 1988;122:22–27. doi: 10.1210/endo-122-1-22. [DOI] [PubMed] [Google Scholar]
- 17.Wergedal JE, Mohan S, Lundy M, Baylink DJ. Skeletal growth factor and other growth factors known to be present in bone matrix stimulate proliferation and protein synthesis in human bone cells. J Bone Miner Res. 1990;5(2):179–186. doi: 10.1002/jbmr.5650050212. [DOI] [PubMed] [Google Scholar]
- 18.Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH. Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocr Rev. 2001;22:153–183. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- 19.Borgatti P, Martelli AM, Bellacosa A, Casto R, Massari L, Capitani S, Neri LM. Translocation of Akt/PKB to the nucleus of osteoblast-like MC3T3-E1 cells exposed to proliferative growth factors. FEBS Lett. 200(477):27–32. doi: 10.1016/s0014-5793(00)01758-0. [DOI] [PubMed] [Google Scholar]
- 20.Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- 21.Franke TF, Kaplan DR, Cantley LC. PI3K: Downstream AKTion blocks apoptosis. Cell. 1997;88:435–437. doi: 10.1016/s0092-8674(00)81883-8. [DOI] [PubMed] [Google Scholar]
- 22.Zheng B, Clemmons DR. Blocking ligand occupancy of the αVβ3 integrin inhibits insulin-like growth factor I signaling in vascular smooth muscle cells. Proc Natl Acad Sci USA. 1998;95:11217–11222. doi: 10.1073/pnas.95.19.11217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clemmons DR, Horvitz G, Engleman W, Nichols T, Moralez A, Nickols GA. Synthetic αVβ3 antagonists inhibit insulin-like growth factor-I-stimulated smooth muscle cell migration and replication. Endocrinology. 1999;140:4616–4621. doi: 10.1210/endo.140.10.7027. [DOI] [PubMed] [Google Scholar]
- 24.Wierzbicka-Patynowski I, Niewiarowski S, Marcinkiewicz C, Calvete JJ, Marcinkiewicz MM, McLane MA. Structural requirements of echistatin for the recognition of αVβ3 and α5β1 integrins. J Biol Chem. 1999;274(53):37809–37814. doi: 10.1074/jbc.274.53.37809. [DOI] [PubMed] [Google Scholar]
- 25.O’Sullivan S, Naot D, Callon K, Porteous F, Horne A, Wattie D, Watson M, Cornish J, Browett P, Grey A. Imatinib promotes osteoblast differentiation by inhibiting PDGFR signaling and inhibits osteoclastogenesis by both direct and stromal cell-dependent mechanisms. J Bone Miner Res. 2007;22(11):1679–89. doi: 10.1359/jbmr.070719. [DOI] [PubMed] [Google Scholar]
- 26.Centrella M, McCarthy TL, Canalis E. Platelet-derived growth factor enhances DNA and collagen synthesis in osteoblast-enriched cultures from fetal rat parietal bone. Endocrinology. 1989;125:13–19. doi: 10.1210/endo-125-1-13. [DOI] [PubMed] [Google Scholar]
- 27.Howes R, Bowness JM, Grotendorst GR, Martin GR, Reddi AH. Platelet-derived growth factor enhances demineralized bone matrix-induced cartilage and bone formation. Cal Tiss Intl. 1988;42:34–38. doi: 10.1007/BF02555836. [DOI] [PubMed] [Google Scholar]
- 28.Rasubala L, Yoshikawa H, Nagata K, Iijima T, Ohishi M. Platelet-derived growth factor and bone morphogenetic protein in the healing of mandibular fractures in rats. Br J Oral Maxillofac Surg. 2003;41(3):173–8. doi: 10.1016/s0266-4356(03)00075-5. [DOI] [PubMed] [Google Scholar]
- 29.Ahdjoudj S, Kaabeche K, Holy X, Fromigue O, Modrowski, Zerath E, Marie PJ. Transforming growth factor-B inhibits CCAAT/enhancer-binding protein expression and PPARg activity in unloaded bone marrow stromal cells. Exp Cell Res. 2005;303:138–147. doi: 10.1016/j.yexcr.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 30.Canalis E, McCarthy TL, Centrella M. Effects of desamino-(1–3)-insulin-like growth factor I on bone cell function in rat calvarial cultures. Endocrinology. 1991;129(1):534–41. doi: 10.1210/endo-129-1-534. [DOI] [PubMed] [Google Scholar]
- 31.Giancotti FG, Ruoslahti E. Integrin signaling. Science. 1999;285:1028–1032. doi: 10.1126/science.285.5430.1028. [DOI] [PubMed] [Google Scholar]
- 32.Hughes DE, Salter DM, Dedhar S, Simpson R. Integrin expression in human bone. J Bone Miner Res. 1993;8(5):527–533. doi: 10.1002/jbmr.5650080503. [DOI] [PubMed] [Google Scholar]
- 33.Gronthos S, Stewart K, Graves SE, Hay S, Simmons PJ. Integrin expression and function on human osteoblast-like cells. J Bone Miner Res. 1997;12(8):1189–1197. doi: 10.1359/jbmr.1997.12.8.1189. [DOI] [PubMed] [Google Scholar]
- 34.Halloran BP, Bikle DD, Cone CM, Morey-Holton E. Glucocorticoids and inhibition of bone formation induced by skeletal unloading. Am J Phys. 1988;255:E875–879. doi: 10.1152/ajpendo.1988.255.6.E875. [DOI] [PubMed] [Google Scholar]
- 35.Bikle DD, Harris J, Halloran BP, Morey-Holton ER. Skeletal unloading induces resistance to insulin-like growth factor I. J Bone Miner Res. 1994;9(11):1789–1796. doi: 10.1002/jbmr.5650091116. [DOI] [PubMed] [Google Scholar]
- 36.Kostenuik PJ, Harris J, Halloran BP, Turner RT, Morey-Holton ER, Bikle DD. Skeletal unloading causes resistance of osteoprogenitor cells to parathyroid hormone and to insulin-like growth factor-I. J Bone Miner Res. 1999;14(1):21–31. doi: 10.1359/jbmr.1999.14.1.21. [DOI] [PubMed] [Google Scholar]
- 37.Kapur S, Mohan S, Baylink DJ, Lau KHW. Fluid shear stress synergizes with insulin-like growth factor-I on osteoblast proliferation through integrin-dependent activation of IGF-I mitogenic signaling pathway. J Biol Chem. 2005;280(20):20163–70. doi: 10.1074/jbc.M501460200. [DOI] [PubMed] [Google Scholar]
- 38.Saegusa J, Yamaji S, Ieguchi K, Wu C, Lam KS, Liu F, Takada YK, Takada Y. The direct binding of insulin-like growth factor-I to integrin αVβ3 is involved in IGF-I signaling. J Biol Chem. 2009;284(36):24106–14. doi: 10.1074/jbc.M109.013201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hauck CR, Hsia DA, Schlaepfer DD. Focal adhesion kinase facilitates platelet-derived growth factor-BB-stimulated ERK2 activation required for chemotaxis migration of vascular smooth muscle cells. J Biol Chem. 2000;275(52):41092–9. doi: 10.1074/jbc.M005450200. [DOI] [PubMed] [Google Scholar]
- 40.Sieg DJ, Hauck CR, Ilic D, Klingbeil CK, Schaefer E, Damsky CH, Schlaepfer DD. FAK integrates growth-factor and integrin signals to promote cell migration. Nat Cell Biol. 2000;2(5):249–56. doi: 10.1038/35010517. [DOI] [PubMed] [Google Scholar]
- 41.Sekimoto H, Eipper-Mains J, Pond-Tor S, Boney CM. αVβ3 integrin and Pyk2 mediate insulin-like growth factor I activation of Src and mitogen-activated protein kinase in 3T3-L1 cells. Mol Endocrinol. 2005;19(7):1859–1867. doi: 10.1210/me.2004-0481. [DOI] [PubMed] [Google Scholar]
- 42.Baron V, Calleja V, Ferrari P, Alengrin F, VanObberghen E. p125FAK focal adhesion kinase is a substrate for the insulin and insulin-like growth factor I tyrosine kinase receptors. J Biol Chem. 1998;273(12):7162–7168. doi: 10.1074/jbc.273.12.7162. [DOI] [PubMed] [Google Scholar]
- 43.Goel HL, Fornaro M, Moro L, Teider N, Rhim JS, King M, Languino LR. Selective modulation of type 1 insulin-like growth factor receptor signaling and functions by β1 integrins. J Cell Biol. 2004;166(3):407–418. doi: 10.1083/jcb.200403003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goel HL, Breen M, Zhang J, Das I, Aznavoorian-Cheshire S, Greenberg NM, Elgavish A, Languino LR. β1A integrin expression is required for type 1 insulin-like growth factor receptor mitogenic and transforming activities and localization to focal contacts. Cancer Res. 2005;65(15):6692–6700. doi: 10.1158/0008-5472.CAN-04-4315. [DOI] [PubMed] [Google Scholar]
- 45.Mehrotra M, Krane SM, Walters K, Pilbeam C. Differential regulation of platelet-derived growth factor stimulated migration and proliferation in osteoblastic cells. J Cell Biochem. 2004;93(4):741–52. doi: 10.1002/jcb.20138. [DOI] [PubMed] [Google Scholar]
- 46.Mitlak BH, Finkelman RD, Hill EL, Li J, Martin B, Smith T, D’Andrea M, Antoniades HN, Lynch SE. The effect of systemically administered PDGF-BB on the rodent skeleton. J Bone Min Res. 1996;11(2):238–47. doi: 10.1002/jbmr.5650110213. [DOI] [PubMed] [Google Scholar]