

Abstract
Self-assembling peptide amphiphiles (PAs) have been extensively used in the development of novel biomaterials. Due to their propensity to form cylindrical micelles, their use is limited in applications where small spherical micelles are desired. Here we present a platform method for controlling the self-assembly of biofunctional PAs into spherical 50 nm particles using dendrimers as shape-directing scaffolds. This templating approach results in biocompatible, stable protein-like assemblies displaying peptides with native secondary structure and biofunctionality.
Keywords: Peptide amphiphile, dendrimer, templating, protein analog, self-assembly, biomaterials
Globular proteins are ubiquitous in nature and possess an extraordinary breadth of functionalities that are fundamental to life. Although great efforts have been directed toward the understanding of protein composition, function, and folding, it remains a challenge to design and fabricate proteins employing a bottom-up approach. Peptide amphiphile (PA) micelles are self-assembled aggregates that exhibit a structural resemblance to proteins by having folded bioactive peptides displayed on the exterior of a hydrophobic core. PA supramolecular assembly often results in high aspect ratio cylindrical micelles, making spherical PA nanoparticles difficult to access. To address this challenge, we report a modular and versatile approach to control the self-assembly of PAs into spherical nanoparticles by exploiting dendrimer templates, with an overarching goal of developing globular protein analogs. The stable nanoparticles presented in this study display peptides bearing native biological secondary structure and function, and have potential in applications that necessitate the delivery of protein-like therapeutics, diagnostics, and targeting agents.
Self-assembly provides a robust and reliable strategy for creating highly ordered, modular, and multifunctional structures, which can be exploited to make biomaterials with defined and tunable physicochemical properties1. Peptide amphiphiles (PAs), common building blocks for self-assembly2–4, are synthesized by conjugating peptides to hydrophobic moieties, typically fatty acids or lipids. The amphiphilic nature enables self-assembly into micelles above a critical aggregation concentration (CAC), and the structural constraints imposed on the peptides in the aggregated state often induce folding into their native secondary structure5–7. The recent increase in attention garnered by PAs focuses mainly on cylindrical micelles, which have been heavily exploited for a wide range of biomimetic applications from biomineralization and templating to tissue engineering and therapeutics8–9. Although cylindrical micelles are versatile in function, there are many biological processes that favor low aspect ratio nanoparticles. For example, spherical particles are preferred in applications where efficient cell internalization and trafficking is required. Previous in vitro studies found that spherical nanoparticles approximately 50 nm in diameter exhibit the most efficient cell internalization10–11. In vivo studies show that particles less than 100 nm are ideal for transport into tissues12–13, although higher aspect ratio particles, such as filomicelles, can have long circulation times14.
Current strategies to prepare spherical PA micelles use chemical modifications that avert their natural tendency towards cylindrical morphologies. Modifications include the addition of large polymer spacers or bulky pendant groups near the core of micelles and interfering with backbone hydrogen bonding by substituting modified amino acids into the sequence15–19. Given the extensive array of applications for PAs and the sensitivity between chemical composition and function, a facile method for controlling their self-assembly without interfering with their inherent chemical structure is highly desirable.
This study describes a versatile strategy for controlling the self-assembly of biorelevant PAs into spherical structures by using dendrimers as supramolecular templates (Fig. 1a). The template used was a 5th generation dendrimer synthesized by a rapid, orthogonal AB2-CD2 approach employing thiol-ene and azide-alkyne cycloaddition chemistries20 (see Supplemental Information). Dendrimers were selected over inorganic nanoparticles and other polymers for their inherent modularity and the ability to control their size precisely. Our template was designed with a hydrophobic periphery to interact with the tails of the PAs. The model PAs used in this investigation were di-C16-bZip, a DNA binding PA sequence adapted from the alpha helical yeast transcription factor GCN4, which self-assembles into elongated micelles (Fig. 1b), and di-C16-NLS, a cell internalization PA, which self-assembles into vesicles (Fig. 1c).
Figure 1.

The dendrimers template micelle-forming and vesicle-forming PAs into spherical PRTNs. (a) Schematic of templating process: i. bZip PA cylindrical micelle ii. NLS PA vesicle iii. dendrimer iv. Spherical bZip PRTN v. Spherical NLS PRTN. Cryo-TEM images of self-assemblies: (b) bZip PA cylindrical micelles. (c) NLS PA vesicles. (d) Spherical bZip PRTNs. (e) Spherical NLS PRTNs. Scale bars are 100 nm.
The templating approach was successful for fabricating protein-resembling templated nanopsheres (PRTNs), showing that the dendrimers had a dramatic effect on the self-assembly of the model PAs. In our studies, 50 nm PRTNs were prepared by mixing 20 molar equivalents of PA with one equivalent of dendrimer in a mixture of THF and methanol, which was then solvent exchanged into ultrapure water. This straightforward approach provides a means to tune the size of the aggregates through the concentration and ratio of components, as expected for self-assembled systems21–22 (see Supplemental Information). PRTN and salt concentration adjustments post hydration did not affect PRTN size. Cryo-TEM images in Fig. 1d and 1e show the spherical bZip and NLS PRTNs. Dynamic light scattering (DLS) measured the hydrodynamic radii of bZip and NLS PRTNs as 26.6 nm ± 9.6 nm and 21.5 nm ± 5.1 nm, respectively. This size corresponds to an average functionalization of approximately 500 PAs per PRTN (see Supplemental Information).
Stability is a critical requirement for the successful application of supramolecular assemblies, where non-covalent associations must endure the rigors of biological environments. Here, it was found that the PRTNs had a lower CAC than the PAs alone due to the presence of the hydrophobic dendrimer, which translated to an increase in PRTN stability relative to the PA-only assemblies. The critical aggregation concentration determined with the pyrene fluorescence method23 revealed that the value for the di-C16-NLS PAs was 60.4 μM, whereas the CAC of the PRTNs was an order of magnitude lower at 5.6 μM (see Supplemental Information). Due to the low CAC of di-C16-bZip micelles5, an alternate method had to be employed to determine their stability. The dissociation kinetics of fluorescein labeled di-C16-bZip micelles and PRTNs in the presence of bovine serum albumin were measured using fluorescence quenching experiments24. Fig. 2 shows that after one hour at physiological temperature only 22% of the bZip PRTNs had dissociated, and at any time up to one hour, at least 50% more micelles than PRTNs had disassembled. This enhanced stability of the PRTNs in BSA relative to their micelle counterparts is an early indication of their potential in vivo capacity.
Figure 2.
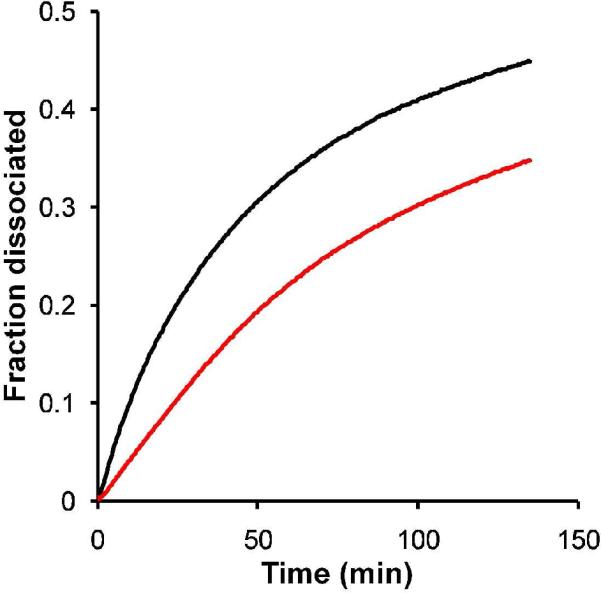
Kinetics experiments show the fraction of aggregates that dissociated over time in the presence of albumin. bZip micelles ( ) dissociate more quickly than bZip PRTNs (
) dissociate more quickly than bZip PRTNs ( ).
).
Another important design consideration for peptide-based constructs is the ability of the peptide to retain its function, which is often correlated with its secondary structure. Small peptide sequences isolated from parent proteins generally lose their secondary structure, which can be detrimental to their activity25–26. As observed with bZip, the peptide loses its native secondary structure but regains some degree of helical content after lipid conjugation, and exhibits greatly enhanced folding in the headgroup of micelles 5,27. Interestingly, circular dichroism (CD) of di-C16-bZip showed that the PAs within the PRTNs possessed enhanced α-helical content, resembling that of micelles, with minima at 208 nm and 222 nm (Fig. 3a). The α-helical bZip PRTNs were then examined for their ability to bind short DNA strands, which was monitored by the increase in fluorescence anisotropy of rhodamine-labeled DNA strands that occurred on binding. The PA only and PRTN anisotropy data in Fig. 3b diverged significantly as the concentration of bZip was increased, which suggests that the intact PRTN was binding DNA. If the PAs were desorbing from the PRTN to bind DNA, the data would overlay rather than diverge. In addition, no precipitate was observed in the solution to indicate that the hydrophobic dendrimer surface was exposed to the aqueous solution. The anisotropy value for the PA increased with concentration as the DNA strands bound by free PAs were concurrently incorporated into growing micelles. The anisotropy for the bZip PRTNs began to plateau once a PRTN was bound to the DNA and those sites could no longer be occupied. The binding of the bZip PRTNs to long calf-thymus DNA (ctDNA) strands was studied to supplement the anisotropy experiments. Previous work showed that bZip micelles rearranged to form lamellar sheets upon complexation with ctDNA5, whereas atomic force microscopy (AFM) shows that the bZip PRTNs maintained their integrity upon complexation (Fig. 3d–f). The bZip PRTNs were not only able to bind ctDNA but also remained intact along the strands, which further demonstrated the increased stability of the bZip PRTNs relative to the PA micelles.
Figure 3.

bZip secondary structure and DNA binding. (a) Circular dichroism of bZip peptide, bZip PA below and above the CMC, and bZip PRTNs. The lipidation of the bZip peptide provides a slight increase in helicity over the free peptide, while aggregation into micelles or on the surface of a dendrimer results in drastic increase in α-helicity indicated by the CD minima at 208 nm and 222 nm. (b) An increase in anisotropy corresponds to an increase in binding of bZip PAs (□) and bZip PRTNs ( ) to rhodamine labeled 30bp DNA strands. The bZip PA curve has higher anisotropy at higher concentrations because the DNA strands are bound by long micelles rather than small PTRNs. Data points are the mean of 10 measurements, and error bars are standard deviation. (c) AFM height image of free ctDNA (3 μm frame depicted). (d) AFM height image of bZip PRTNs (spherical objects indicated by black arrows) bound to ctDNA (6 μm frame). PRTNs are not present on the surface but only appear in areas of DNA, indicating they are bound to the DNA and not attached to free mica. (e, f) AFM height and phase image of PRTNs bound to DNA (3 μm frame),where the spherical geometry is clearly seen. Additional AFM images available in the Supplemental Information.
) to rhodamine labeled 30bp DNA strands. The bZip PA curve has higher anisotropy at higher concentrations because the DNA strands are bound by long micelles rather than small PTRNs. Data points are the mean of 10 measurements, and error bars are standard deviation. (c) AFM height image of free ctDNA (3 μm frame depicted). (d) AFM height image of bZip PRTNs (spherical objects indicated by black arrows) bound to ctDNA (6 μm frame). PRTNs are not present on the surface but only appear in areas of DNA, indicating they are bound to the DNA and not attached to free mica. (e, f) AFM height and phase image of PRTNs bound to DNA (3 μm frame),where the spherical geometry is clearly seen. Additional AFM images available in the Supplemental Information.
The modularity afforded by self-assembled systems provides the means to produce multifunctional particles by mixing different PA monomers. Mixed PRTNs were prepared using fluorescently labeled di-C16-bZip-fluorescein and di-C16-NLS-rhodamine PAs, and were characterized by fluorescence resonance energy transfer (FRET). The results show that mixing equimolar amounts of the two different PAs with dendrimers led to a decrease in fluorescein fluorescence relative to the control at 530 nm and a concomitant increase in rhodamine fluorescence at 575 nm (Fig. 4). The FRET observed here indicates that mixed PRTNs possessing both DNA binding and cell internalizing functions were formed, as energy transfer suggests that the PAs reside on the same particle. It should be noted that DLS measurements confirmed that mixing PAs did not affect the size of the PRTNs, with the mixed nanostructures having a similar size of 28.3 nm ± 6.2 nm (see Supplemental Information). The multifunctionality of the PRTNs, combined with the increased stability, may help increase the efficacy of peptide-based therapeutic vectors where variable functions such as targeting agents, drugs, and therapeutics can be packaged and delivered in a single nanoscale system.
Figure 4.
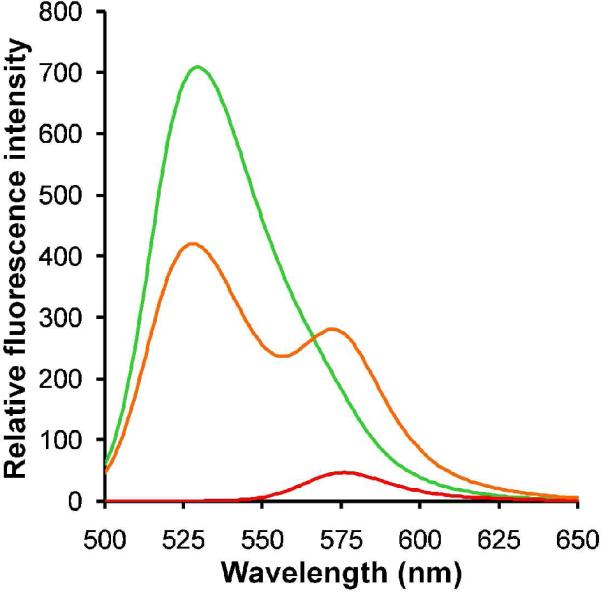
Formation of multifunctional PRTNs. FRET was observed for the co-assembly of 1:1 ratio of bZip:NLS PAs with dendrimer ( ), confirming that the PAs formed “mixed' PRTNs. Fluorescein-tagged bZip PRTNs (
), confirming that the PAs formed “mixed' PRTNs. Fluorescein-tagged bZip PRTNs ( ) and rhodamine-labeled NLS PRTNs (
) and rhodamine-labeled NLS PRTNs ( ) were used as controls. These experiments were performed with an excitation wavelength of 475 nm.
) were used as controls. These experiments were performed with an excitation wavelength of 475 nm.
Given the overarching goal of developing biocompatible protein analogs, in vitro cell association of the PRTNs was examined to gain insight into their biocompatibility. Mixed PRTNs and micelles, composed of a 9:1 ratio of di-C16-bZip-fluorescein and di-C16-NLS PAs, were each incubated with HeLa cells for two hours and then qualitatively examined through fluorescence microscopy (Fig. 5a and 5b). Distinct fluorescent spots were visible in both the micelle and PRTN images, indicative of internalization via endocytosis. In the case of the micelles a diffuse fluorescence was also observed, suggesting that bZip micelles came apart upon membrane association and were subsequently internalized as PA monomers, complying with a previously reported mechanism28. PRTN fluorescence in the cells remained punctate (spotted), which suggests their entrapment in the endosomes and also that they remained intact (PA monomers did not escape into the cytoplasm). The NLS did not appear to aid in endosomal escape at this concentration. The cell experiments show that the micelles and PRTNs are likely internalized through different mechanisms, although the details cannot be extracted from these experiments alone. Our future work includes elucidating the mechanisms of PRTN internalization.
Figure 5.
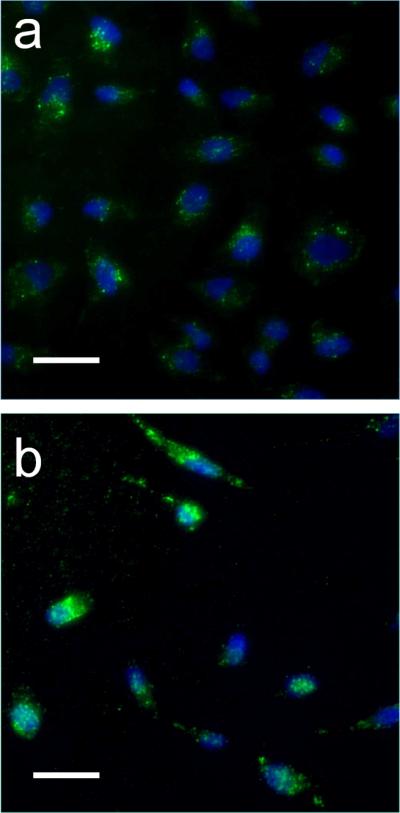
In vitro cell uptake of micelles and PRTNs. The green color in the images represents the fluorescein labeled bZip PAs, while the blue is Hoechst nuclear stain. Micelles and PRTNs are both internalized by HeLa cells. (a) Mixed PRTNs (90% bZip-FAM PA and 10% NLS PA) display punctate fluorescence, suggesting they are trapped in endosomes. The fluorescence is located in the bulk volume of the cell near the nucleus, and not observed throughout the cytoplasm to indicate endosomal escape. Intact PRTNs show low fluorescence, presumably due to fluorescence quenching. (b) Mixed micelles (90% bZip-FAM PA and 10% NLS PA) appear to be internalized via endocytosis as seen through the punctate fluorescence, but also show diffuse fluorescence, indicating PA distribution throughout the cell. The exact mechanisms of internalization are still under investigation. Scale bars are 50 μm.
In summary, we have demonstrated a general method for controlling the self-assembly of peptide amphiphiles to make well-defined spherical particles that resemble proteins in architecture and function. The simple combination of robust, molecularly precise dendrimer templates and PA building blocks results in PRTNs that possess the multivalent and multifunctional qualities inherent in natural systems. These supramolecular assemblies display peptides in their native conformation, and are sufficiently stable to remain intact during interactions with their biomolecular targets and internalization by cells. We envision that the controlled bottom-up templating approach presented here will aid in the future design of sophisticated protein-analogous constructs for in vivo diagnostics, imaging, targeting, and therapeutics.
Supplementary Material
Acknowledgments
This work was supported by the MRSEC Program of the NSF under Award No. DMR05-20415, the National Institutes of Health as a Program of Excellence in Nanotechnology under Award Nos. NIH1U01 HL080718, HHSN268201000046C, the CCNE Program of the National Institutes of Health under Award No. NIH1U54 CA119335, and the UC Berkeley College of Engineering. M.J.R thanks the DOE Office of Science for a Graduate Fellowship. The authors thank P. Allen for the artistic renderings, the R. Segalman lab for use of their DLS, and the T. Xu lab for use of their AFM.
Footnotes
Additional information The authors declare no competing financial interests. Supplementary information accompanies this paper. Correspondence and requests for materials should be addressed to L.M.C. or M.V.T.
Supporting Information Available This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Zhang SG. Nat Biotechnol. 2003;21:1171. doi: 10.1038/nbt874. [DOI] [PubMed] [Google Scholar]
- (2).Cavalli S, Albericio F, Kros A. Chem Soc Rev. 2010;39:241. doi: 10.1039/b906701a. [DOI] [PubMed] [Google Scholar]
- (3).Versluis F, Marsden HR, Kros A. Chem Soc Rev. 2010;39:3434. doi: 10.1039/b919446k. [DOI] [PubMed] [Google Scholar]
- (4).Hamley IW. Soft Matter. 2011;7:4122. [Google Scholar]
- (5).Tu RS, Marullo R, Pynn R, Bitton R, Bianco-Peled H, Tirrell MV. Soft Matter. 2010;6:1035. [Google Scholar]
- (6).Yu YC, Berndt P, Tirrell M, Fields GB. J Am Chem Soc. 1996;118:12515. [Google Scholar]
- (7).Missirlis D, Farine M, Kastantin M, Ananthanarayanan B, Neumann T, Tirrell M. Bioconjugate Chem. 2010;21:465. doi: 10.1021/bc900383m. [DOI] [PubMed] [Google Scholar]
- (8).Webber MJ, Kessler JA, Stupp SI. J Intern Med. 2010;267:71. doi: 10.1111/j.1365-2796.2009.02184.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Hartgerink JD, Beniash E, Stupp SI. Science. 2001;294:1684. doi: 10.1126/science.1063187. [DOI] [PubMed] [Google Scholar]
- (10).Jiang W, Kim BYS, Rutka JT, Chan WCW. Nat Nanotechnol. 2008;3:145. doi: 10.1038/nnano.2008.30. [DOI] [PubMed] [Google Scholar]
- (11).Chithrani DB. Mol Membr Biol. 2010;27:299. doi: 10.3109/09687688.2010.507787. [DOI] [PubMed] [Google Scholar]
- (12).Mitragotri S, Lahann J. Nat Mater. 2009;8:15. doi: 10.1038/nmat2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Schluep T, Hwang J, Hildebrandt IJ, Czernin J, Choi CHJ, Alabi CA, Mack BC, Davis ME. P Natl Acad Sci USA. 2009;106:11394. doi: 10.1073/pnas.0905487106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Geng Y, Dalhaimer P, Cai SS, Tsai R, Tewari M, Minko T, Discher DE. Nat Nanotechnol. 2007;2:249. doi: 10.1038/nnano.2007.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Muraoka T, Koh CY, Cui HG, Stupp SI. Angew Chem Int Edit. 2009;48:5946. doi: 10.1002/anie.200901524. [DOI] [PubMed] [Google Scholar]
- (16).Accardo A, Tesauro D, Del Pozzo L, Mangiapia G, Paduano L, Morelli G. J Pept Sci. 2008;14:903. doi: 10.1002/psc.1024. [DOI] [PubMed] [Google Scholar]
- (17).Paramonov SE, Jun HW, Hartgerink JD. J Am Chem Soc. 2006;128:7291. doi: 10.1021/ja060573x. [DOI] [PubMed] [Google Scholar]
- (18).Peters D, Kastantin M, Kotamraju VR, Karmali PP, Gujraty K, Tirrell M, Ruoslahti E. P Natl Acad Sci USA. 2009;106:9815. doi: 10.1073/pnas.0903369106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Torchilin VP, Lukyanov AN, Gao ZG, Papahadjopoulos-Sternberg B. P Natl Acad Sci USA. 2003;100:6039. doi: 10.1073/pnas.0931428100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Antoni P, Robb MJ, Campos L, Montanez M, Hult A, Malmstrom E, Malkoch M, Hawker CJ. Macromoiecuies. 2010;43:6625. [Google Scholar]
- (21).Li F, Prevost S, Schweins R, Marcelis ATM, Leermakers FAM, Stuart MAC, Sudholter EJR. Soft Matter. 2009;5:4169. [Google Scholar]
- (22).Bull SR, Palmer LC, Fry NJ, Greenfield MA, Messmore BW, Meade TJ, Stupp SI. J Am Chem Soc. 2008;130:2742. doi: 10.1021/ja710749q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Astafieva I, Khougaz K, Eisenberg A. Macromolecules. 1995;28:7127. [Google Scholar]
- (24).Kastantin M, Missirlis D, Black M, Ananthanarayanan B, Peters D, Tirrell M. J Phys Chem B. 2010;114:12632. doi: 10.1021/jp1001786. [DOI] [PubMed] [Google Scholar]
- (25).Bernal F, Tyler AF, Korsmeyer SJ, Walensky LD, Verdine GL. J Am Chem Soc. 2007;129:5298. doi: 10.1021/ja0693587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Missirlis D, Krogstad DV, Tirrell M. Mol Pharm. 2010;7:2173. doi: 10.1021/mp100193h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Bitton R, Schmidt J, Biesalski M, Tu R, Tirrell M, Bianco-Peled H. Langmuir. 2005;21:11888. doi: 10.1021/la051811p. [DOI] [PubMed] [Google Scholar]
- (28).Missirlis D, Khant H, Tirrell M. Biochemistry. 2009;48:3304. doi: 10.1021/bi802356k. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.