

Abstract
The Ts65Dn mouse is the best-studied animal model for Down syndrome. In the experiments described here, NMDA-mediated or mGluR-mediated LTD was induced in the CA1 region of hippocampal slices from Ts65Dn and euploid control mice by bath application of 20 µM NMDA for 3 min and 50 µM DHPG for 5 min, respectively. We found that Ts65Dn mice display exaggerated NMDA-induced, but not mGluR-induced, LTD in the CA1 region of the hippocampus compared with euploid control animals. In addition, this abnormal level of LTD can be pharmacologically rescued by the NMDA receptor antagonist memantine.
Down syndrome (DS) and fragile X syndrome (FXS) are, respectively, the most common genetically defined and the most commonly inherited causes of intellectual disability. Together, these two genetic disorders account for over half a million affected persons in the United States (Canfield et al. 2006; Hagerman 2008). Biologically, the two syndromes are generally thought to have very little in common. DS is caused by a supernumerary chromosome 21 and consequent overexpression of the genes located in this chromosome (Lejeune 1959). Whereas FXS is typically produced by an expansion of a CGG triplet repeat sequence upstream of the FMR1 gene, which results in transcriptional silencing of FMRP, the fragile X mental retardation protein (Fu et al. 1991).
The work by Huber et al. (2002) has been an important step toward the understanding of the pathophysiology of the cognitive deficits associated with FXS. These investigators have shown that a null mutant mouse lacking FMRP displays exaggerated metabotropic glutamate receptor (mGluR)-dependent long-term synaptic depression (LTD) in area CA1 of the hippocampus, but no alteration on the levels of N-methyl-D-aspartate (NMDA) receptor (NMDAR)-dependent LTD. These findings are consistent with the so-called “mGluR hypothesis of FXS,” which posits that FMRP normally regulates protein synthesis downstream from Group I mGluRs (mGluR1 and mGluR5) and, in its absence, mGluR-dependent protein synthesis becomes abnormal (Bear et al. 2004). Interestingly, exaggerated LTD has also been found in the same area of the hippocampus of the Ts65Dn mouse (Siarey et al. 1999), which is the most widely studied animal model for DS (for review, see Patterson and Costa 2005; Costa 2011; Liu et al. 2011). However, to date, no attempt has been made to pharmacologically identify whether there is any neurotransmitter receptor specificity to this phenomenon, similar to what has been done for the FMR1 null mutant mouse. Therefore, the present study was designed to fill this critical knowledge gap.
Our research team has found previously that the Ts65Dn mouse displays pharmacological responses consistent with dysfunction in molecular pathways coupled to the gating of NMDARs (Costa et al. 2008; Siddiqui et al. 2008). Proteins encoded by nine genes located on chromosome 21 and in the Ts65Dn trisomic segment (APP, TIAM1, BACH1, SOD1, SYNJ1, ITSN1, RCAN1, DYRK1A, and PCP4) have been shown to interact either directly with NMDARs, or indirectly via the protein phosphatase calcineurin (CaN) (Siddiqui et al. 2008; Gardiner 2010). Inhibition of CaN activity alters NMDAR gating kinetics (Lieberman and Mody 1994) and causes increased sensitivity to the stimulatory locomotor actions of the NMDAR antagonist MK-801 (Miyakawa et al. 2003). The Ts65Dn and Ts1Cje mouse models of DS display similar hypersensitivity to these psychotomimetic actions of MK-801 (Costa et al. 2008; Siddiqui et al. 2008), which supports the hypothesis that CaN is inhibited in these animal models and that, consequently, NMDAR function may indeed be altered. In addition, we also have found that acute doses of the low-affinity uncompetitive NMDAR antagonist memantine rescue learning and memory deficits in Ts65Dn mice in a fear-conditioning test (Costa et al. 2008). More recently, two separate research groups have confirmed memantine's memory/learning enhancing effects on Ts65Dn mice (Lockrow et al. 2010; Rueda et al. 2010). Therefore, we have hypothesized that, differently from the FMR1 null mutant mouse, an NMDAR-dependent mechanism might underlie the exaggerated LTD seen in Ts65Dn mice. Moreover, we have hypothesized that the drug memantine might be able to rescue this altered level of synaptic plasticity in these animals.
The original production of the segmental trisomy Ts65Dn has been well described in the literature (Reeves et al. 1995; Costa et al. 1999). Experimental mice used here were generated by repeated back-crossing of Ts65Dn females to C57BL/6JEiJ × C3Sn.BLiA-Pde6b + /D F1 hybrid males (as described by Costa et al. 2010) in colonies at the University of Colorado Anschutz Medical Campus or The Jackson Laboratory. The euploid littermates of Ts65Dn mice were used as controls. Animals from the same litter and sex were housed in the same cage and maintained in a 12:12 h light/dark schedule (lights on at 7:00 am) with free access to food and water. Only males (aged 6–8 mo) were used in this study. The experimental methods have received approval of the University of Colorado Anschutz Medical Campus Animal Care and Use Committee.
Adult Ts65Dn and euploid littermate control mice aged 6–8 mo were used in this experiment. Mice were decapitated under halothane anesthesia, and the brains were rapidly harvested in ice-cold dissecting artificial cerebral spinal fluid (aCSF) (in mM: 210 Sucrose, 2.5 KCl, 1 CaCl2, 7 MgSO4, 1.5 NaH2PO4, 26 NaHCO3, and 10 D-glucose, saturated with 95% O2 and 5% CO2) and cut with a vibrating blade microtome (VT 1000s, Leica) into transverse slices 400-µm thick. The hippocampus was then dissected out and the CA3 region was removed. Slices were moved to a holding chamber containing aCSF (in mM: 120 NaCl, 3.5 KCl, 2.5 CaCl2, 1.3 MgSO4, 1.25 NaH2PO4, 26 NaHCO3, and 10 D-glucose, saturated with 95% O2 and 5% CO2) and allowed to recover for at least 1 h at room temperature. Half of the slices were then moved to another holding chamber with aCSF containing 1 µM memantine. Both sets of hippocampus slices were allowed to recover for an additional 4 h before being placed in the recording chamber. The concentration of memantine used in this study was derived from published data indicating that at steady-state (chronic treatment for several weeks) and therapeutic doses (typically 20–30 mg/day), plasma levels of memantine are in a range of from 0.4 to 1 µM (Kornhuber and Quack 1995; Danysz et al. 1997; Periclou et al. 2006). Also, at the concentration of 1 µM memantine, Chen and Lipton (1997) demonstrated ∼50% inhibition of NMDA-gated whole-cell currents recorded from retinal ganglion cells, voltage clamped at −60 mV, and elicited by a saturating (200 µM) concentration step of NMDA applied through a fast superfusion system (flow pipes). Thus, at 1 µM memantine, it is unlikely that the occupancy of NMDARs will be close to saturation levels.
In the present study, a customized electrophysiology setup was designed to allow for simultaneous recordings of treated (memantine) and untreated hippocampal slices from the same animal. In all experiments, we used only one hippocampal slice per animal per condition and, consistently, n = 12. Field excitatory postsynaptic potentials (fEPSP) were recorded with nichrom recording electrodes through thin-walled borosilicate glass micropipettes (3–5 MΩ resistance, 1.5 mm, WPI) filled with aCSF. The micropipette was inserted in to the apical dendritic layer of the CA1 region of the hippocampus. Two fine bipolar Pl/Ir electrodes (FHC) were inserted in the CA1 stratum radiatum on opposite sides of the recording pipette (electrodes were designated S1 and S2). These electrodes provided alternative ways of stimulating the Schaffer collateral pathway and testing the consistency of the fEPSP recordings (the data shown here were produced via stimulation of the S1 electrode; see Supplemental data for S2-generated recordings). A stable baseline of synaptic transmission was established for 20 min prior to induction of LTD. Stimulation intensity was adjusted to 40%–50% of the amplitude required to producing population spikes. Signals from the recording electrode were amplified 2000–5000 times (Brownlee Precision Electrophysiology Amplifier with signal conditioner, Model 440), low-pass filtered at 2 kHz, and digitized by a Digidata digitizer (1322A, Axon Instruments) into a PC microcomputer. PCLAMP software (Version 8.1, Axon Instruments) was used for data acquisition, and analysis was performed off-line. After establishing a stable baseline, either 20 µM NMDA was bath applied for 3 min to induce NMDA-mediated LTD, or 50 µM group I mGluR agonist [3,5-RS] dihydroxyphenylhydrazine (DHPG) was bath applied for 5 min to induce mGluR-mediated LTD, and fEPSP's were recorded for an additional 60 min. Data for time points 0–14 min in Figure 1 (and Supplemental Fig. 1) have been omitted to help make the graphs easier to read. Analysis of time points for 0–3 min for NMDA-mediated LTD, and 0–5 min for mGluR-mediated LTD did not indicate significant differences in the dynamic parameters of LTD induction between control and Ts65Dn mice (supplemental Tables 1 and 2).
Figure 1.

LTD induced by bath application of either NMDA (5 µM, 3 min) or DHPG (20 µM, 5 min). (A,B) Normalized fEPSP slopes before and after bath application of NMDA. Notice that the hippocampal slices from Ts65Dn mice displayed larger levels of NMDA-induced LTD than those from the euploid control animals, and that preincubation with memantine (1 µM, 4 h) rescued this phenotype. (C,D) Normalized fEPSP slopes before and after bath application of DHPG. Here, the levels of LTD in Ts65Dn mice and euploid control animals are indistinguishable with or without preincubation with memantine. (In A–D, calibration bars represent 1 mV and 10 msec.) (E) Summary graph representation of the ANOVA of the LTD data (mean fEPSP depression ± SEM; with fEPSP depression = the average baseline fEPSP slope—average fEPSP slope during the last 10 min of recording for each hippocampus slice) in A and C showing which differences reached significance in the post hoc analyses. (F) Summary graph representation of the ANOVA of the LTD data (mean ± SEM) in B and D. Statistical significance is expressed in this figure: (**) P < 0.01.
Figure 1A depicts LTD produced by a 3-min bath application of 20 µM NMDA on the hippocampal slices. The resulting data showed that slices from Ts65Dn mice displayed an exaggerated LTD compared with euploid control animals. In contrast, we found that a 5-min bath application of the 50 µM group I mGluR agonist DHPG produced indistinguishable levels of LTD in slices of Ts65Dn and control mice (Fig. 1B). Interestingly, additional experiments showed that the exaggerated level of NMDA-induced LTD could be rescued pharmacologically by preincubation of the slices in 1 µM memantine (Fig. 1C). However, preincubation with memantine at this same concentration did not produce any significant change in the level of DHPG-induced LTD in slices from either Ts65Dn or littermate control mice (Fig. 1D). The bar graphs in Figure 1E summarize Analysis of Variance (ANOVA) results for the mean levels of NMDA-induced LTD (expressed as the mean percentage of the baseline level ± SEM) in four groups of hippocampal slices shown in Figure 1A and C: (1) untreated control; (2) untreated Ts65Dn; (3) control preincubated with memantine; and (4) Ts65Dn preincubated with memantine. We found a significant group effect (F(3,44) = 4.077, P = 0.0122), and Fisher's PLSD post hoc comparisons revealed a significant difference between untreated control and untreated Ts65Dn slices without memantine (P = 0.0027), but no significant difference between untreated control slices and Ts65Dn slices preincubated in memantine (P = 0.7422). Figure 1F summarizes the ANOVA of DHPG-induced LTD data (i.e., the results shown in Fig. 1B,D). As can be inferred from the inspection of the figures, we found no significant group effect (F(3,44) = 0.827, P = 0.4862) in these experiments.
In order to investigate other potential effects of memantine on basic synaptic function of Ts65Dn and euploid control mice, we performed assessments of input/output (I/O) relationships and paired-pulse facilitation (PPF) for the four groups of hippocampal slices, i.e., untreated control, untreated Ts65Dn, control preincubated with memantine, and Ts65Dn preincubated with memantine (n = 12 for each group). Analysis of the I/O function plot by repeated measures ANOVA showed no significant group differences (F(3,44) = 0.8243; P = 0.7115) (Fig. 2A), nor did it indicate interaction between group and stimulus intensity (F(27,396) = 0.5447; P = 0.9709). However, as expected, there was a significant stimulus intensity effect (F(9,396) = 139.09; P < 0.001). We also investigated PPF, which is a form of short-lived plasticity sensitive to presynaptic changes. In a subset of slices, PPF was produced by delivering two stimuli of identical strength at different interpulse intervals to assess the integrity of presynaptic mechanisms in Ts65Dn mice. PPF was calculated as the slope ratio between the second and first stimuli. Analysis of the data showed no significant group effect (F(3,44) = 0.431; P = 0.7315) (Fig. 2B) or interaction between genotype and interpulse interval (F(10,440) = 0.747; P = 0.8335) in the range tested. A strongly significant interpulse interval effect was found (F(10,440) = 138.011; P < 0.001).
Figure 2.

Preincubation with memantine had no significant effect on basal synaptic transmission in slices from either euploid control or Ts65Dn mice. (A) Input–output function at the CA3–CA1 hippocampal synapse for untreated euploid control and Ts65Dn hippocampal slices and control, and Ts65Dn slices pre-incubated with memantine. Input intensities were set at 10, 20, 30, 40, 50, 60, 70, 80, 90, and 100 µA. Repeated measures ANOVA showed no significant group effect. (B) Paired-pulse facilitation was assessed at 11 different interpulse intervals: 25, 50, 75, 100, 125, 150, 175, 200, 300, 400, and 500 msec in the four groups of slices (i.e., untreated control, untreated Ts65Dn, control preincubated with memantine, and Ts65Dn preincubated with memantine). Again, repeated measures ANOVA failed to reveal a significant group effect. (n = 12 for each group in all experiments.)
The finding of an exaggerated NMDA-mediated LTD (but not mGluR-mediated LTD) in the CA1 region of the hippocampus of the mouse model of DS Ts65Dn may have interesting consequences in terms of shaping the way we think of the pathophysiology of genetic disorders leading to intellectual disability. As illustrated in Figure 3, one can picture a scenario in which two different genetic insults can lead to essentially the same phenotypic outcome, i.e., exaggerated LTD (likely due to increased internalization of AMPA receptors). However, this common outcome may be reached through very different mechanisms in DS vs. FXS. For DS, it would be the consequence of dysfunction in molecular pathways associated with the induction of NMDA-dependent LTD and, for FXS, it is the result of altered mGluR-dependent protein synthesis.
Figure 3.
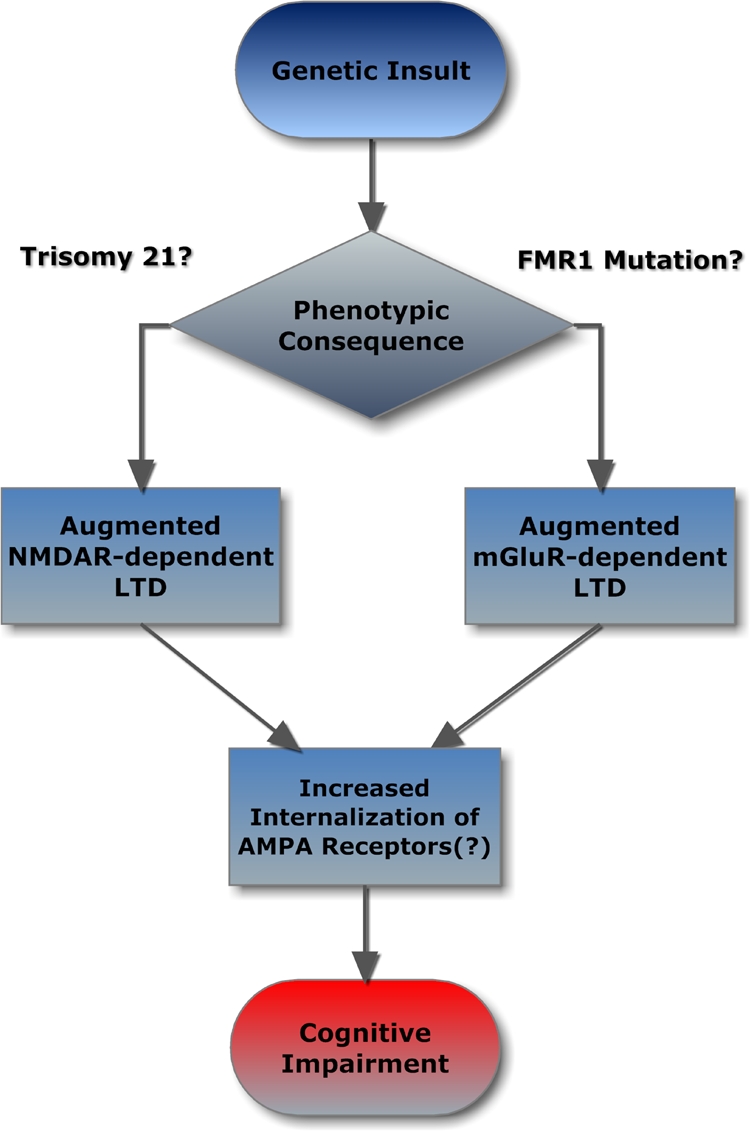
Flowchart describing how different genetic insults (trisomy 21 or FMR1 gene mutation) can produce functionally similar phenotypes (exaggerated LTD) through unrelated, parallel mechanisms.
Post-translational modification of AMPAR leading to decreases in open channel probability and increased AMPAR internalization in response to LTD-inducing stimuli have firmly been implicated in NMDA-dependent LTD for more than a decade (for classic reviews, see Malinow and Malenka 2002; Malenka and Bear 2004). Although there are clear examples of LTP and LTD of NMDAR-mediated currents in the literature (Selig et al. 1995; Montgomery et al. 2005; Morishita et al. 2005), it is unlikely that changes in the NMDAR component of fEPSPs could account for the altered levels of LTD in the Ts65Dn mice described here. This is simply due to the fact that the non-NMDA current component is responsible for the vast majority of fEPSPs under typical electrical stimulation frequencies and intensities and physiological concentrations of Mg2+ (Nishikawa and Maclver 2000). Obviously, further studies specifically designed to quantify receptor post-translational modification and internalization will be needed to test whether altered NMDAR activity-mediated internalization of AMPA receptors in Ts65Dn mice is indeed responsible for the augmented levels of LTD seen in these mice in the present study.
The overarching hypothesis behind the present work is that the exaggerated NMDAR-dependent LTD observed in hippocampal slices from Ts65Dn mice is likely to be the result of direct and indirect interactions of key proteins encoded by trisomic chromosome 21 homologous genes with NMDAR, leading to increased channel opening probability. An alternative, but not mutually exclusive hypothesis would involve altered NMDAR subunit composition in Ts65Dn mice. In support of this hypothesis is the recently reported increased NR2A expression in the cerebellum of the TgDyrk1A mouse model of DS (Altafaj et al. 2008). In addition, Roberson et al. (2008) proposed a potential role for impaired trafficking of the NR2B subunit in Ts65Dn mice based on their observation of a significant decrease in the levels of the kinesin motor protein, KIF17, compared with euploid control mice. However, these authors, as well as a more recent and more comprehensive study by Fernandez et al. (2009), reported no significant differences in either NR2A or NR2B levels in Ts65Dn mice compared with euploid control mice. Finally, studies by Kleschevnikov et al. (2004), Costa and Grybko (2005), and Fernandez et al. (2007) have provided evidence of increased γ-aminobutyric acid (GABA) receptor A (GABAAR)-mediated inhibition and/or altered plasticity of the inhibitory circuitry in Ts65Dn mice compared with euploid control animals. Under these circumstances, one could hypothesize that such enhanced GABAAR -mediated synaptic activity might shift neuroplasticity thresholds in favor of LTD at the expense of LTP in Ts65Dn mice. However, such a hypothesis is in direct contradiction with the experimentally observed finding here that the increased levels of LTD in Ts65Dn mice can be rescued pharmacologically by the NMDAR antagonist memantine.
In the present work, we propose that the pharmacological rescue of the exaggerated level of LTD in Ts65Dn mice by memantine, which has no effect on the level of DHPG-induced LTD, provides a potential mechanism by which memantine exerts its memory/learning enhancing actions in Ts65Dn mice. Furthermore, these data have their own consequences in terms of rationales for potential pharmacotherapeutic interventions in DS vis-à-vis FXS. For example, these findings provide additional support to the idea that memantine may have disorder-specific therapeutic value on enhancing the cognitive abilities of persons with DS (which we are exploring currently through a single-site investigator-initiated pilot clinical trial; NCT01112683 at http://www.clinicaltrials.gov). In contrast, findings from a small clinical trial in FXS with the Novartis mGluR5 antagonist, AFQ056, have been published (Jacquemont et al. 2011). Although no significant effects of treatment on the primary outcome measure were found, post hoc analysis of the data showed that a subgroup of seven of the patients with full FMR1 promoter methylation and no detectable FMR1 mRNA in blood cells showed statistically significant improvement in several measures after AFQ056 treatment when compared with placebo.
One should notice that the underlying biology of either DS or FXS is very complex. For example, it is estimated that up to 4% of brain mRNAs may bind FMRP (Brown et al. 2001), whereas trisomy 21 results in the overexpression of 1% of the genes in the entire genome (Sturgeon and Gardiner 2011). Therefore, the same way that the “mGluR hypothesis of FXS” is likely to only account for a portion of the neurodevelopmental phenotype associated with FXS, NMDAR-dependent mechanisms are sure to represent only one of many important factors underlying the neurodevelopmental and neurodegenerative aspects of DS.
Acknowledgments
Support was provided by a CCTSI TL1 fellowship, Linda Crnic Institute, Coleman Institute, and NIH grant HD056235.
Footnotes
[Supplemental material is available for this article.]
References
- Altafaj X, Ortiz-Abalia J, Fernandez M, Potier MC, Laffaire J, Andreu N, Dierssen M, Gonzalez-Garcia C, Cena V, Marti E, et al. 2008. Increased NR2A expression and prolonged decay of NMDA-induced calcium transient in cerebellum of TgDyrk1A mice, a mouse model of Down syndrome. Neurobiol Dis 32: 377–384 [DOI] [PubMed] [Google Scholar]
- Bear MF, Huber KM, Warren ST 2004. The mGluR theory of fragile X mental retardation. Trends Neurosci 27: 370–377 [DOI] [PubMed] [Google Scholar]
- Brown V, Jin P, Ceman S, Darnell JC, O'Donnell WT, Tenenbaum SA, Jin X, Feng Y, Wilkinson KD, Keene JD, et al. 2001. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell 107: 477–487 [DOI] [PubMed] [Google Scholar]
- Canfield MA, Honein MA, Yuskiv N, Xing J, Mai CT, Collins JS, Devine O, Petrini J, Ramadhani TA, Hobbs CA, et al. 2006. National estimates and race/ethnic-specific variation of selected birth defects in the United States, 1999–2001. Birth Defects Res A Clin Mol Teratol 76: 747–756 [DOI] [PubMed] [Google Scholar]
- Chen HS, Lipton SA 1997. Mechanism of memantine block of NMDA-activated channels in rat retinal ganglion cells: Uncompetitive antagonism. J Physiol 499: 27–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa AC 2011. On the promise of pharmacotherapies targeted at cognitive and neurodegenerative components of Down syndrome. Dev Neurosci. 10.1159/000330861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa AC, Grybko MJ 2005. Deficits in hippocampal CA1 LTP induced by TBS but not HFS in the Ts65Dn mouse: A model of Down syndrome. Neurosci Lett 382: 317–322 [DOI] [PubMed] [Google Scholar]
- Costa AC, Walsh K, Davisson MT 1999. Motor dysfunction in a mouse model for Down syndrome. Physiol Behav 68: 211–220 [DOI] [PubMed] [Google Scholar]
- Costa AC, Scott-McKean JJ, Stasko MR 2008. Acute injections of the NMDA receptor antagonist memantine rescue performance deficits of the Ts65Dn mouse model of Down syndrome on a fear conditioning test. Neuropsychopharmacology 33: 1624–1632 [DOI] [PubMed] [Google Scholar]
- Costa AC, Stasko MR, Schmidt C, Davisson MT 2010. Behavioral validation of the Ts65Dn mouse model for Down syndrome of a genetic background free of the retinal degeneration mutation Pde6b(rd1). Behav Brain Res 206: 52–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danysz W, Parsons CG, Kornhuber J, Schmidt WJ, Quack G 1997. Aminoadamantanes as NMDA receptor antagonists and antiparkinsonian agents–preclinical studies. Neurosci Biobehav Rev 21: 455–468 [DOI] [PubMed] [Google Scholar]
- Fernandez F, Morishita W, Zuniga E, Nguyen J, Blank M, Malenka RC, Garner CC 2007. Pharmacotherapy for cognitive impairment in a mouse model of Down syndrome. Nat Neurosci 10: 411–413 [DOI] [PubMed] [Google Scholar]
- Fernandez F, Trinidad JC, Blank M, Feng DD, Burlingame AL, Garner CC 2009. Normal protein composition of synapses in Ts65Dn mice: A mouse model of Down syndrome. J Neurochem 110: 157–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu YH, Kuhl DP, Pizzuti A, Pieretti M, Sutcliffe JS, Richards S, Verkerk AJ, Holden JJ, Fenwick RG Jr, Warren ST, et al. 1991. Variation of the CGG repeat at the fragile X site results in genetic instability: Resolution of the Sherman paradox. Cell 67: 1047–1058 [DOI] [PubMed] [Google Scholar]
- Gardiner KJ 2010. Molecular basis of pharmacotherapies for cognition in Down syndrome. Trends Pharmacol Sci 31: 66–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman PJ 2008. The fragile X prevalence paradox. J Med Genet 45: 498–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber KM, Gallagher SM, Warren ST, Bear MF 2002. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci 99: 7746–7750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemont S, Curie A, des Portes V, Torrioli MG, Berry-Kravis E, Hagerman RJ, Ramos FJ, Cornish K, He Y, Paulding C, et al. 2011. Epigenetic modification of the FMR1 gene in fragile X syndrome is associated with differential response to the mGluR5 antagonist AFQ056. Sci Transl Med 3: 64ra61 10.1126/scitranslmed.3001708 [DOI] [PubMed] [Google Scholar]
- Kleschevnikov AM, Belichenko PV, Villar AJ, Epstein CJ, Malenka RC, Mobley WC 2004. Hippocampal long-term potentiation suppressed by increased inhibition in the Ts65Dn mouse, a genetic model of Down syndrome. J Neurosci 24: 8153–8160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornhuber J, Quack G 1995. Cerebrospinal fluid and serum concentrations of the N-methyl-D-aspartate (NMDA) receptor antagonist memantine in man. Neurosci Lett 195: 137–139 [DOI] [PubMed] [Google Scholar]
- Lejeune J 1959. Le mongolism: Premier exemple d'aberration autosomique humaine. Ann Genet 1: 1–49 [Google Scholar]
- Lieberman DN, Mody I 1994. Regulation of NMDA channel function by endogenous Ca(2+)-dependent phosphatase. Nature 369: 235–239 [DOI] [PubMed] [Google Scholar]
- Liu C, Belichenko PV, Zhang L, Fu D, Kleschevnikov AM, Baldini A, Antonarakis SE, Mobley WC, Yu YE 2011. Mouse models for Down syndrome-associated developmental cognitive disabilities. Dev Neurosci. 10.1159/000329422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockrow J, Boger H, Bimonte-Nelson H, Granholm AC 2010. Effects of long-term memantine on memory and neuropathology in Ts65Dn mice, a model for Down syndrome. Behav Brain Res. 221: 610–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Bear MF 2004. LTP and LTD: An embarrassment of riches. Neuron 44: 5–21 [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC 2002. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci 25: 103–126 [DOI] [PubMed] [Google Scholar]
- Miyakawa T, Leiter LM, Gerber DJ, Gainetdinov RR, Sotnikova TD, Zeng H, Caron MG, Tonegawa S 2003. Conditional calcineurin knockout mice exhibit multiple abnormal behaviors related to schizophrenia. Proc Natl Acad Sci 100: 8987–8992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery JM, Selcher JC, Hanson JE, Madison DV 2005. Dynamin-dependent NMDAR endocytosis during LTD and its dependence on synaptic state. BMC Neurosci 6: 48 10.1186/1471-2202-6-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishita W, Marie H, Malenka RC 2005. Distinct triggering and expression mechanisms underlie LTD of AMPA and NMDA synaptic responses. Nat Neurosci 8: 1043–1050 [DOI] [PubMed] [Google Scholar]
- Nishikawa K, MacIver MB 2000. Excitatory synaptic transmission mediated by NMDA receptors is more sensitive to isoflurane than are non-NMDA receptor-mediated responses. Anesthesiology 92: 228–236 [DOI] [PubMed] [Google Scholar]
- Patterson D, Costa AC 2005. Down syndrome and genetics - a case of linked histories. Nat Rev Genet 6: 137–147 [DOI] [PubMed] [Google Scholar]
- Periclou A, Ventura D, Rao N, Abramowitz W 2006. Pharmacokinetic study of memantine in healthy and renally impaired subjects. Clin Pharmacol Ther 79: 134–143 [DOI] [PubMed] [Google Scholar]
- Reeves RH, Irving NG, Moran TH, Wohn A, Kitt C, Sisodia SS, Schmidt C, Bronson RT, Davisson MT 1995. A mouse model for Down syndrome exhibits learning and behaviour deficits. Nat Genet 11: 177–184 [DOI] [PubMed] [Google Scholar]
- Roberson R, Toso L, Abebe D, Spong CY 2008. Altered expression of KIF17, a kinesin motor protein associated with NR2B trafficking, may mediate learning deficits in a Down syndrome mouse model. Am J Obstet Gynecol 198: 313.e1–313.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueda N, Llorens-Martin M, Florez J, Valdizan E, Banerjee P, Trejo JL, Martinez-Cue C 2010. Memantine normalizes several phenotypic features in the Ts65Dn mouse model of Down syndrome. J Alzheimers Dis 21: 277–290 [DOI] [PubMed] [Google Scholar]
- Selig DK, Hjelmstad GO, Herron C, Nicoll RA, Malenka RC 1995. Independent mechanisms for long-term depression of AMPA and NMDA responses. Neuron 15: 417–426 [DOI] [PubMed] [Google Scholar]
- Siarey RJ, Carlson EJ, Epstein CJ, Balbo A, Rapoport SI, Galdzicki Z 1999. Increased synaptic depression in the Ts65Dn mouse, a model for mental retardation in Down syndrome. Neuropharmacology 38: 1917–1920 [DOI] [PubMed] [Google Scholar]
- Siddiqui A, Lacroix T, Stasko MR, Scott-McKean JJ, Costa AC, Gardiner KJ 2008. Molecular responses of the Ts65Dn and Ts1Cje mouse models of Down syndrome to MK-801. Genes Brain Behav 7: 810–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturgeon X, Gardiner KJ 2011. Transcript catalogs of human chromosome 21 and orthologous chimpanzee and mouse regions. Mamm Genome 22: 261–271 [DOI] [PubMed] [Google Scholar]