

Abstract
Inactivation of inward-rectifying K+ channels (IK,in) by a rise in cytosolic free [Ca2+] ([Ca2+]i) is a key event leading to solute loss from guard cells and stomatal closure. However, [Ca2+]i action on IK,in has never been quantified, nor are its origins well understood. We used membrane voltage to manipulate [Ca2+]i (A. Grabov and M.R. Blatt [1998] Proc Natl Acad Sci USA 95: 4778–4783) while recording IK,in under a voltage clamp and [Ca2+]i by Fura-2 fluorescence ratiophotometry. IK,in inactivation correlated positively with [Ca2+]i and indicated a Ki of 329 ± 31 nm with cooperative binding of four Ca2+ ions per channel. IK,in was promoted by the Ca2+ channel antagonists Gd3+ and calcicludine, both of which suppressed the [Ca2+]i rise, but the [Ca2+]i rise was unaffected by the K+ channel blocker Cs+. We also found that ryanodine, an antagonist of intracellular Ca2+ channels that mediate Ca2+-induced Ca2+ release, blocked the [Ca2+]i rise, and Mn2+ quenching of Fura-2 fluorescence showed that membrane hyperpolarization triggered divalent release from intracellular stores. These and additional results point to a high signal gain in [Ca2+]i control of IK,in and to roles for discrete Ca2+ flux pathways in feedback control of the K+ channels by membrane voltage.
Ca2+ underlies many fundamental regulatory processes in plants, including adaptive responses to abiotic environmental stress (Knight et al., 1996; Russell et al., 1996; McAinsh et al., 1997) and programmed cell death evoked by pathogen attack (Low and Merida, 1996; Hammondkosack and Jones, 1997). Coordination of changes in [Ca2+]i and its integration with downstream response elements are central in coupling stimulus input to cellular response in these processes.
In stomatal guard cells, the best characterized higher-plant cell model, major downstream targets of [Ca2+]i and their roles in stomatal function have been identified. Increasing [Ca2+]i is known to inactivate IK,in and to activate Cl− channels, events that bias plasma membrane transport for net efflux of osmotically active solute and a loss of turgor, which drives stomatal closure (Blatt and Grabov, 1997). Furthermore, changes in [Ca2+]i are associated with ABA, CO2, and the growth hormone auxin (Blatt and Grabov, 1997; McAinsh et al., 1997). These [Ca2+]i signals have been observed to oscillate (McAinsh et al., 1995; Webb et al., 1996), characteristics that may constitute “Ca2+ signatures” to encode specific downstream responses (Berridge, 1996). Yet, despite the evidence for [Ca2+]i signaling in guard cells, surprisingly little detail is known about the link between [Ca2+]i changes and ion channel activity at the plasma membrane or about the mechanisms mediating such [Ca2+]i changes. To our knowledge, in no instance have the characteristics of ion channel regulation by Ca2+ been quantified directly in any higher-plant cell.
We recently described the coupling of membrane voltage to [Ca2+]i, demonstrating that hyperpolarization, whether under a voltage clamp or in the presence of low [K+]o, evoked [Ca2+]i increases in guard cells, and that the voltage threshold for [Ca2+]i rise was profoundly altered by ABA (Grabov and Blatt, 1998). Our observations indicated a link to Ca2+ influx across the plasma membrane and raised questions about the efficacy of [Ca2+]i in inactivating IK,in and about the contributions of intracellular Ca2+ release to the [Ca2+]i signal. We have used membrane voltage to experimentally manipulate [Ca2+]i and report that IK,in is strongly dependent on [Ca2+]i, consistent with a cooperative binding of four Ca2+ ions to effect inactivation. Additional experiments indicate that voltage-evoked [Ca2+]i increases depend both on Ca2+ influx and on release of Ca2+ from intracellular stores. These results underscore the role of [Ca2+]i as a high-gain “switch” in the control of IK,in, and implicate [Ca2+]i in feedback control linking membrane voltage to the activity of the K+ channels.
MATERIALS AND METHODS
Plant Material
Broad bean (Vicia faba L. cv Bunyan, Bunyard Exhibition) plants were grown and epidermal strips prepared as described previously (Blatt and Armstrong, 1993). All operations were carried out on a Zeiss Axiovert microscope fitted with Nomarski differential interference contrast optics with strips bathed in rapidly flowing solutions (10 mL/min [approximately 20 chamber volumes/min]) at 20°C to 22°C. The standard medium was prepared with 5 mm Mes titrated to its pKa (= 6.1) with Ca(OH)2 (final [Ca2+] approximately 1 mm). KCl and other compounds were included as required. Buffers and salts were from Sigma. All agonists/antagonists were from Calbiochem. In some experiments, Mes buffer was titrated with KOH, and CaCl2 or MnCl2 were added separately as required.
Electrophysiology and Photometry
Electrical recordings and iontophoretic injections were achieved with three-barreled microelectrodes coated with paraffin to reduce electrode capacitance and, unless noted, microelectrode barrels were filled with 200 mm potassium acetate to minimize salt leakage and salt-loading artifacts associated with the Cl− anion (Blatt and Armstrong, 1993). Connection to the amplifier headstage was via a 1 m KCl/Ag-AgCl half-cell, and a matching half-cell and 1 m KCl-agar bridge served as the reference (bath) electrode. Membrane currents were measured by voltage clamp under microprocessor control (μLAB/μLAN, WyeScience, Wye, UK) using three-pulse protocols (sampling frequency, 2 kHz) and bipolar staircase duty cycles (Blatt and Armstrong, 1993). Voltage and current were also sampled at low frequency (64 Hz) concurrently with measurements of [Ca2+]i.
[Ca2+]i was determined by ratio fluorescence with a microphotometer (Cairn, Faversham, UK) using the dye Fura-2 (Molecular Probes, Eugene, OR) excited at 340, 360, and 390 nm (10-nm half-bandwidth filter, Schott, Yonkers, NY). Fluorescence was recorded through a slit diaphragm after filtering with a 480-nm long-pass filter (Schott) and excluded microelectrode fluorescence. Dye loading was by iontophoresis (Blatt and Armstrong, 1993) and was judged successful by visual checks for cytoplasmic dye distribution and by stabilization of the fluorescence ratio signal. Measurements were calibrated (Grabov and Blatt, 1997), and experiments were generally carried out within the first 20 to 30 min after Fura-2 loading to avoid difficulties associated with bleaching and decay of the fluorescence signals.
Numerical Analysis
Data analysis was carried out by nonlinear, least-squares fitting (Marquardt, 1963) and, where appropriate, results are reported as the means ± se of (n) observations.
RESULTS
K+ Channel Inactivation Is a Consequence of [Ca2+]i Elevation
Although current through IK,in was previously reported not to inactivate with time, past studies were generally limited by voltage-clamp steps to periods of 2 to 4 s (Blatt and Grabov, 1997; Thiel and Wolf, 1997) or, over longer times, were carried out with [Ca2+]i buffers present at the cytosolic face of the membrane after exchange with patch-pipette solutions (Schroeder, 1988). By contrast, concurrent measurements of [Ca2+]i and K+ channel current in vivo showed a pronounced inactivation of IK,in evident only after the first 2 to 4 s of 20-s voltage steps to −200 mV and suggested a close link to moderate increases in [Ca2+]i (Grabov and Blatt, 1998).
To relate IK,in inactivation to the voltage threshold initiating a [Ca2+]i rise, guard cells loaded with Fura-2 were driven through slow, 2-min voltage ramps from +20 to −200 mV. f340, f360, and f390 light, as well as the f340/f390 ratio as a measure of [Ca2+]i, were recorded concurrently with a membrane current under voltage clamp. Figure 1 shows the voltage, current, and fluorescence ratios recorded from one guard cell, with the time course of the [Ca2+]i rise in Figure 1B calculated from f340/f390 and the individual fluorescence signals shown in Figure 1D. The [Ca2+]i signal increased appreciably, but only once the voltage was driven more negative than at about −120 mV (Grabov and Blatt, 1998). Note that no change was seen in the f360 trace with the voltage clamp in contrast to f340 and f390, indicating that the changes in fluorescence were a consequence of changes in [Ca2+]i rather than an effect of voltage on dye leakage or redistribution. Under similar voltage-clamp conditions (n = 55 cells), membrane hyperpolarization to −200 mV was accompanied by increases in [Ca2+]i from a mean resting value of 202 ± 23 nm to values often in excess of 1 μm (Fig. 2C; mean ± se, 703 ± 98 nm), and depolarizations were followed by recovery of [Ca2+]i to the initial resting values.
Figure 1.

Voltage ramps demonstrate a voltage threshold for increases in [Ca2+]i and consequent inactivation of current carried by IK,in. Concurrent records of voltage (A), [Ca2+]i (B), and clamp current (C) are shown, along with the raw Fura-2 fluorescence recorded on f340, f360, and f390 (D). Data are from one guard cell bathed in 5 mm Ca2+-Mes, pH 6.1, with 10 mm KCl. Slowly ramping membrane voltage from +20 to −200 mV under voltage clamp was accompanied by an appreciable rise in [Ca2+]i at voltages negative of about −120 mV. The outward (positive) current at the start of the voltage ramp is associated with IK,out (Blatt and Grabov, 1997). Activation of inward (negative) current at voltages negative of −120 mV, carried predominantly by IK,in (Blatt and Grabov, 1997), was followed by a near-complete decay in current amplitude during the final 10 s of the ramp and coincident with the [Ca2+]i rise near and above 400 nm. Gradual decay in fluorescence recorded on excitation at all three wavelengths (D) is characteristic of progressive photobleaching of Fura-2 under these conditions. Note the absence any influence of the voltage ramp on the fluorescence trajectory recorded at the isobestic wavelength f360.
Figure 2.

Inactivation of current through IK,in is correlated with [Ca2+]i elevation. Voltage steps of 20 s duration from −50 to −200 mV leading to limited (A) and profound (B) increases in [Ca2+]i (bottom trace, note different scales) in two broad bean guard cells. The clamp current (top trace, note different scales) shows the characteristic time course for IK,in activation (A) in the absence of an extensive rise in [Ca2+]i, and activation followed by a decay in current amplitude (B) with the more pronounced rise in [Ca2+]i. C, Summary of relative IK,in inactivation as a function of the change in [Ca2+]i (Δ[Ca2+]i) evoked by 20-s voltage steps from −50 to −200 mV recorded in 52 independent experiments (solid points). Histograms show the means ± se of measurements binned in successive pools of 10 or 11 experiments. Inactivation of IK,in was calculated from the ratio (Imax − Ifinal)/Imax, with Imax determined at maximum IK,in amplitude and Ifinal taken as the final current amplitude without correction for instantaneous current. Δ[Ca2+]i values were determined from the mean [Ca2+]i recorded over periods of 1 s immediately before and at the end of the voltage steps. Note that the analysis does not account for measurements in which [Ca2+]i was initially high, nor does it account for measurements in which clamp steps yielded little inward current. The distribution is therefore probably skewed to the right along the x axis, but nonetheless shows that current inactivation was associated with the rise in [Ca2+]i. D, [Ca2+]i elevation (Δ[Ca2+]i) after 20-s steps to −200 mV is dependent on the resting [Ca2+]i level. Data from C plotted as a function of [Ca2+]i before voltage steps to −200 mV. Histograms show the means ± se of measurements binned in successive pools with [Ca2+]i ≤ 80 nm, 80 nm < [Ca2+]I ≤ 300 nm, and [Ca2+]I > 300 nm at rest. Note the logarithmic abscissa. The decline in the mean Δ[Ca2+]i from high starting [Ca2+]i values is not consistent with saturation of the Fura-2 signal.
The voltage-clamp record in Figure 1 shows that the rise in [Ca2+]i was accompanied by a decline in the amplitude of inward membrane current. The initial clamp step to +20 mV in Figure 1C showed a large, outward (positive) current corresponding to the activation of IK,out at this voltage. As the voltage was driven to values between approximately −50 and −130 mV (at which, with 10 mm K+ outside, IK,out is inactive) the current declined to close to 0. At voltages negative of about −140 mV a large, inward current was observed, consistent with the activation of the Ca2+-sensitive IK,in (Lemtiri-Chlieh and MacRobbie, 1994; Grabov and Blatt, 1997). However, this inward current decayed in magnitude in this and in the other recordings as the voltage approached −200 mV, coincident with the rise in [Ca2+]i.
To quantify the effect of prolonged membrane hyperpolarization on IK,in, measurements were compiled from 52 independent experiments in which the membrane voltage was driven stepwise to −200 mV for 20 s or longer. Again, the characteristics of IK,in were related to [Ca2+]i through concurrent recordings of Fura-2 fluorescence. Examples of clamp-current and [Ca2+]i recordings from two guard cells are shown in Figure 2, A and B. The voltage step for the cell in Figure 2A led to a small increase only in [Ca2+]i (bottom trace). In this case, and in cells showing a similar [Ca2+]i response, the inward current exhibited prolonged activation, with an apparent half-time near 100 ms, typical of IK,in. This current remained stable or increased slowly thereafter during the 20-s period (Fig. 2A, top trace). In contrast, experiments in which hyperpolarization led to an appreciable increase in [Ca2+]i (Fig. 2B, bottom trace) also showed a pronounced biphasic rise with a subsequent decline in the clamp-current amplitude after the first 2 to 4 s at −200 mV (Fig. 2B, top trace). A summary of all 52 experiments shows the positive correlation between the maximum change in [Ca2+]i during clamp steps and the relative inactivation of the current measured at the end of the 20-s clamp step compared with that recorded 2 s after its start (Fig. 2C). We also noted a dependence of voltage-evoked [Ca2+]i increases on the resting [Ca2+]i level before stimulation (Fig. 2D). The analysis shows that the greatest rise in [Ca2+]i was evoked from resting values around a median of 140 nm, whereas cells with resting [Ca2+]i near and below 80 nm and above 300 nm generally showed less sensitivity to the voltage stimulus.
The causal relationship of IK,in inactivation to the [Ca2+]i increases was confirmed in single guard cells using a standard two-pulse protocol with eight cycles of a 0.5-s conditioning step to −100 mV and 2-s steps to test voltages from +20 to −200 mV, by introducing additional 0.5-s voltage steps between cycles to manipulate [Ca2+]i. Figure 3 shows the results of two consecutive voltage-clamp protocols with the additional steps either to −250 mV (a) or to −30 mV (b). The current records are overlaid in Figure 3A in each case and show, in successive test pulses, the time-dependent outward current of IK,out evoked on positive voltage steps and IK,in on steps negative of −120 mV. Intervening steps to −250 mV yielded a cumulative rise in [Ca2+]i (Fig. 3B, bottom trace) and gave rise to the final time-dependent inward current (a, numbered 1–8, corresponding to the numbered steps in Fig. 3B, top trace) that declined in magnitude with each cycle coincident with the rise in [Ca2+]i. However, intervening steps to −30 mV, which did not evoke a rise in [Ca2+]i, had little effect on [Ca2+]i or on the K+ current.
Figure 3.

Inactivation of current through IK,in is evoked by [Ca2+]i elevation. Data are from one guard cell bathed in 5 mm Ca2+-Mes, pH 6.1, with 10 mm KCl. A, Clamp current recorded using standard two-pulse protocols of eight cycles with the addition of a third, 0.5-s intervening step at the end of each cycle to −250 mV (a) or to −30 mV (b). Clamp cycles: 0.5-s conditioning step, −100 mV; 2-s test steps (8) from +20 to −200 mV. B, Voltage (V, top trace) and [Ca2+]i (bottom trace) records with intervening voltage steps numbered according to the cycle (1–8, cross-referenced to currents in A). The mean [Ca2+]i during the final three cycles in each protocol is indicated by the solid lines overlaid on the [Ca2+]i record. C, Steady-state current-voltage characteristic determined from the currents recorded at the end of the test voltage steps in protocols a and b. The curves have not been corrected for the background (“instantaneous”) current.
A cursory view of the last three cycles shows that IK,in evoked by the test voltage steps was greatly reduced in protocol a compared with the same steps in protocol b, coincident with the mean [Ca2+]i elevation (solid bars over [Ca2+]i trace) near 320 nm compared with 170 nm, respectively. In this case, analysis of the steady-state current showed a 73% reduction of IK,in after accounting for background (instantaneous) currents (Fig. 3C). Current at 0 mV, which is associated with IK,out, was recorded before a significant rise in [Ca2+]i and is known to be insensitive to [Ca2+]i (Hosoi et al., 1988; Lemtiri-Chlieh and MacRobbie, 1994; Grabov and Blatt, 1997).
K+ Channel Activity Shows a Steep Dependence on [Ca2+]i
To quantify the [Ca2+]i sensitivity of IK,in, a similar strategy of repeated hyperpolarizations was used to manipulate [Ca2+]i, and both current and Fura-2 fluorescence were measured concurrently. In this case, 20-s steps from a holding voltage of −50 to −200 mV were used to raise [Ca2+]i and the intervals between steps decreased from 90 to 20 s to give an elevated background of [Ca2+]i at the start of each subsequent step. IK,in was characterized after subtracting the background (instantaneous) current from measurements during the first 2 s of each step.
Figure 4A shows the results of measurements from one guard cell obtained at four different [Ca2+]i values (in μm on right). The steady-state IK,in from these data are included in Figure 4B (solid symbols) along with the means ± se of IK,in from another 52 guard cells binned over [Ca2+]i intervals of 80 nm. The results demonstrate a steep dependence of IK,in on [Ca2+]i that was most pronounced over the range from 200 to 500 nm [Ca2+]i. When subjected to nonlinear, least-squares fitting (Marquardt, 1963), the data could not be accommodated satisfactorily with a simple titration function of a single Ca2+-binding site. Therefore, fittings were carried out using a formulation of the Hill equation (Hill, 1910):
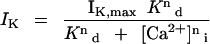 |
where IK and IK,max are the current and maximum current at −200 mV, respectively; Kd is the dissociation constant; and n is the cooperativity (Hill) coefficient and corresponds to the apparent number of Ca2+ ions binding per channel. Best fittings were obtained with a Ki of 329 ± 31 nm. Analyses carried out on a cell-by-cell basis (not shown) gave a similar Ki of 346 ± 22 nm. In every case, the analyses also yielded values for n close to 4 (mean ± se, 4.1 ± 0.5 on a cell-by-cell basis), consistent with the cooperative action of at least four Ca2+ ions to inactivate IK,in. Thus, the data indicate a profound sensitivity of the channels to relatively small changes in [Ca2+]i above normal resting values, a point we will return to below.
Figure 4.

Inactivation of IK,in shows a steep dependence on [Ca2+]i above resting [Ca2+]i levels. A, Data are from one guard cell bathed in 5 mm Ca2+-Mes, pH 6.1, with 10 mm KCl. IK,in recorded during the first 2 s of 20-s steps to −200 mV with [Ca2+]i elevated by successively decreasing the interstep interval from 90 to 20 s. Data fitted to single-exponential activation curves and points are shown at 100-ms intervals for clarity. [Ca2+]i is on the right (in μm). B, Summary of steady-state IK,in from A (solid symbols) along with means ± se of data from 52 guard cells (open symbols) binned in pools of eight to nine experiments with increasing [Ca2+]i. [Ca2+]i values were determined from the mean [Ca2+]i recorded over the final 1 s of voltage steps after prior stimulation to raise [Ca2+]i (see A) or from equivalent measurements from resting [Ca2+]i conditions. The solid line is the result of nonlinear least-squares fitting of the means to the Hill equation (Eq. 1). Fitted parameters: Ki, 329 ± 31 nm; n (cooperativity coefficient), 4.1 ± 0.5. Statistically equivalent results were obtained when the data were fitted without binning (not shown).
Ca2+ Influx and the [Ca2+]i Rise Are Independent of IK,in
Previous experiments showed that the amplitude of voltage-evoked [Ca2+]i increases were linearly related to the external Ca2+ concentration and were associated with Fura-2 quench when Mn2+ was substituted for external Ca2+ (Grabov and Blatt, 1998). These results and the dependence of [Ca2+]i on negative membrane voltages suggested a specific Ca2+ influx triggered by the voltage across the plasma membrane. However, because IK,in also activates at these voltages, albeit over a much shorter time, it could be argued that the Ca2+ influx occurred through the K+ channels. Ca2+ entry through the K+ channels in guard cells has been proposed, based on tail-current-reversal analyses (Fairley-Grenot and Assmann, 1992).
To distinguish between these two possibilities, we recorded [Ca2+]i and the channel current under voltage clamp during challenge with Ca2+ and K+ channel antagonists. Figure 5A shows that adding 0.1 mm Gd3+, a Ca2+ channel blocker (Klusener et al., 1995; Sedbrook et al., 1996), outside virtually eliminated the [Ca2+]i transients evoked by subsequent voltage steps to −200 mV (bottom trace), while promoting the current associated with IK,in (top trace). Similar results were obtained from an additional four independent experiments with the Ca2+ channel blocker. Because Gd3+ may permeate the plant plasma membrane (Klusener et al., 1995), albeit slowly, we also explored the effects of higher-Mr peptide toxins that show specificity for plasma membrane Ca2+ channels in animals.
Figure 5.

Voltage-evoked [Ca2+]i rise is sensitive to extracellular Ca2+ channel blockers. A, [Ca2+]i increases (trace below) and clamp current (trace above) during 20-s steps from −50 to −200 mV (⊔, above) before and after adding 0.1 mm GdCl3 to the bath. Period of GdCl3 exposure is indicated by the open bar. Data are from one guard cell in 5 mm Ca2+-Mes, pH 6.1, with 10 mm KCl. Inset, Clamp current during steps to −200 mV (⊔, above) replotted on expanded time scale shows characteristic activation of the IK,in current (Grabov and Blatt, 1997) when the [Ca2+]i rise is suppressed in the presence of Gd3+ (fine line) and its time-dependent inactivation when [Ca2+]i rises in the absence of Gd3+ (solid line). B, [Ca2+]i increases evoked during 20-s steps from −50 to −200 mV (⊔, above) before and after adding 0.5 μm calcicludine to the bath. Data are from one guard cell in 5 mm Ca2+-Mes, pH 6.1, with 10 mm KCl. Period of exposure to the Ca2+ channel blocker is indicated by an open bar. Time scale (below), 2 min.
Exposures to 0.5 μm calcicludine suppressed the [Ca2+]i rise and relieved IK,in inactivation at −200 mV (Fig. 5B). Calcicludine is a 6.9-kD peptide toxin derived from Dendroaspis augusticeps venom and preferentially blocks L-type Ca2+ channels in animal tissues (Schweitz et al., 1994). The toxin is a highly charged, soluble protein and is therefore unlikely to pass across the plasma membrane. In contrast (not shown), the voltage-evoked [Ca2+]i rise was unaffected by a peptide toxin, 1 μm ω-conotoxin (GVIIIa), and a specific N-type Ca2+ channel antagonist (Mori et al., 1991; Leveque et al., 1994). Substitution of external K+ with Cs+, which blocks the current through IK,in (Thiel and Wolf, 1997), had no measurable effect on the voltage-evoked [Ca2+]i rise in each of the six experiments (not shown), and the [Ca2+]i rise was insensitive to changes in external K+ between 0.1 and 10 mm (Grabov and Blatt, 1998). These latter results, and the insensitivity of IK,in to Gd3+ and calcicludine compared with the [Ca2+]i transients, implicate a unique class of Ca2+-permeable channels at the plasma membrane that activate on hyperpolarization to facilitate Ca2+ entry. The data also argue against a predominance of the Ca2+ flux through the K+ channels (Fairley-Grenot and Assmann, 1992), a point we comment on in Discussion.
Evoked [Ca2+]i Increases Are Coupled to Cytosolic Ca2+ Release
Although the data above point to entry across the plasma membrane as the initial source of Ca2+ for the voltage-evoked [Ca2+]i transients, intracellular Ca2+ stores have also been indicated in evoked [Ca2+]i signals in guard cells (Blatt and Grabov, 1997; McAinsh et al., 1997; Grabov and Blatt, 1998). Analogous patterns of Ca2+ entry and subsequent release from intracellular stores are well known in animal cells and result from positive feedback of Ca2+ that activates intracellular Ca2+ release channels. Such CICR rapidly amplifies [Ca2+]i signals and facilitates [Ca2+]i spikes and oscillations (Clapham, 1995; Berridge, 1996).
To assess the contribution of intracellular Ca2+ release to the voltage-evoked [Ca2+]i transients, we used a strategy of Fura-2 fluorescence quenching with Mn2+, which passes through many Ca2+-permeable channels (Fasolato et al., 1993; Pineros and Tester, 1995; Schofield and Mason, 1996; Striggow and Ehrlich, 1996). Its binding causes a loss of Fura-2 fluorescence that can be separated from the effects of [Ca2+]i by recording the Fura-2 fluorescence emission at f360, the isobestic wavelength. Thus, if the [Ca2+]i rise depended on divalent release from intracellular stores, we reasoned that hyperpolarization should be followed by a stimulation of Fura-2 quenching once guard cells were loaded with Mn2+.
For these experiments guard cells were exposed to 2 mm Mn2+ for 8 h to load intracellular Ca2+ stores with Mn2+ (Fasolato et al., 1993). Before the start of recordings, the guard cells were washed in Ca2+-Mes buffer with 10 mm KCl to remove external Mn2+. After impalements, membrane voltage was clamped to −50 mV and the cells were loaded with Fura-2 in the usual manner. Thereafter, Fura-2 fluorescence was recorded after excitation at 360 nm and the cells were challenged with membrane voltage steps to −200 mV. Results from one guard cell are shown in Figure 6A. Voltage steps to −200 mV resulted in a rise, and then a more rapid fall in f360 when Ca2+ was present outside. However, when Ca2+ was removed from the bath to prevent Ca2+ entry, triggering intracellular release, only a stepwise increase in f360 was seen and then only during the period that the voltage was clamped to −200 mV. In fact, comparing f360 quench immediately after the voltage steps showed that a 5.5-fold greater rate of dye quenching followed the stimulus in the presence of external Ca2+ (Fig. 6A, inset). Equivalent results were obtained in each of six separate experiments, including four experiments with and without external Ca2+. On one occasion f360 was seen to decay even before the end of the voltage step in the presence of external Ca2+. In every case, the quenching during the first 30 s following voltage steps was accelerated in the presence of external Ca2+ by comparison with measurements in the absence of external Ca2+ (mean acceleration ± se: +Ca2+, 6.2 ± 0.5-fold; −Ca2+, 0.9 ± 0.1-fold).
Figure 6.

Evoked [Ca2+]i rise is facilitated by Ca2+ release from intracellular stores. A, Fura-2 fluorescence quenching recorded on f360. Data are from one guard cell recorded after intracellular loading with Mn2+ in 20 mm K+-Mes, pH 6.1, and 2 mm MnCl2. Voltage steps of 20 s to −200 mV (⊔, above) applied with the cell bathed in 20 mm K+-Mes, pH 6.1, plus 2 mm CaCl2, then in the same buffer without CaCl2 (open bar), and finally with 2 mm MnCl2 (striped bar). Inset, f360 during the first 20 s after voltage steps without (○) and with (•) Ca2+ outside after normalizing to the fluorescence signal at the start of each voltage step. Note the rapid decay in f360 after voltage stimulus in the presence of external Ca2+. B to D, [Ca2+]i rise was suppressed by 10 μm ryanodine (B) and augmented by 100 μm heparin (C) and 1 mm neomycin sulfate (D). Data are from three guard cells in 5 mm Ca2+-Mes, pH 6.1, with 10 mm KCl. Membrane voltages were clamped to −50 mV in each case. Voltage steps to −200 mV (B) and −180 mV (C and D) are indicated above (⊔). Times of treatments with ryanodine, heparin, and neomycin sulfate are indicated by the open bars in each case. Note the prolonged secondary rise in [Ca2+]i in the presence of heparin and neomycin sulfate.
We interpret this behavior and the increase in fluorescence during the voltage steps to reflect the characteristics of the two divalent flux events. In the presence of external Ca2+, the voltage stimulus evokes a Ca2+ influx and, with the Mn2+ electrochemical gradient directed out of the cell in the absence of external Mn2+, simultaneously a Mn2+ efflux across the plasma membrane, which leads to an increase in Fura-2 fluorescence. The entry of Ca2+ then triggers Mn2+ release from intracellular stores and a consequent quenching of the fluorescence. That quenching is potentiated beyond the period of the voltage step implies that Mn2+ release continues after the voltage step and is wholly consistent with the continued rise in [Ca2+]i and prolonged high [Ca2+]i recorded even after voltage steps (compare Figs. 2 and 3; also see Grabov and Blatt, 1998). By contrast, in the absence of external Ca2+ no intracellular release is evoked and only the initial increase in fluorescence associated with Mn2+ efflux during the voltage step is observed.
We also tested the effects of neomycin sulfate and heparin (Fig. 6, C and D), antagonists of inositol-1,4,5-trisphosphate-mediated Ca2+ release, and ryanodine (Fig. 6B), which blocks cADPR (ryanodine-receptor)-activated Ca2+ release channels (Ehrlich et al., 1994; Clapham, 1995). Of these antagonists, we found that only ryanodine antagonized the [Ca2+]i response evoked by the voltage steps (Fig. 6B). In the presence of 10 μm ryanodine [Ca2+]i transients recorded on negative voltage steps were reduced to 46% ± 8% (n = 5) of the control before treatments. Ryanodine also affected the rate of the [Ca2+]i rise in parallel with the reduction in [Ca2+]i peak amplitude (Fig. 6B). By contrast, evoked [Ca2+]i transients were 1.3- to 2.5-fold greater than those recorded before treatments in the presence of 1 mm neomycin sulfate and 100 μm heparin (Mr 3000; Fig. 6, C and D), concentrations consistent with previous studies of guard cells and pollen (Armstrong and Blatt, 1995; Franklin-Tong et al., 1996). Furthermore, in the presence of these compounds the [Ca2+]i transients in every case failed to recover fully and were followed by prolonged secondary increases in [Ca2+]i. Because neomycin sulfate and heparin can interact with ryanodine-sensitive Ca2+ channels (Ehrlich et al., 1994; Wang et al., 1996), these results implicate a homologous pathway for [Ca2+]i release in facilitating these [Ca2+]i transients.
DISCUSSION
Our demonstration of K+ channel control by [Ca2+]i establishes the [Ca2+]i signal as a pivotal intermediate in feedback coupling between membrane voltage and the K+ channels. Previous studies (Grabov and Blatt, 1998) showed that prolonged membrane hyperpolarization within the normal physiological voltage range led to a rise in [Ca2+]i that was coupled to Ca2+ influx across the plasma membrane. The results summarized above show that, in vivo, this elevation of [Ca2+]i leads to an inactivation of IK,in (Figs. 1–3). Remarkably, the action of [Ca2+]i on the K+ channels was realized at free concentrations only just above normal resting values (apparent Ki, 329 ± 31 nm). Furthermore, the [Ca2+]i sensitivity of the current indicated a concerted action of at least four Ca2+ ions, leading to K+ channel inactivation (Fig. 4). Such exquisite sensitivity to [Ca2+]i has the effect of a very high gain and, hence, a narrow dynamic range for control of IK,in. In other words, relatively small changes in [Ca2+]i are able to act as a regulatory “on/off switch,” transferring the K+ channels between active and inactive states.
To our knowledge such cooperativity has not previously been documented in vivo in plant cells. Previous studies broadly defined only the limits of [Ca2+]i action on IK,in in guard cells between 100 nm and 1 μm [Ca2+]i (Schroeder and Hagiwara, 1989; Lemtiri-Chlieh and MacRobbie, 1994; Grabov and Blatt, 1997). Nonetheless, a similar degree of [Ca2+]i sensitivity may be characteristic of the KCO1 K+ channel recently cloned from Arabidopsis (Czempinski et al., 1997). It is interesting, too, that a 4-fold cooperativity is known in Ca2+-signaling processes in animals (Clapham, 1995; Berridge, 1996; Cheng et al., 1996; Dolphin, 1996), including cGMP cyclase activation in vertebrate rods (Stryer and Koch, 1988). This situation, however, contrasts with pHi control of IK,in that is graded over a broad range of [H+]i and appears to depend on the binding of only one H+ ion per channel (Grabov and Blatt, 1997).
In light of earlier studies of [Ca2+]i action on IK,in, it is worth noting the difficulties of quantifying the effects of [Ca2+]i in vivo and of Ca2+ buffering, especially in patch-electrode experiments. In general, whole-cell patch recording results in a rapid exchange of the cytosol with the solution in the patch electrode and leads to the loss of cytosolic regulatory factors and Ca2+ buffering (Pusch and Neher, 1988) that may be replaced in part by Ca2+ buffers included in the patch pipette. Kelly et al. (1995) reported that [Ca2+]i elevation suppresses IK,in when buffered with 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA) but not with EGTA. They ascribed the difference to buffer efficacy, the slower kinetics of Ca2+-EGTA binding, and H+ competition with Ca2+ for binding to EGTA. However, because their measurements were carried out under steady-state [Ca2+]i and with pHi held constant, the discrepancy in Ca2+ buffer actions remains difficult to explain. In contrast, our use of intracellular microelectrodes has the advantage that measurements are made without inclusion of Ca2+ buffers and with a minimum of disturbance to the cell and cytosol, because diffusional exchange with intracellular microelectrodes is restricted by the bulk access resistance of the microelectrode tip and shank (Purves, 1981).
In fact, the consequences of voltage-evoked [Ca2+]i changes for the current through the K+ channels are likely to be appreciable, as the microelectrode recordings indicate. Guard cells commonly show two states of membrane voltage that may often be separated by 100 mV or more (Thiel et al., 1992; Gradmann et al., 1993), and spontaneous membrane hyperpolarization can increase [Ca2+]i from approximately 100 to 200 nm to steady-state values near 500 nm (Grabov and Blatt, 1998). Under these conditions, the positive effect of membrane hyperpolarization in activating IK,in will be largely overshadowed with inactivation of the current as the steady-state [Ca2+]i becomes elevated. In numerical terms, hyperpolarization from −140 mV to voltages near and negative of −200 mV may still limit steady-state IK,in to values of approximately 10 μA cm−2 in the face of the [Ca2+]i rise, despite the increase in electrochemical driving force and the effect of voltage otherwise on the open-channel probability (Figs. 3 and 4; see also Grabov and Blatt, 1997).
How is this rise in [Ca2+]i achieved? Although the increase in [Ca2+]i is triggered by Ca2+ influx across the plasma membrane and at voltages essentially parallel to those effective in activating IK,in (Grabov and Blatt, 1998), present evidence argues against Ca2+ entry through the K+ channels themselves (Fairley-Grenot and Assmann, 1992). We found that the Ca2+ channel antagonist Gd3+ blocked the voltage-evoked rise in [Ca2+]i, and yet the same experiments showed that the antagonist actually promoted current through the K+ channels (Fig. 5). The specificity of Gd3+ for Ca2+ channels is known to be relatively poor (Alexandre and Lassalles, 1991; Zou et al., 1991). However, the [Ca2+]i rise was also sensitive to calcicludine (Fig. 5), a peptide toxin that shows high specificity for L-type Ca2+ channels (Schweitz et al., 1994). In contrast, voltage-evoked [Ca2+]i increases were unaffected by another venom toxin, ω-conotoxin, that blocks N-type Ca2+ channels (Leveque et al., 1994). In complementary experiments we observed that the [Ca2+]i rise was independent of extracellular K+ (Grabov and Blatt, 1998) and was unaffected by block of the K+ current with Cs+. Were a significant flux of Ca2+ to pass through the K+ channels, both competition with K+ and Cs+ block might have been expected to reduce the [Ca2+]i rise. So, the simplest interpretation is that Ca2+ entry across the plasma membrane occurs through hyperpolarization-activated Ca2+ channels. In fact, a sensitivity to membrane hyperpolarization may be a common feature of many Ca2+ channels in the plant plasma membrane, although it remains to be seen whether hyperpolarization-activated Ca2+ channels of higher plants are found predominantly in specialized cells, such as guard cells. Hyperpolarization-activated Ca2+ channels have also been reported in tomato plasma membrane (Gelli and Blumwald, 1997), and there are indications of similar Ca2+ channels in Mimosa pudica (Stoeckel and Takeda, 1995).
Elevation of [Ca2+]i by membrane hyperpolarization also appears to depend on Ca2+ release from intracellular stores. We observed an accelerated quenching of Fura-2 fluorescence by intracellular (sequestered) Mn2+ following voltage steps when guard cells were preloaded with the divalent cation, and the results of studies with Ca2+ release antagonists lead to a similar conclusion (Fig. 6). Block of the voltage-evoked [Ca2+]i rise by ryanodine, and its stimulation by heparin and neomycin sulfate, suggests a parallel to ryanodine-receptor Ca2+ channels that mediate Ca2+ release in neuromuscular tissues (Ehrlich et al., 1994; Clapham, 1995). The fact that ryanodine block was incomplete may reflect the relatively short exposures that could be achieved within the time frame of these experiments, or it may indicate a significant contribution of external Ca2+ influx to the rise in [Ca2+]i. However, we cannot rule out additional contributions to [Ca2+]i release via inositol-1,4,5-trisphosphate-sensitive or other unrelated pathways (Cheek et al., 1994; Jouaville et al., 1995). We also noted a dependence of voltage-evoked [Ca2+]i increases on the resting [Ca2+]i level before stimulation (Fig. 2D). A similar sensitivity to [Ca2+]i is common to many Ca2+-release events in animals that show a bell-shaped curve characteristic of potentiation by [Ca2+]i (Callamaras and Parker, 1994; Bezprozvanny and Ehrlich, 1995; Cheng et al., 1996; Thorn et al., 1996).
These parallels to Ca2+ release events in animals do raise questions about the nature of the guard cell Ca2+-release pathway and the location of the Ca2+ stores. It is possible, for example, that [Ca2+]i changes are mediated by spatially distinct Ca2+ stores with different characteristics for Ca2+ release (Bootman and Berridge, 1996; Plieth et al., 1998). Thus, conceivably, short-term dynamic control of IK,in might be more closely coupled to [Ca2+]i and Ca2+ release events close to the plasma membrane, in contrast to the current characteristics determined under quasi-steady-state [Ca2+]i, as described above. In this context, we note that ryanodine affects Ca2+ release from plant microsomes but acts to stimulate Ca2+ release even at micromolar concentrations (Allen et al., 1995; Muir and Sanders, 1996), in contrast to our observations (Fig. 6B). Furthermore, despite the presumed role for vacuolar Ca2+ in plant cell (and guard cell) signaling (Ward and Schroeder, 1994; Allen and Sanders, 1995, 1996; Johannes and Sanders, 1995), one recent study indicated that evoked Ca2+ release in the alga Chara does not depend on vacuolar Ca2+ stores (Plieth et al., 1998). Therefore, it is conceivable that the [Ca2+]i signal in the guard cells draws on more than one Ca2+-release pathway, each with differing pharmacological characteristics and, possibly, separate Ca2+ stores. Such issues aside, these and our previous observations (Grabov and Blatt, 1998) implicate the coordination of Ca2+ influx and Ca2+ release from intracellular stores that parallels CICR events in animal cells (Clapham, 1995; Berridge, 1996). Again, the data also underscore at least one important difference from the animal models. The [Ca2+]i rise in the guard cells was evoked at negative voltages, whereas voltage-evoked CICR in neuromuscular tissues is normally triggered by membrane depolarization that activates Ca2+ influx through pharmacologically distinct, L-type Ca2+ channels (Schweitz et al., 1994; Chavis et al., 1996).
What are the physiological roles for coupling IK,in to membrane voltage through [Ca2+]i and for the high gain inherent in IK,in inactivation by [Ca2+]i? One clue may lie in voltage oscillations that have been observed in guard cells (Thiel et al., 1992; Gradmann et al., 1993). These oscillations occur spontaneously, they are potentiated by ABA and auxin that effect stomatal movements (Thiel et al., 1992; Blatt and Thiel, 1994), and they may be aperiodic or exhibit periodicities of 10 to 20 s (Gradmann et al., 1993) to many minutes (Thiel et al., 1992; Blatt and Thiel, 1994). These events arise through fluctuations in the activities of the K+ and anion channels (Blatt and Thiel, 1994) similar to those of the action potentials in Characean algae (Beilby, 1986). The effect is to drive the membrane between the hyperpolarized state, characterized by K+ uptake via IK,in and balanced by H+ efflux through the H+-ATPase, and the depolarized state, in which efflux of K+ and anions dominate. Significantly, [Ca2+]i elevation leads to inhibition of the H+-ATPase (Kinoshita et al., 1995) and IK,in (Figs. 3 and 4), as well as activation of anion channels (Hedrich et al., 1990; Schroeder and Keller, 1992). Inhibition of the H+-ATPase shows an apparent Kd of approximately 300 nm, similar to what we found for IK,in (Fig. 4). We (Grabov and Blatt, 1998) previously demonstrated the coupling of [Ca2+]i to membrane voltage and suggest now that its function may be to provide the necessary feedback to entrain K+ and anion channels as a response “cassette” for osmotic balance, switching the membrane between states for net uptake and net loss of these solutes (Gradmann et al., 1993).
Coupling to membrane voltage may also serve to “condition” [Ca2+]i signals that are triggered in response to hormones and other stimuli (Grabov and Blatt, 1998). Downstream of the initial stimulus, effective control of the ion channels and solute flux requires that second-messenger “signatures” correctly reflect the needs for change in solute flux dictated by the prior transport status of the cell. Thus, a second function for voltage coupling may lie in adapting the [Ca2+]i signal output on stimulation to the prevailing requirements for solute flux. In the simplest sense, if the purpose of a rise in [Ca2+]i, for example, in response to ABA (Blatt and Grabov, 1997; Thiel and Wolf, 1997), is to trigger membrane depolarization when the voltage is situated well negative of the K+ equilibrium voltage EK, the same [Ca2+]i rise will be superfluous should the membrane already be situated at a voltage positive of EK and, hence, biased for solute loss. In fact, ABA does not evoke significant [Ca2+]i increases when the membrane voltage is clamped near −50 mV but only when the voltage is situated near or negative of −100 mV (Grabov and Blatt, 1998). These ideas lend a further dimension to concepts of frequency encoding (Campbell et al., 1996; Knight et al., 1996) and to our understanding of [Ca2+]i oscillations in plants (McAinsh et al., 1995; Webb et al., 1996), and they raise issues relating to the spatiokinetic mechanics that couple membrane voltage, Ca2+ influx, and Ca2+ release at the subcellular level.
In conclusion, we find that inactivation of IK,in by [Ca2+]i appears to depend on the cooperative action of four Ca2+ ions, conferring on the K+ channels a very steep dependence on [Ca2+]i in the free-concentration range just above normal resting values. Such [Ca2+]i increases in broad bean guard cells can be evoked on membrane hyperpolarization within the normal physiological voltage range and thus couple IK,in activity to membrane voltage in a negative-feedback control loop. We also find that voltage-evoked [Ca2+]i increases depend on intracellular Ca2+ release and on Ca2+ influx, the latter occurring via a pathway that is pharmacologically distinct from IK,in. These observations implicate a process of CICR with characteristics that differ markedly from the conventional neuromuscular models and suggest a function in coordinate control of K+, H+, and anion transport for osmotic balance.
Abbreviations:
- [Ca2+]i
cytosolic free [Ca2+]
- CICR
Ca2+-induced Ca2+ release
- f340
f360, f390, fluorescence excited by 340, 360, and 390 nm light, respectivelyIK,
- in
IK,out, inward- and outward-rectifying K+ channels, respectively
Footnotes
This work was supported by grants from the Gatsby Charitable Foundation, the Royal Society, Human Frontiers Science Program (no. RG95/303 M), and the European Community Biotech (no. CT96-0062). A.G. was supported by the British Biotechnology and Biological Sciences Research Council (grant no. 32/C098-1).
LITERATURE CITED
- Alexandre J, Lassalles JP. Hydrostatic and osmotic pressure activated channel in plant vacuole. Biophys J. 1991;60:1326–1336. doi: 10.1016/S0006-3495(91)82170-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen GJ, Muir SR, Sanders D. Release of Ca2+ from individual plant vacuoles by both insp(3) and cyclic ADP-ribose. Science. 1995;268:735–737. doi: 10.1126/science.7732384. [DOI] [PubMed] [Google Scholar]
- Allen GJ, Sanders D. Calcineurin, a type 2B protein phosphatase, modulates the Ca2+-permeable slow vacuolar ion channel of stomatal guard cells. Plant Cell. 1995;7:1473–1483. doi: 10.1105/tpc.7.9.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen GJ, Sanders D. Control of ionic currents in guard cell vacuoles by cytosolic and luminal calcium. Plant J. 1996;10:1055–1069. doi: 10.1046/j.1365-313x.1996.10061055.x. [DOI] [PubMed] [Google Scholar]
- Armstrong F, Blatt MR. Evidence for K+ channel control in Vicia guard cells coupled by G-proteins to a 7TMS receptor. Plant J. 1995;8:187–198. [Google Scholar]
- Beilby MJ. Potassium channels and different states of the Chara plasmalemma. J Membr Biol. 1986;89:241–249. [Google Scholar]
- Berridge MJ. Microdomains and elemental events in calcium signaling. Cell Calcium. 1996;20:95–96. doi: 10.1016/s0143-4160(96)90098-6. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I, Ehrlich BE. The inositol 1,4,5-trisphosphate (insp3) receptor. J Membr Biol. 1995;145:205–216. doi: 10.1007/BF00232713. [DOI] [PubMed] [Google Scholar]
- Blatt MR, Armstrong F. K+ channels of stomatal guard cells: abscisic acid-evoked control of the outward rectifier mediated by cytoplasmic pH. Planta. 1993;191:330–341. [Google Scholar]
- Blatt MR, Grabov A. Signalling gates in abscisic acid-mediated control of guard cell ion channels. Physiol Plant. 1997;100:481–490. [Google Scholar]
- Blatt MR, Thiel G. K+ channels of stomatal guard cells: bimodal control of the K+ inward-rectifier evoked by auxin. Plant J. 1994;5:55–68. doi: 10.1046/j.1365-313x.1994.5010055.x. [DOI] [PubMed] [Google Scholar]
- Bootman MD, Berridge MJ. Subcellular Ca2+ signals underlying waves and graded responses in HeLa cells. Curr Biol. 1996;6:855–865. doi: 10.1016/s0960-9822(02)00609-7. [DOI] [PubMed] [Google Scholar]
- Callamaras N, Parker I. Inositol 1,4,5-trisphosphate receptors in Xenopus laevis oocytes—localization and modulation by Ca2+ Cell Calcium. 1994;15:66–78. doi: 10.1016/0143-4160(94)90105-8. [DOI] [PubMed] [Google Scholar]
- Campbell AK, Trewavas AJ, Knight MR. Calcium imaging shows differential sensitivity to cooling and communication in luminous transgenic plants. Cell Calcium. 1996;19:211–218. doi: 10.1016/s0143-4160(96)90022-6. [DOI] [PubMed] [Google Scholar]
- Chavis P, Fagni L, Lansman JB, Bockaert J. Functional coupling between ryanodine receptors and L-type calcium channels in neurons. Nature. 1996;382:719–722. doi: 10.1038/382719a0. [DOI] [PubMed] [Google Scholar]
- Cheek TR, Berridge MJ, Moreton RB, Stauderman KA, Murawsky MM, Bootman MD. Quantal Ca2+ mobilization by ryanodine receptors is due to all-or-none release from functionally discrete intracellular stores. Biochem J. 1994;301:879–883. doi: 10.1042/bj3010879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Lederer MR, Xiao RP, Gomez AM, Zhou YY, Ziman B, Spurgeon H, Lakatta EG, Lederer WJ. Excitation-contraction coupling in heart—new insights from Ca2+ sparks. Cell Calcium. 1996;20:129–140. doi: 10.1016/s0143-4160(96)90102-5. [DOI] [PubMed] [Google Scholar]
- Clapham DE. Calcium signaling. Cell. 1995;80:259–268. doi: 10.1016/0092-8674(95)90408-5. [DOI] [PubMed] [Google Scholar]
- Czempinski K, Zimmermann S, Ehrhardt T, MullerRober B. New structure and function in plant K+ channels: KCO1, an outward rectifier with a steep Ca2+ dependency. EMBO J. 1997;16:2565–2575. doi: 10.1093/emboj/16.10.2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolphin AC. Facilitation of Ca2+ current in excitable cells. Trends Neurosci. 1996;19:35–43. doi: 10.1016/0166-2236(96)81865-0. [DOI] [PubMed] [Google Scholar]
- Ehrlich BE, Kaftan E, Bezprozvannaya S, Bezprozvanny I. The pharmacology of intracellular Ca2+-release channels. Trends Pharmacol Sci. 1994;15:145–149. doi: 10.1016/0165-6147(94)90074-4. [DOI] [PubMed] [Google Scholar]
- Fairley-Grenot KA, Assmann SM. Permeation of Ca2+ through K+ channels in the plasma membrane of Vicia faba guard cells. J Membr Biol. 1992;128:103–113. doi: 10.1007/BF00231883. [DOI] [PubMed] [Google Scholar]
- Fasolato C, Hoth M, Matthews G, Penner R. Ca2+ and Mn2+ influx through receptor-mediated activation of nonspecific cation channels in mast cells. Proc Natl Acad Sci USA. 1993;90:3068–3072. doi: 10.1073/pnas.90.7.3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklintong VE, Drobak BK, Allan AC, Watkins PAC, Trewavas AJ. Growth of pollen tubes of Papaver rhoeas is regulated by a slow-moving calcium wave propagated by inositol 1,4,5-trisphosphate. Plant Cell. 1996;8:1305–1321. doi: 10.1105/tpc.8.8.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelli A, Blumwald E. Hyperpolarization-activated Ca2+-permeable channels in the plasma membrane of tomato cells. J Membr Biol. 1997;155:35–45. doi: 10.1007/s002329900156. [DOI] [PubMed] [Google Scholar]
- Grabov A, Blatt MR. Parallel control of the inward-rectifier K+ channel by cytosolic-free Ca2+ and pH in Vicia guard cells. Planta. 1997;201:84–95. [Google Scholar]
- Grabov A, Blatt MR. Membrane voltage initiates Ca2+ waves and potentiates Ca2+ increases with abscisic acid in stomatal guard cells. Proc Natl Acad Sci USA. 1998;95:4778–4783. doi: 10.1073/pnas.95.8.4778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradmann D, Blatt MR, Thiel G. Electrocoupling of ion transporters in plants. J Membr Biol. 1993;136:327–332. doi: 10.1007/BF00233671. [DOI] [PubMed] [Google Scholar]
- Hammondkosack KE, Jones JDG. Plant disease resistance genes. Annu Rev Plant Physiol Plant Mol Biol. 1997;48:575–607. doi: 10.1146/annurev.arplant.48.1.575. [DOI] [PubMed] [Google Scholar]
- Hedrich R, Busch H, Raschke K. Ca2+ and nucleotide dependent regulation of voltage dependent anion channels in the plasma membrane of guard cells. EMBO J. 1990;9:3889–3892. doi: 10.1002/j.1460-2075.1990.tb07608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill AV. The possible effects of the aggregation of the molecules of hemoglobin on its dissociation curves. J Physiol. 1910;40:4–7. [Google Scholar]
- Hosoi S, Iino M, Shimazaki K. Outward-rectifying K+ channels in stomatal guard cell protoplasts. Plant Cell Physiol. 1988;29:907–911. [Google Scholar]
- Johannes E, Sanders D. The voltage-gated Ca2+ release channel in the vacuolar membrane of sugar beet resides in 2 activity states. FEBS Lett. 1995;365:1–6. doi: 10.1016/0014-5793(95)00424-8. [DOI] [PubMed] [Google Scholar]
- Jouaville LS, Ichas F, Holmuhamedov EL, Camacho P, Lechleiter JD. Synchronization of calcium waves by mitochondrial substrates in Xenopus laevis oocytes. Nature. 1995;377:438–441. doi: 10.1038/377438a0. [DOI] [PubMed] [Google Scholar]
- Kelly WB, Esser JE, Schroeder JI. Effects of cytosolic calcium and limited, possible dual, effects of G-protein modulators on guard cell inward potassium channels. Plant J. 1995;8:479–489. [Google Scholar]
- Kinoshita T, Nishimura M, Shimazaki KI. Cytosolic concentration of Ca2+ regulates the plasma membrane H+-ATPase in guard cells of fava bean. Plant Cell. 1995;7:1333–1342. doi: 10.1105/tpc.7.8.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klusener B, Boheim G, Liss H, Engelberth J, Weiler EW. Gadolinium-sensitive, voltage-dependent calcium release channels in the endoplasmic reticulum of a higher-plant mechanoreceptor organ. EMBO J. 1995;14:2708–2714. doi: 10.1002/j.1460-2075.1995.tb07271.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight H, Trewavas AJ, Knight MR. Cold calcium signaling in Arabidopsis involves 2 cellular pools and a change in calcium signature after acclimation. Plant Cell. 1996;8:489–503. doi: 10.1105/tpc.8.3.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemtiri-Chlieh F, MacRobbie EAC. Role of calcium in the modulation of Vicia guard cell potassium channels by abscisic acid: a patch-clamp study. J Membr Biol. 1994;137:99–107. doi: 10.1007/BF00233479. [DOI] [PubMed] [Google Scholar]
- Leveque C, Elfar O, Martinmoutot N, Sato K, Kato R, Takahashi M, Seagar MJ. Purification of the N-type calcium-channel associated with syntaxin and synaptotagmin—a complex impli-cated in synaptic vesicle exocytosis. J Biol Chem. 1994;269:6306–6312. [PubMed] [Google Scholar]
- Low PS, Merida JR. The oxidative burst in plant defense—function and signal transduction. Physiol Plant. 1996;96:533–542. [Google Scholar]
- Marquardt D. An algorithm for least-squares estimation of nonlinear parameters. J Soc Ind Appl Math. 1963;11:431–441. [Google Scholar]
- McAinsh MR, Brownlee C, Hetherington AM. Calcium ions as second messengers in guard cell signal transduction. Physiol Plant. 1997;100:16–29. [Google Scholar]
- McAinsh MR, Webb AAR, Taylor JE, Hetherington AM. Stimulus-induced oscillations in guard cell cytosolic-free calcium. Plant Cell. 1995;7:1207–1219. doi: 10.1105/tpc.7.8.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori Y, Friedrich T, Kim M-S, Mikami A, Nakai J, Ruth P, Bosse E, Hofmann F, Flockerzi V, Furuichi T and others. Primary structure and functional expression from complementary DNA of a brain calcium channel. Nature. 1991;350:398–402. doi: 10.1038/350398a0. [DOI] [PubMed] [Google Scholar]
- Muir SR, Sanders D. Pharmacology of Ca2+ release from red beet microsomes suggests the presence of ryanodine receptor homologs in higher-plants. FEBS Lett. 1996;395:39–42. doi: 10.1016/0014-5793(96)01000-9. [DOI] [PubMed] [Google Scholar]
- Pineros M, Tester M. Characterization of a voltage-dependent Ca2+-selective channel from wheat roots. Planta. 1995;195:478–488. [Google Scholar]
- Plieth C, Sattelmacher B, Hansen UP, Thiel G. The action potential in Chara: Ca2+ release from internal stores visualized by Mn2+-induced quenching of fura-dextran. Plant J. 1998;13:167–175. [Google Scholar]
- Purves JD (1981) Microelectrode Methods for Intracellular Recording and Ionophoresis, Vol 1. Academic Press, London, pp 1–146
- Pusch M, Neher E. Rates of diffusional exchange between small cells and a measuring patch pipette. Pflugers Archiv Eur J Physiol. 1988;411:204–214. doi: 10.1007/BF00582316. [DOI] [PubMed] [Google Scholar]
- Russell AJ, Knight MR, Cove DJ, Knight CD, Trewavas AJ, Wang TL. The moss, Physcomitrella patens, transformed with apoaequorin cDNA responds to cold shock, mechanical perturbation and pH with transient increases in cytoplasmic calcium. Transgenic Res. 1996;5:167–170. doi: 10.1007/BF01969705. [DOI] [PubMed] [Google Scholar]
- Schofield GG, Mason MJ. A Ca2+ current activated by release of intracellular Ca2+ stores in rat basophilic leukemia cells (RBL-1) J Membr Biol. 1996;153:217–231. doi: 10.1007/s002329900125. [DOI] [PubMed] [Google Scholar]
- Schroeder JI. K+ transport properties of K+ channels in the plasma membrane of Vicia faba guard cells. J Gen Physiol. 1988;92:667–683. doi: 10.1085/jgp.92.5.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder JI, Hagiwara S. Cytosolic calcium regulates ion channels in the plasma membrane of Vicia faba guard cells. Nature. 1989;338:427–430. [Google Scholar]
- Schroeder JI, Keller BU. Two types of anion channel currents in guard cells with distinct voltage regulation. Proc Natl Acad Sci USA. 1992;89:5025–5029. doi: 10.1073/pnas.89.11.5025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweitz H, Heurteaux C, Bois P, Moinier D, Romey G, Lazdunski M. Calcicludine, a venom peptide of the Kunitz-type protease inhibitor family, is a potent blocker of high-threshold Ca2+ channels with a high-affinity for L-type channels in cerebellar granule neurons. Proc Natl Acad Sci USA. 1994;91:878–882. doi: 10.1073/pnas.91.3.878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedbrook JC, Kronebusch PJ, Borisy CG, Trewavas AJ, Masson PH. Transgenic aequorin reveals organ-specific cytosolic Ca2+ responses to anoxia in Arabidopsis thaliana seedlings. Plant Physiol. 1996;111:243–257. doi: 10.1104/pp.111.1.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeckel H, Takeda K. Calcium-sensitivity of the plasmalemmal delayed rectifier potassium current suggests that calcium influx in pulvinar protoplasts from Mimosa pudica l can be revealed by hyperpolarization. J Membr Biol. 1995;146:201–209. doi: 10.1007/BF00238009. [DOI] [PubMed] [Google Scholar]
- Striggow F, Ehrlich BE. The inositol 1,4,5-trisphosphate receptor of cerebellum—Mn2+ permeability and regulation by cytosolic Mn2+ J Gen Physiol. 1996;108:115–124. doi: 10.1085/jgp.108.2.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stryer L, Koch K-W. Highly cooperative feedback control of retinal rod guanylate cyclase by calcium ions. Nature. 1988;334:64–66. doi: 10.1038/334064a0. [DOI] [PubMed] [Google Scholar]
- Thiel G, MacRobbie EAC, Blatt MR. Membrane transport in stomatal guard cells: the importance of voltage control. J Membr Biol. 1992;126:1–18. doi: 10.1007/BF00233456. [DOI] [PubMed] [Google Scholar]
- Thiel G, Wolf AH. Operation of K+ channels in stomatal movement. Trends Plant Sci. 1997;2:339–345. [Google Scholar]
- Thorn P, Moreton R, Berridge M. Multiple, coordinated Ca2+-release events underlie the inositol trisphosphate-induced local Ca2+ spikes in mouse pancreatic acinar cells. EMBO J. 1996;15:999–1003. [PMC free article] [PubMed] [Google Scholar]
- Wang JP, Needleman DH, Seryshev AB, Aghdasi B, Slavik KJ, Liu SQ, Pedersen SE, Hamilton SL. Interaction between ryanodine and neomycin binding sites on Ca2+ release channel from skeletal muscle sarcoplasmic reticulum. J Biol Chem. 1996;271:8387–8393. doi: 10.1074/jbc.271.14.8387. [DOI] [PubMed] [Google Scholar]
- Ward JM, Schroeder JI. Calcium-activated K+ channels and calcium-induced calcium release by slow vacuolar ion channels in guard-cell vacuoles implicated in the control of stomatal closure. Plant Cell. 1994;6:669–683. doi: 10.1105/tpc.6.5.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb AAR, McAinsh MR, Mansfield TA, Hetherington AM. Carbon dioxide induces increases in guard cell cytosolic free calcium. Plant J. 1996;9:297–304. [Google Scholar]
- Zou XL, Stumpf MA, Hoch HC, Kung C. A mechanosensitive channel in whole cells and in membrane patches of the fungus Uromyces. Science. 1991;253:1415–1417. doi: 10.1126/science.1716786. [DOI] [PubMed] [Google Scholar]