

Abstract
Intraprotein electron transfer (IET) from flavin mononucleotide (FMN) to heme is an essential step in nitric oxide (NO) synthesis by NO synthase (NOS). The IET kinetics in neuronal and inducible NOS (nNOS and iNOS) holoenzymes have been previously determined in our laboratories by laser flash photolysis [reviewed in: C.J. Feng, G. Tollin, Dalton Trans., (2009) 6692-6700]. Here we report the kinetics of the IET in a bovine endothelial NOS (eNOS) holoenzyme in the presence and absence of added calmodulin (CaM). The IET rate constant in the presence of CaM is estimated to be ~ 4.3 s-1. No IET was observed in the absence of CaM, indicating that CaM is the primary factor in controlling the FMN–heme IET in the eNOS enzyme. The IET rate constant value for the eNOS holoenzyme is approximately 10 times smaller than those obtained for the iNOS and CaM-bound nNOS holoenzymes. Possible mechanisms underlying the difference in IET kinetics among the NOS isoforms are discussed. Because the rate-limiting step in the IET process in these enzymes is the conformational change from input state to output state, a slower conformational change (than in the other isoforms) is most likely to cause the slower IET in eNOS.
Keywords: Nitric oxide synthase, Kinetics, Electron transfer, Mechanism
1. Introduction
Nitric oxide (NO), synthesized by the NO synthase (NOS) enzyme (EC 1.14.13.39), is a ubiquitous signaling molecule (at low nM concentrations) and a cytotoxin (at higher μM concentrations) [1, 2]. Aberrant NO synthesis by NOS is implicated in an increasing number of human pathologies, including stroke and cancer [2, 3]. Selective NOS modulators are required for therapeutic intervention because of the ubiquitous nature of NO in mammalian physiology, and the fact that multiple NOS isoforms are each capable of producing NO in vivo. Recent advances in understanding the structural and functional mechanisms of this enzyme class have led to identification of new agents that are designed to selectively modulate the various NOS isoforms [4-7].
There are three mammalian NOS isoforms: endothelial, neuronal and inducible NOS (eNOS, nNOS and iNOS, respectively). Under physiological conditions, NO produced by eNOS regulates vascular tone, insulin secretion, and smooth muscle tension, and NO produced by nNOS functions as a neurotransmitter [8]. These ‘constitutive’ isoforms (cNOSs) are Ca2+/calmodulin (CaM) regulated [9], generating NO as an intercellular messenger [2]. On the other hand, iNOS binds CaM irreversibly and is transcriptionally regulated. Two important differences for the CaM regulated isoforms of nNOS and eNOS, when compared to the Ca2+ insensitive isoform iNOS, are the presence of internal control elements [10], such as a unique autoregulatory insert within the FMN domain [11], and the sizes of their C-terminal tails [12].
Mammalian NOS catalyzes the oxidation of l-arginine (Arg) to NO and l-citrulline with NADPH and O2 as co-substrates [13]. NOS is a homodimeric flavo-hemoprotein comprised of an N-terminal oxygenase domain (containing a catalytic heme active site) and a C-terminal reductase domain, with a calmodulin (CaM) binding region between the two domains [10, 14]. The substrate, l-Arg, and a cofactor, (6R)-5,6,7,8 tetrahydrobiopterin (H4B), both bind near the heme center in the oxygenase domain [15].
The interdomain electron transfer (IET) processes are key steps in NO synthesis by coupling reactions between the flavin and heme domains (Figure 1) [10, 13, 16]. Uncoupled or partially coupled NOS results in production of reactive oxygen species such as superoxide and peroxynitrite [17]. Specifically, the CaM-controlled intersubunit FMN–heme IET is essential in coupling electron transfer in the reductase domain with NO synthesis in the heme domain by delivery of electrons required for O2 activation at the catalytic heme site [18]. It is generally accepted that CaM-binding has little or no effect on the thermodynamics of redox processes in NOS [19-22], indicating that the regulation of the IET processes within the enzyme by CaM binding is accomplished dynamically through controlling conformational changes required for effective IET. Because of their biomedical significance, it is of current interest to study the CaM-modulated FMN–heme IET in the NOS isoforms [16, 23, 24].
Figure 1.
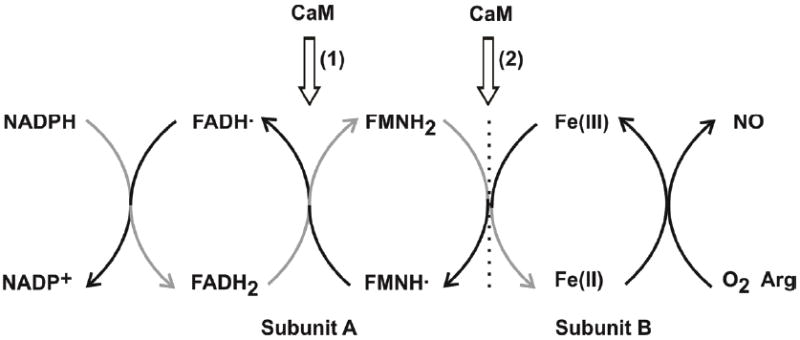
The electron flow in the dimeric NOS enzyme goes from NADPH through FAD and FMN in the reductase domain of one subunit (A) to the heme iron in the oxygenase domain of another subunit (B). The gray arrows indicate the direction of electron flow. CaM-binding to eNOS and nNOS isoforms facilitates two IET reactions: (1) IET between FAD and FMN within the reductase domain, and (2) inter-subunit IET between FMN and heme. The FMN–heme IET process (2) is physiologically essential in NO synthesis, and is of particular interest in this study. Adapted from ref. [29].
A laser flash photolysis approach, recently developed in our laboratories [25], has been used for direct determination of kinetics of the FMN–heme IET in the bi-domain oxygenase/FMN constructs [25-28] and in the full-length nNOS and iNOS enzymes [27, 29]. In this study the kinetics of the FMN–heme IET process in an eNOS holoenzyme was determined, and the kinetics results have been compared with our previous data of nNOS and iNOS holoenzymes.
2. Experimental procedures
2.1. Expression and purification of bovine eNOS holoenzyme
The original vector coding for bovine eNOS cloned into pCWOri was a generous gift to J.G.G. from Dr. Bettie Sue Masters (University of Texas Health Science Center at San Antonio, San Antonio, TX, USA). To assist in the purification, the expression vector was modified to contain an N-terminal polyhistidine tail cloned upstream from its start codon (vector peNOShisAmp). The bovine eNOS was coexpressed with GroESL (vector pGroESLChlor) in E. coli BL21 (DE3) as previously described [30]. The overall purification of the bovine eNOS enzyme involved the use of ammonium sulfate precipitation, followed by metal chelation chromatography, and finally 2’5’ ADP sepharose chromatography [31]
2.2. Laser flash photolysis
CO photolysis experiments were performed at room temperature, and the sample was kept in ice between flashes to stabilize the protein [25, 26, 29]. The laser apparatus and associated visible absorbance detection system have been described previously [32]. Briefly, a pulsed N2 laser (Photochemical Research Associates (PRA), London, Ontario, Canada) was used to pump a dye sample to give a nanosecond light pulse near 450 nm, which was focused onto the sample cuvette, and used to trigger the photochemical reactions in the sample. A 0.3 ml solution containing 20 μM 5-deazariboflavin (dRF) and 5 mM fresh semicarbazide in pH 7.6 buffer (40 mM Bis-Tris propane, 400 mM NaCl, 2 mM l-Arg, 20 μM H4B, 1 mM Ca2+ and 10 % glycerol) was degassed in a laser photolysis cuvette by a mixture of Ar and CO (with a ratio of ~ 3:1). l-Arg was added to keep oxidized heme in the catalytically relevant high spin state. Ca2+ was present in all the experiments involving subsequent addition of CaM (extracted from bovine brain, Sigma-Aldrich). The absorption change at 510 nm due to dRF semiquinone (dRFH•) formation and disappearance was monitored by using a pulse of 400 nm laser light (to check the degassing efficiency), prior to addition of microliter volumes of concentrated proteins. Aliquots of concentrated eNOS (170 μM) and CaM (2.2 mM) proteins were subsequently injected through a septum to achieve the desired concentrations, and the solution was kept in ice and further purged by passing the Ar/CO mixture over the solution surface for 45 min to remove minor oxygen contamination before being subjected to illumination. The solution in cuvette was then illuminated with steady white light for a certain period of time to obtain a partially reduced [Fe(II)–CO][FMNH•] form. The sample was subsequently flashed with 450 nm laser excitation to dissociate CO from Fe(II)–CO, and to generate a transient Fe(II) species that is able to transfer one electron to the FMNH• to produce FMNhq and Fe(III). This latter IET process was followed by the time-resolved loss of absorbance of FMNH• at 580 nm. Data from ~ 20 laser flashes were averaged, and transient absorbance changes were analyzed using OriginPro 8.5 (OriginLab).
3. Results and Discussion
3.1. Photochemical reduction of the oxidized bovine eNOS holoenzyme by dRF semiquinone
The UV-vis absorption spectrum of the oxidized eNOS sample in the presence of l-Arg (Figure 2a) has maximum absorbance at 395 nm, typical of a high spin penta-coordinated heme protein, and typical flavin peaks at 450 and 480 nm and a characteristic high spin ferriheme charge transfer band at 650 nm. The oxidized eNOS holoenzyme in the presence of dRF and CO was exposed to steady white light illumination for various periods of time to photoreduce the protein. The steady-state difference spectrum (inset of Figure 2b) has the characteristic peak of Fe(II)–CO at 446 nm and the broad band of flavin semiquinone around 580 nm. The UV–vis spectrum of the partially reduced eNOS possesses the characteristic absorption band of oxidized flavin at 480 nm (Figure 2b), although with reduced intensity owing to reduction of flavins. It is unlikely for the reduced protein to have oxidized FMN in the presence of FADH• because the midpoint potentials of the FMN/FMNH• and FAD/FADH• couples in the eNOS holoenzyme have been determined to be -105 mV and -230 mV, respectively [21]. We thus assigned the 480 nm band in Figure 2b to oxidized FAD. This showed that the eNOS holoenzyme is readily reduced to the [Fe(II)–CO][FMNH•] form by photo-illumination in the presence of dRF and CO under anaerobic conditions. Similar difference spectra were previously observed for the photo-reduced nNOS [29] and iNOS [27] holoenzymes. We are aware that since the sample does not contain any mediator, the di-semiquinone form, [Fe(II)-CO][FMNH•-FADH•] also may be formed under the experimental conditions. Due to the spectral similarities, it is difficult to distinguish between the FAD and FMN semiquinones. In what follows, we will assume that the major semiquinone species is that due to FMN.
Figure 2.
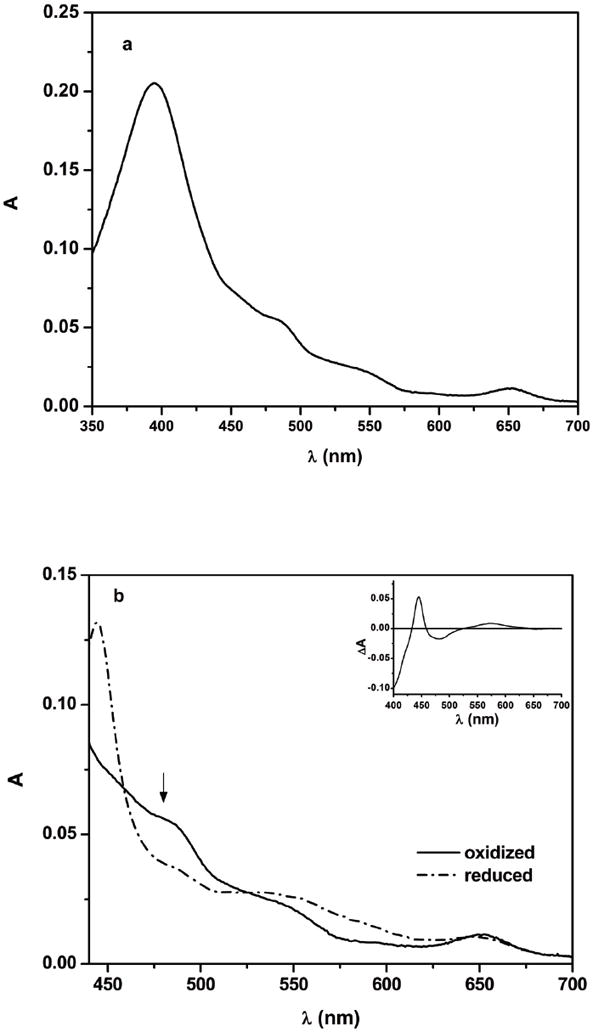
(a) Absorption spectrum of as-isolated bovine eNOS holoenzyme and (b) absorption spectra of the eNOS holoenzyme in the presence of dRF obtained before and after approximately 1 min of steady light illumination. b Solid line oxidized eNOS, dash dot line partially reduced eNOS; note the characteristic absorption of oxidized flavin at 480 nm, as indicated by an arrow. Inset of panel b, difference spectrum obtained after the light illumination. Anaerobic solutions contained 7.5 μM eNOS, ~ 20 μM dRF and 5 mM fresh semicarbazide in pH 7.6 buffer (40 mM Bis-Tris propane, 400 mM NaCl, 2 mM l-Arg, 20 μM H4B, 1 mM Ca2+, and 10 % glycerol). The sample was well degassed by Ar/CO (3:1) before illumination.
3.2. Electron transfer between the heme and FMN domains in the partially reduced CaM-bound eNOS holoenzyme
The partially reduced eNOS [Fe(II)–CO][FMNH•] sample with added CaM was then flashed by a 450 nm laser excitation pulse to dissociate CO from the Fe(II)–CO complex and form a transient Fe(II) species. Note that the loss of absorbance due to FMNH• reduction dominates the absorption at 580 nm [33]. Indeed, the flash-induced difference spectra (Figure 3) show that 580 nm gives the largest signal change in this region. Therefore this wavelength was selected to monitor the IET kinetics (see below for further discussion). Upon the 450 nm laser flash, the absorption at 580 nm decays below the pre-flash baseline (Figure 4a), with a rate constant of 4.3 ± 1.0 s-1, followed by a slower recovery toward the initial baseline (Figure 4b). Our previous studies showed that CO photolysis of the [Fe(II)–CO][FMNH•] form of iNOS holoenzyme [27] and CaM-bound rat nNOS holoenzyme [29] give a similar decay at 580 nm (although with more rapid kinetics) followed by a slower recovery (due to CO rebinding). Based on these observations, we have assigned the absorbance decay at 580 nm (Figure 4) to the following IET process between the catalytically significant redox couples of FMN and heme in the eNOS holoenzyme:
| eq 1 |
In eq 1, FMNhq stands for the FMN hydroquinone species generated by the IET reaction. Since the IET reaction is an equilibrium process, we are in fact measuring it in both directions; note that the CO photolysis technique follows the IET process in the reverse direction of the enzymatic turnover.
Figure 3.
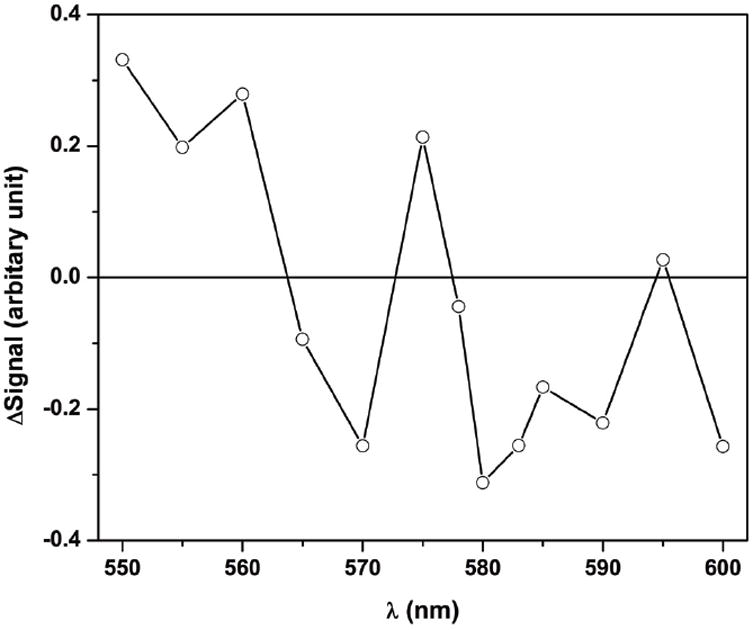
Laser flash-induced difference spectrum (550-600 nm) of the partially reduced eNOS holoenzyme (with added CaM) obtained 980 ms after the 450 nm laser pulse. Anaerobic solutions contained 14 μM eNOS, 35 μM CaM, ~ 20 μM dRF and 5 mM fresh semicarbazide in pH 7.6 buffer (40 mM Bis-Tris propane, 400 mM NaCl, 2 mM l-Arg, 20 μM H4B, 1 mM Ca2+ and 10 % glycerol). The sample was well degassed by Ar/CO (3:1).
Figure 4.
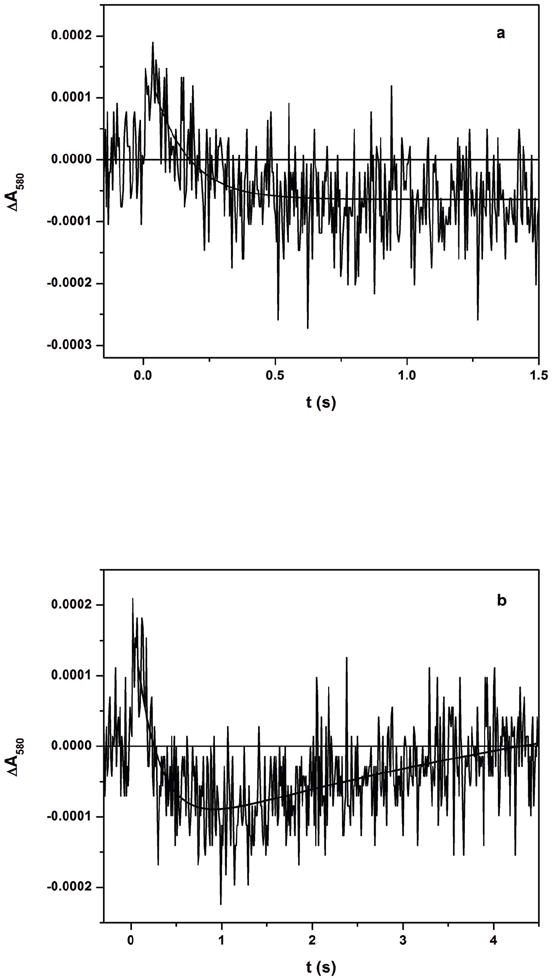
Transient traces at 580 nm at (a) 0 – 2 s and (b) 0 – 5 s obtained for [Fe(II)-CO][FMNH•] form of a bovine eNOS holoenzyme with added CaM flashed by 450 nm laser excitation. The traces in panels (a) and (b) were fitted with single exponential and biphasic models, respectively. Experimental conditions were the same as Figure 3.
Because the IET and CO rebinding phases have comparable rates (see Figures 4a and 4b), there is some overlap between these two phases in the traces. The fitting of the 2 s traces with a single exponential decay function gave 5.6 ± 0.9 s-1 (Figure 4a), while the 5 s traces fitting with a (two-exponential) biphasic model gave 3.4 ± 0.9 s-1 for the first phase (Figure 4b). These two rate constant values are similar, and a final value for the IET rate constant (4.3 ± 1.0 s-1) was obtained by averaging the values from both fittings (averaged over 10 individual traces). We did not attempt to estimate the rate constant for the second phase in the 5 s trace at 580 nm (i.e. the CO rebinding process) due to the significant overlap between the two phases. Instead, the CO rebinding rate was determined to be 1.02 ± 0.02 s-1 by fitting of the absorbance change at 455 nm where the Fe(II)–CO species dominates the absorbance (data not shown). The comparable IET and CO rebinding rates most likely contributes to the observed small amplitude of the kinetics trace at 580 nm because the two processes compete for free Fe(II) species, and their absorption changes at 580 nm go in opposite directions [27, 29]. Also note that the signal noise is nearly as large as the absorbance change, with a signal-to-noise ratio (S/N) ~ 1; see Figure 4. This will certainly affect the confidence in the obtained value of the rate constant. With a S/N value of 1, the absolute value of the rate constant could be uncertain by as much as ± 100 %; the value of ± 23 % given above reflects only the standard error in the computer fit to the data.
Miller et al. have reported a heme reduction rate of 2.3 s-1 in a bovine eNOS holoenzyme under similar conditions [35]. This rate obtained by stopped flow is comparable to the IET value in the present study (4.3 s-1). In light of the steady-state NO production activity of eNOS (kcat ~ 0.3 s-1) [10, 36], the FMN–heme IET is not the rate-limiting step in the NOS turnover, which is similar to the nNOS [29] and iNOS [27] holoenzymes.
The midpoint potentials for the FMNH•/FMNhq and FAD/FADH• couples in eNOS holoenzyme are nearly iso-potential (-240 mV and -230 mV, respectively) [21], and IET from FMNhq to FAD (after the FMN–heme IET) is thus also feasible. As such, the [Fe(III)][FMNhq-FAD] and [Fe(III)][FMNH•-FADH•] forms exist in an equilibrium distribution containing ~ 50% of each species, in which the formation of the bi-radical causes an increase of the absorbance at 580 nm. The rate of electron transfer from FMNhq to FAD may be relatively slow, however, because this IET occurs in the input state and the conversion of the output state back to the input state is expected to be a slow process (based on a gated conformational sampling mechanism for NOS IET [37]). Therefore, in the case of eNOS, the IET rate constant between heme and flavin may be modified by the extent and speed of the formation of bi-radical [FMNH•-FADH•]. This could complicate accurate measurements of rate constant of the FMN–heme IET in eNOS. However, this possibility seems unlikely for eNOS because: (i) the midpoint potentials for the FMNH•/FMNH2 and FAD/FADH• couples in eNOS and iNOS holoenzymes are similar [21], but their IET rates are different by ~10-fold, and (ii) the rate of flavin reduction in eNOS is quite fast compared to the observed IET rate for eNOS in this study. Furthermore, if this were the case one might expect biphasic kinetics for the IET process, which is not observed. The rate of flavin reduction correlates in a positive manner with the rate of heme reduction [35]. This is suggested by the different rates of NO synthesis exhibited by the three NOS isoforms, as determined by the rate of electron transfer between FAD and FMN or between FMN and the heme [38].
We are aware that in eNOS the conversion of Fe(II) to Fe(III) (eq 1 above) also causes loss of absorbance at 580 nm [34], and consequently the total amount of ΔA580 is increased. However, since these changes both proceed from the same IET reaction (eq 1), this will not affect the measured rate constant value, and only the amplitude of the absorbance change will change. To avoid any possible complications, it would be ideal to use the isosbestic points of the eNOS heme redox reaction, such as 532 and 615 nm [34]. However, the absorbance change due to the FMNH• reduction (eq 1) is negligibly small at these wavelengths [39]. Thus, 580 nm is an optimal choice for monitoring the eNOS IET kinetics. Also note that in nNOS the absorbance change at 580 nm due to the Fe(II)-to-Fe(III) conversion is also in the same direction as in eNOS, and 580 nm is not an isosbestic point for the nNOS heme re-oxidation process either [40].
3.3. Laser flash photolysis of the partially reduced eNOS holoenzyme in the absence of CaM
The spectral “transition” in Figure 4b (i.e. a reversal in direction of absorption changes over time) in the 580 nm trace was absent without added CaM (Figure 5), indicating the absence of FMNH• consumption, i.e. no FMN–heme IET occurred in the absence of CaM (also note that the trace does not go below the pre-flash baseline). Therefore IET between the FMN and heme domains in the eNOS holoenzyme was only observed in the presence of added CaM. This is consistent with our previous findings that CaM is the primary factor in controlling the FMN–heme IET in the NOS holoenzymes [28, 29].
Figure 5.
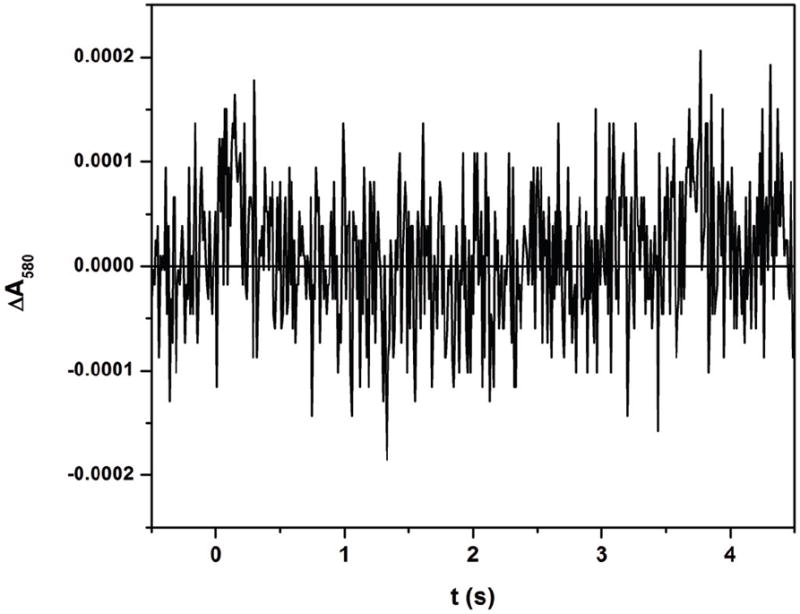
Transient trace at 580 nm at 0–5 s for [Fe(II)-CO][FMNH•] form of eNOS (10 μM) without added CaM flashed by 450 nm laser excitation. Note that the trace stays above the baseline, in contrast to Figure 4b. Experimental conditions were the same as Figure 3.
3.4. Comparison of the FMN–heme IET rate constants of the NOS isoforms
Table 1 lists the IET rate constants of iNOS and CaM-bound eNOS/nNOS holoenzymes. These IET kinetics were all determined by laser flash photolysis under the same conditions at room temperature. Note that the eNOS value is about ten times smaller than those of both nNOS and iNOS, and that the value for the nNOS holoenzyme is similar to that of iNOS. A similar order has been reported previously [14], although the kinetics was determined indirectly by a stopped-flow approach at 10 °C. The midpoint potentials of the Fe(III)/Fe(II) and FMNH•/FMNhq couples of the three NOS isoforms are similar [21]. Therefore the difference in the IET rate constants is very unlikely to be due to variation in the thermodynamic driving force. We discuss below the possible mechanisms underlying the difference in the IET kinetics of the NOS isoforms.
Table 1.
Rates of the FMN–heme IET of NOS holoenzymes determined by CO photolysis a
The IET kinetics were determined under the same conditions at room temperature; buffer: 40 mM Bis-Tris propane, 400 mM NaCl, 2 mM l-Arg, 20 μM H4B, 1 mM Ca2+ and 10 % glycerol, pH 7.6.
The absolute value of the rate constant could be uncertain by ± 100 % (due to small S/N). The error value of ± 1.0 s-1 given in the Table reflects only the standard error in the computer fit to the data.
Biological electron transfer systems depend on proper interactions between proteins/domains that shuttle electron(s) [41-43]. A “tethered shuttle” model has been originally proposed by Salerno and Ghosh [26, 44] and supported by recent kinetics studies [16, 24, 45], in which CaM activates NO synthesis in eNOS and nNOS through a conformational change of the FMN domain from its shielded electron-accepting (input) state to a new electron-donating (output) state, and bound CaM is required for proper alignment of the FMN and heme domains. Therefore several mechanisms can limit the dynamic FMN–heme IET process [46]: (i) the intrinsic rate constant may be slow in the output state due to imperfect alignment; (ii) the enzyme may spend only a small percentage of time in the docked state; and (iii) the conformational change is slow and gates the IET. Our previous studies indicate that in the holoenzyme the rate-limiting step in the FMN–heme IET is the conversion of the shielded electron-accepting (input) state to a new electron-donating (output) state [27, 29], i.e., the FMN–heme IET is a conformationally-gated process [37]. The observed IET rate may thus be the same as the conformation conversion rate, and therefore a slower conformational change is most likely the cause of the slower IET in eNOS (i.e. factor (iii) above). As noted above, the very small absorbance change observed in the IET experiments (Figure 4) is a consequence of this slow rate which overlaps the rate of the CO rebinding process. However, another factor might be that very little of the enzyme is in a state where the FMN and heme domains are in electrical communication (i.e. factor (ii) above), which could lead to small amount of IET. Further clarification may be obtained by determining the flavin fluorescence lifetime of NOS which may provide insight into the dynamics of the FMN domain conformational change [47]. Such experimental information may be useful to determine to what extent factor (iii) above is involved in the difference in IET kinetics among the NOS isoforms.
The interdomain contacts between the heme and FMN domains of NOS must be highly specific because if the FMN domain of nNOS is replaced by its counterpart from the P450 reductase, the resultant chimeric enzymes no longer support electron transfer from FMN to heme, despite their similar ET functions [48]. Crystal structures [49] and mutational studies [50, 51] have indicated that complementary electrostatic interaction between the FMN and heme domains is critical in guiding the interdomain docking in the nNOS isoform. However, the heme domain and FMN domain surfaces of nNOS and eNOS isoforms are similarly charged (based on protein sequence alignment). This implies that electrostatic interactions guiding the docking of FMN domain to the heme domain may be similar in the two isoforms, and may not directly cause the difference in the IET kinetics.
Importantly, CaM binding is critical in activating the IET, and the interactions between CaM and NOS domains are important in aligning the FMN and heme domains. In the absence of NOS holoenzyme crystal structures, it is difficult to envisage how CaM interacts with the NOS domains, and if the interactions are different in the NOS isoforms. It is thus important to utilize other techniques to provide insight into the interdomain FMN/heme alignment in the isoforms. We have recently used a pulsed EPR technique to directly measure the FMN⋯heme distance in iNOS [23]. This is significant because protein electron transfer is primarily controlled by the distance and orientation between redox centers [52]. Thus, a comparative study of the interdomain FMN⋯heme distance in the NOS isoforms is needed to determine if the FMN/heme alignment differs significantly in the NOS isoforms.
Additionally, it is likely that the NOS output state is populated only transiently [24, 53], and that this may limit the rate of heme reduction (factor (ii) above). Such a conformational sampling mechanism has been shown to limit electron transfer in a range of enzymes [54]. Spectroscopic approaches such as NMR and pulsed EPR may shed light on the conformational equilibrium of the FMN domain in the NOS isoforms.
In conclusion, the kinetics of the IET between the catalytically significant redox couples of the FMN and heme domains in eNOS holoenzyme has been directly determined by laser flash photolysis. The IET is activated by CaM binding to eNOS, supporting a critical role of CaM in alignment of the FMN and heme domains. The eNOS IET is distinctly slower than in the nNOS and iNOS isoforms, and potential mechanisms underlying the difference have been discussed. This work suggests the need for new efforts to carry out comparative structural and spectroscopic studies of the FMN/heme complexes in the NOS isoforms.
Research Highlights.
Intraprotein electron transfer (IET) between FMN and heme centers is essential for NOS catalysis.
The FMN–heme IET kinetics in eNOS holoenzyme was directly determined by laser flash photolysis.
The IET was not observed without added calmodulin (CaM), showing that CaM binding activates the IET.
The IET rate value is about 10 times smaller than those of the iNOS and CaM-bound nNOS holoenzymes.
Acknowledgments
This work was supported by grants from the National Institutes of Health (GM081811 and HL091280 to C.F.) and AHA Grant-in-Aid (09GRNT2220310 to C.F.) and the Natural Science and Engineering Research Council of Canada (18351 to JGG). The project described was also supported by Grant Number P20RR016480 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health. CF acknowledges the support of UNM HSC RAC grant. We thank helpful comments from a reviewer.
Abbreviations
- NO
nitric oxide
- NOS
nitric oxide synthase
- eNOS
endothelial NOS
- iNOS
inducible NOS
- nNOS
neuronal NOS
- CaM
calmodulin
- IET
intraprotein interdomain electron transfer
- FMN
flavin mononucleotide
- FMNH•
FMN semiquinone
- FMNhq
FMN hydroquinone
- FAD
flavin adenine dinucleotide
- FADH•
FAD semiquinone
- dRF
5-deazariboflavin
- dRFH•
5-deazariboflavin semiquinone
- H4B
(6R)-5,6,7,8-tetrahydrobiopterin
- l-Arg
l-arginine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schmidt H, Walter U. NO at work. Cell. 1994;78:919–925. doi: 10.1016/0092-8674(94)90267-4. [DOI] [PubMed] [Google Scholar]
- 2.Moncada S, Higgs EA. The discovery of nitric oxide and its role in vascular biology. Br J Pharmacol. 2006;147:S193–S201. doi: 10.1038/sj.bjp.0706458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lancaster JR, Xie KP. Tumors face NO problems? Cancer Res. 2006;66:6459–6462. doi: 10.1158/0008-5472.CAN-05-2900. [DOI] [PubMed] [Google Scholar]
- 4.Xue F, Li H, Delker SL, Fang J, Martásek P, Roman LJ, Poulos TL, Silverman RB. Potent, highly selective, and orally bioavailable gem-difluorinated monocationic inhibitors of neuronal nitric oxide synthase. J Am Chem Soc. 2010;132:14229–14238. doi: 10.1021/ja106175q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Delker SL, Ji H, Li H, Jamal J, Fang J, Xue F, Silverman RB, Poulos TL. Unexpected binding modes of nitric oxide synthase inhibitors effective in the prevention of a cerebral palsy phenotype in an animal model. J Am Chem Soc. 2010;132:5437–5442. doi: 10.1021/ja910228a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Silverman RB. Design of selective neuronal nitric oxide synthase inhibitors for the prevention and treatment of neurodegenerative diseases. Acc Chem Res. 2009;42:439–451. doi: 10.1021/ar800201v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garcin ED, Arvai AS, Rosenfeld RJ, Kroeger MD, Crane BR, Andersson G, Andrews G, Hamley PJ, Mallinder PR, Nicholls DJ, St-Gallay SA, Tinker AC, Gensmantel NP, Mete A, Cheshire DR, Connolly S, Stuehr DJ, Aberg A, Wallace AV, Tainer JA, Getzoff ED. Anchored plasticity opens doors for selective inhibitor design in nitric oxide synthase. Nat Chem Biol. 2008;4:700–707. doi: 10.1038/nchembio.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Salerno JC, Ghosh DK. Space, time and nitric oxide - neuronal nitric oxide synthase generates signal pulses. FEBS J. 2009;276:6677–6688. doi: 10.1111/j.1742-4658.2009.07382.x. [DOI] [PubMed] [Google Scholar]
- 9.Abu-Soud HM, Stuehr DJ. Nitric-oxide synthases reveal a role for calmodulin in controlling electron transfer. Proc Natl Acad Sci U S A. 1993;90:10769–10772. doi: 10.1073/pnas.90.22.10769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roman LJ, Martasek P, Masters BSS. Intrinsic and extrinsic modulation of nitric oxide synthase activity. Chem Rev. 2002;102:1179–1189. doi: 10.1021/cr000661e. [DOI] [PubMed] [Google Scholar]
- 11.Salerno JC, Harris DE, Irizarry K, Patel B, Morales AJ, Smith SME, Martasek P, Roman LJ, Masters BSS, Jones CL, Weissman BA, Lane P, Liu Q, Gross SS. An autoinhibitory control element defines calcium-regulated isoforms of nitric oxide synthase. J Biol Chem. 1997;272:29769–29777. doi: 10.1074/jbc.272.47.29769. [DOI] [PubMed] [Google Scholar]
- 12.Roman LJ, Martasek P, Miller RT, Harris DE, de la Garza MA, Shea TM, Kim JJP, Masters BSS. The C termini of constitutive nitric-oxide synthases control electron flow through the flavin and heme domains and affect modulation by calmodulin. J Biol Chem. 2000;275:29225–29232. doi: 10.1074/jbc.M004766200. [DOI] [PubMed] [Google Scholar]
- 13.Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: Structure, function and inhibition. Biochem J. 2001;357:593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stuehr DJ, Santolini J, Wang ZQ, Wei CC, Adak S. Update on mechanism and catalytic regulation in the NO synthases. J Biol Chem. 2004;279:36167–36170. doi: 10.1074/jbc.R400017200. [DOI] [PubMed] [Google Scholar]
- 15.Wei CC, Crane BR, Stuehr DJ. Tetrahydrobiopterin radical enzymology. Chem Rev. 2003;103:2365–2383. doi: 10.1021/cr0204350. [DOI] [PubMed] [Google Scholar]
- 16.Feng CJ, Tollin G. Regulation of interdomain electron transfer in the NOS output state for NO production. Dalton Trans. 2009:6692–6700. doi: 10.1039/b902884f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen CA, Druhan LJ, Varadharaj S, Chen YR, Zweier JL. Phosphorylation of endothelial nitric-oxide synthase regulates superoxide generation from the enzyme. J Biol Chem. 2008;283:27038–27047. doi: 10.1074/jbc.M802269200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Panda K, Ghosh S, Stuehr DJ. Calmodulin activates intersubunit electron transfer in the neuronal nitric-oxide synthase dimer. J Biol Chem. 2001;276:23349–23356. doi: 10.1074/jbc.M100687200. [DOI] [PubMed] [Google Scholar]
- 19.Dunford AJ, Rigby SEJ, Hay S, Munro AW, Scrutton NS. Conformational and thermodynamic control of electron transfer in neuronal nitric oxide synthase. Biochemistry. 2007;46:5018–5029. doi: 10.1021/bi7001339. [DOI] [PubMed] [Google Scholar]
- 20.Noble MA, Munro AW, Rivers SL, Robledo L, Daff SN, Yellowlees LJ, Shimizu T, Sagami I, Guillemette JG, Chapman SK. Potentiometric analysis of the flavin cofactors of neuronal nitric oxide synthase. Biochemistry. 1999;38:16413–16418. doi: 10.1021/bi992150w. [DOI] [PubMed] [Google Scholar]
- 21.Gao YT, Smith SME, Weinberg JB, Montgomery HJ, Newman E, Guillemette JG, Ghosh DK, Roman LJ, Martasek P, Salerno JC. Thermodynamics of oxidation-reduction reactions in mammalian nitric-oxide synthase Isoforms. J Biol Chem. 2004;279:18759–18766. doi: 10.1074/jbc.M308936200. [DOI] [PubMed] [Google Scholar]
- 22.Daff S, Noble MA, Craig DH, Rivers SL, Chapman SK, Munro AW, Fujiwara S, Rozhkova E, Sagami I, Shimizu T. Control of electron transfer in neuronal NO synthase. Biochem Soc Trans. 2001;29:147–152. doi: 10.1042/0300-5127:0290147. [DOI] [PubMed] [Google Scholar]
- 23.Astashkin AV, Elmore BO, Fan W, Guillemette JG, Feng C. Pulsed EPR determination of the distance between heme iron and FMN centers in a human inducible nitric oxide synthase. J Am Chem Soc. 2010;132:12059–12067. doi: 10.1021/ja104461p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stuehr DJ, Tejero J, Haque MM. Structural and mechanistic aspects of flavoproteins: Electron transfer through the nitric oxide synthase flavoprotein domain. FEBS J. 2009;276:3959–3974. doi: 10.1111/j.1742-4658.2009.07120.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feng CJ, Thomas C, Holliday MA, Tollin G, Salerno JC, Ghosh DK, Enemark JH. Direct measurement by laser flash photolysis of intramolecular electron transfer in a two-domain construct of murine inducible nitric oxide synthase. J Am Chem Soc. 2006;128:3808–3811. doi: 10.1021/ja0578606. [DOI] [PubMed] [Google Scholar]
- 26.Feng CJ, Tollin G, Holliday MA, Thomas C, Salerno JC, Enemark JH, Ghosh DK. Intraprotein electron transfer in a two-domain construct of neuronal nitric oxide synthase: The output state in nitric oxide formation. Biochemistry. 2006;45:6354–6362. doi: 10.1021/bi060223n. [DOI] [PubMed] [Google Scholar]
- 27.Feng CJ, Dupont A, Nahm N, Spratt D, Hazzard JT, Weinberg J, Guillemette J, Tollin G, Ghosh DK. Intraprotein electron transfer in inducible nitric oxide synthase holoenzyme. J Biol Inorg Chem. 2009;14:133–142. doi: 10.1007/s00775-008-0431-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feng C, Fan W, Dupont A, Guy Guillemette J, Ghosh DK, Tollin G. Electron transfer in a human inducible nitric oxide synthase oxygenase/FMN construct co-expressed with the N-terminal globular domain of calmodulin. FEBS Lett. 2010;584:4335–4338. doi: 10.1016/j.febslet.2010.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Feng CJ, Tollin G, Hazzard JT, Nahm NJ, Guillemette JG, Salerno JC, Ghosh DK. Direct measurement by laser flash photolysis of intraprotein electron transfer in a rat neuronal nitric oxide synthase. J Am Chem Soc. 2007;129:5621–5629. doi: 10.1021/ja068685b. [DOI] [PubMed] [Google Scholar]
- 30.Newman E, Spratt DE, Mosher J, Cheyne B, Montgomery HJ, Wilson DL, Weinberg JB, Smith SME, Salerno JC, Ghosh DK, Guillemette JG. Differential activation of nitric-oxide synthase isozymes by calmodulin-troponin C chimeras. J Biol Chem. 2004;279:33547–33557. doi: 10.1074/jbc.M403892200. [DOI] [PubMed] [Google Scholar]
- 31.Montgomery HJ, Perdicakis B, Fishlock D, Lajoie GA, Jervis E, Guy Guillemette J. Photo-control of nitric oxide synthase activity using a caged isoform specific inhibitor. Biorg Med Chem. 2002;10:1919–1927. doi: 10.1016/s0968-0896(02)00050-0. [DOI] [PubMed] [Google Scholar]
- 32.Feng C, Tollin G, Enemark JH. Sulfite oxidizing enzymes. Biochim Biophys Acta. 2007;1774:527–539. doi: 10.1016/j.bbapap.2007.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stuehr DJ, Ikeda-Saito M. Spectral characterization of brain and macrophage nitric-oxide synthases - Cytochrome-P-450-like hemeproteins that contain a flavin semiquinone radical. J Biol Chem. 1992;267:20547–20550. [PubMed] [Google Scholar]
- 34.Du M, Yeh HC, Berka V, Wang LH, Tsai AL. Redox properties of human endothelial nitric-oxide synthase oxygenase and reductase domains purified from yeast expression system. J Biol Chem. 2003;278:6002–6011. doi: 10.1074/jbc.M209606200. [DOI] [PubMed] [Google Scholar]
- 35.Miller RT, Martasek P, Omura T, Masters BSS. Rapid kinetic studies of electron transfer in the three isoforms of nitric oxide synthase. Biochem Biophys Res Commun. 1999;265:184–188. doi: 10.1006/bbrc.1999.1643. [DOI] [PubMed] [Google Scholar]
- 36.Martasek P, Liu Q, Liu JW, Roman LJ, Gross SS, Sessa WC, Masters BSS. Characterization of bovine endothelial nitric oxide synthase expressed in E-coli. Biochem Biophys Res Commun. 1996;219:359–365. doi: 10.1006/bbrc.1996.0238. [DOI] [PubMed] [Google Scholar]
- 37.Li W, Fan W, Elmore BO, Feng C. Effect of solution viscosity on intraprotein electron transfer between the FMN and heme domains in inducible nitric oxide synthase. FEBS Lett. 2011 doi: 10.1016/j.febslet.2011.07.022. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nishida CR, de Montellano PRO. Electron transfer and catalytic activity of nitric oxide synthases: Chimeric constructs of the neuronal, inducible, and endothelial isoforms. J Biol Chem. 1998;273:5566–5571. doi: 10.1074/jbc.273.10.5566. [DOI] [PubMed] [Google Scholar]
- 39.Munro AW, Noble MA, Robledo L, Daff SN, Chapman SK. Determination of the redox properties of human NADPH-cytochrome P450 reductase. Biochemistry. 2001;40:1956–1963. doi: 10.1021/bi001718u. [DOI] [PubMed] [Google Scholar]
- 40.Feng CJ, Roman LJ, Hazzard JT, Ghosh DK, Tollin G, Masters BSS. Deletion of the autoregulatory insert modulates intraprotein electron transfer in rat neuronal nitric oxide synthase. FEBS Lett. 2008;582:2768–2772. doi: 10.1016/j.febslet.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tollin G. Interprotein and intraprotein electron transfer mechanisms. Electron Transfer in Chemistry. 2001;IV:202–231. [Google Scholar]
- 42.Leys D, Scrutton NS. Electrical circuitry in biology: Emerging principles from protein structure. Curr Opin Struct Biol. 2004;14:642–647. doi: 10.1016/j.sbi.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 43.Hoffman BM, Celis LM, Cull DA, Patel AD, Seifert JL, Wheeler KE, Wang JY, Yao J, Kurnikov IV, Nocek JM. Differential influence of dynamic processes on forward and reverse electron transfer across a protein-protein interface. Proc Natl Acad Sci U S A. 2005;102:3564–3569. doi: 10.1073/pnas.0408767102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ghosh DK, Salerno JC. Nitric oxide synthases: Domain structure and alignment in enzyme function and control. Front Biosci. 2003;8:D193–D209. doi: 10.2741/959. [DOI] [PubMed] [Google Scholar]
- 45.Welland A, Garnaud PE, Kitamura M, Miles CS, Daff S. Importance of the domain-domain interface to the catalytic action of the NO synthase reductase domain. Biochemistry. 2008;47:9771–9780. doi: 10.1021/bi800787m. [DOI] [PubMed] [Google Scholar]
- 46.Daff S. NO synthase: Structures and mechanisms. Nitric Oxide. 2010;23:1–11. doi: 10.1016/j.niox.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 47.Salerno JC, Ghosh DK, Ray K, Adrados M, Nahm N, Li H, Poulos TI, Lakowicz J. FMN fluorescence in iNOS constructs reveals a series of conformational states involved in the reductase catalytic cycle. Nitric Oxide-Biology and Chemistry. 2010;22:S12–S12. [Google Scholar]
- 48.Roman LJ, McLain J, Masters BSS. Chimeric enzymes of cytochrome P450 oxidoreductase and neuronal nitric-oxide synthase reductase domain reveal structural and functional differences. J Biol Chem. 2003;278:25700–25707. doi: 10.1074/jbc.M212309200. [DOI] [PubMed] [Google Scholar]
- 49.Garcin ED, Bruns CM, Lloyd SJ, Hosfield DJ, Tiso M, Gachhui R, Stuehr DJ, Tainer JA, Getzoff ED. Structural basis for isozyme-specific regulation of electron transfer in nitric-oxide synthase. J Biol Chem. 2004;279:37918–37927. doi: 10.1074/jbc.M406204200. [DOI] [PubMed] [Google Scholar]
- 50.Tejero J, Hannibal L, Mustovich A, Stuehr DJ. Surface charges and regulation of FMN to heme electron transfer in nitric-oxide synthase. J Biol Chem. 2010;285:27232–27240. doi: 10.1074/jbc.M110.138842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Panda K, Haque MM, Garcin-Hosfield ED, Durra D, Getzoff ED, Stuehr DJ. Surface charge interactions of the FMN module govern catalysis by nitric-oxide synthase. J Biol Chem. 2006;281:36819–36827. doi: 10.1074/jbc.M606129200. [DOI] [PubMed] [Google Scholar]
- 52.Page CC, Moser CC, Chen XX, Dutton PL. Natural engineering principles of electron tunnelling in biological oxidation-reduction. Nature. 1999;402:47–52. doi: 10.1038/46972. [DOI] [PubMed] [Google Scholar]
- 53.Ilagan RP, Tejero JS, Aulak KS, Ray SS, Hemann C, Wang Z-Q, Gangoda M, Zweier JL, Stuehr DJ. Regulation of FMN subdomain interactions and function in neuronal nitric oxide synthase. Biochemistry. 2009;48:3864–3876. doi: 10.1021/bi8021087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hay S, Brenner S, Khara B, Quinn AM, Rigby SEJ, Scrutton NS. Nature of the energy landscape for gated electron transfer in a dynamic redox protein. J Am Chem Soc. 2010;132:9738–9745. doi: 10.1021/ja1016206. [DOI] [PubMed] [Google Scholar]