

Abstract
Combined antiretroviral therapy (ART) tremendously improved the lifespan and symptoms associated with AIDS-defining illness in affected individuals. However, chronic ART-treated patients frequently develop age-dependent complications, including dementia, diabetes, and hyperlipidemia: all risk factors of Alzheimer’s disease. Importantly, the effect of ART compounds on amyloid generation and clearance has never been systematically examined. Nine prescribed HIV protease inhibitors were tested for their effect on amyloid-β peptide (Aβ) clearance in primary cultured human monocyte-derived macrophages. Atazanavir, ritonavir, and saquinavir modestly inhibited of Aβ degradation, while lopinavir, nelfinavir, and ritonavir enhanced secretion of undigested Aβ after phagocytosis. Lopinavir, nelfinavir, ritonavir, and saquinavir inhibited endogenous Aβ40 production from primary cultured human cortical neurons, which were associated with reduction in Beta-site APP Converting Enzyme 1 (BACE1) and γ-secretase enzyme activities. However, ARTcompounds showed little inhibition of purified BACE1 activity in vitro, suggesting the indirect effect of ART compounds on BACE1 activity in neurons. Finally, nefinavir or lopinavir/ritonavir (Kaletra) were orally administered for 30 days into APP SCID mice expressing a double mutant form of APP 695 (KM670/671NL + V717F) in homozygosity for the scid allele of Prkdc. There was no difference in beta-amyloidosis by ART drug administration as determined by both immunohistochemistry and ELISA measurements although the therapeutic doses of the ART compounds was present in the brain. These data demonstrated that ART drugs can inhibit Aβ clearance in macrophages and Aβ production in neurons, but these effects did not significantly alter Aβ accumulation in the mouse brain.
Keywords: Alzheimer’s disease, Amyloid-β peptide, Animal model, Antiretroviral therapy, Macrophage, Neuron
Introduction
Alzheimer’s disease (AD), characterized by progressive cognitive decline and disability, is the most common form of senile dementia (Selkoe 2001). It is typified by extensive brain amyloid deposition, neurofibrillary tangle formation, neuroinflammation, and ultimately, synaptic and neuronal loss. To date, there is still no curative treatment for AD, and there is still much more that needs to be understood behind the mechanisms of the disease. One highly regarded mechanism underlying the causes of AD suggests that the accumulation of amyloid-β peptide (Aβ) is a crucial event in the initiation and maintenance of the neuronal degeneration that occurs in AD patients, as evidenced by many studies (Bard et al. 2000; Forman et al. 2004; Hardy and Selkoe 2002; Selkoe 2001).
Aβ is secreted from the endoproteolytic processing of amyloid precursor protein (APP), which is a causative gene of familial AD (Selkoe 2001). APP undergoes initial cleavage by β-site APP-cleaving enzyme (BACE) 1, producing an N-terminal-secreted fragment of APP and a membrane-anchored 11 kDa C-terminal fragment (APPct) (Vassar et al. 1999). APPct is further cleaved at a site within its transmembrane domain by the γ-secretase enzyme complex, leading to the production of various Aβ species, predominantly 40 and 42 residues in length (Hardy and Selkoe, 2002). Alternatively, full-length APP can be cleaved by α-secretase, releasing a soluble fragment extracellularly and precluding Aβ generation (Kojro and Fahrenholz 2005). Aβ peptides are physiologically cleared from the brain via export to the cerebrovasculature, degradation by Aβ-degrading enzymes such as neprilysin, insulin degrading enzyme (IDE), and matrix metalloproteases, or phagocytosis by mononuclear phagocytes (resident microglia and perivascular macrophages) (Xu and Ikezu 2009). Reduction in Aβ clearance leads to aggregation and accumulation of Aβ fibrils, which are mainly composed of Aβ42, a longer version of the Aβ peptide. Mononuclear phagocytes play a key role in the clearance of Aβ fibrils via phagocytosis and intracellular degradation, which are compromised by pro-inflammatory activation, viral infection, or drug treatment (Lan et al. 2011; Yamamoto et al. 2007; Yamamoto et al. 2008).
Human immunodeficiency virus (HIV) infection frequently causes HIV-1-associated neurocognitive disorders (HAND) (Heaton et al. 2011). Since the widespread application of combination antiretroviral therapy (ART), the incidence of acquired immunodeficiency syndrome (AIDS)-defining illnesses, like opportunistic infections and central nervous system neoplasms, has been greatly decreased (Palella et al. 1998). Accordingly, the life span of HIV-infected individuals has been significantly prolonged (Besson et al. 2001), leading to more frequent development of age-dependent complications, neurocognitive disorders, diabetes, cardiovascular disease, bone disease, overall frailty, and non-AIDS malignancies (Llibre et al. 2009). Concern also exists about the increased incidence of AD as the HIV-infected population ages. The impact of ART on neurodegeneration has been controversial. The ART also cause immune reconstitution syndrome or lypodystrophic effects causing hyperlipidemia, diabetes, and coronary artery disease (Xu and Ikezu 2009). In addition, the existence of persistent neurodegeneration was reported in the context of successful viral suppression in ART-treated patients (Brew et al. 2009). This is thought to be a consequence of poor penetration of ART compounds into the central nervous system (CNS), allowing the CNS to function as a viral sanctuary site. The physiology of antiretroviral penetration into the cerebrospinal fluid (CSF) is complex and depends on several factors, such as the molecular characteristics of the drugs, characteristics of the blood brain barrier (BBB) and the presence of active drug transporters that limit the availability of certain drugs (Varatharajan and Thomas 2009). In general, HIV protease inhibitors are highly plasma protein-bound and have poor penetration into CSF. On the other hand, some people have found that HIV-1 protease inhibitors also play a role in neuroprotection. For example, Hisatomi et al reported that some HIV protease inhibitors inhibited mitochondrial apoptosis and thus provided neuroprotection in mice (Hisatomi et al. 2008). This controversy displays the necessity for further study on the relationship between ART and neurodegeneration.
As part of antiretroviral therapy, HIV protease inhibitors have been widely used to treat viral infection. Protease inhibitors prevent viral replication by inhibiting the activity of proteases, e.g. HIV-1 protease, enzymes used by the viruses to cleave nascent proteins for final assembly of new virions. Until now, ten protease inhibitors have been approved by the Food and Drug Administration (FDA), including nelfinavir, lopinavir, saquinavir, ritonavir, indinavir, amprenavir, atazanavir, fosamprenavir, darunavir, and tipranavir. Among those drugs, atazanavir and darunavir are recommended for first-line therapy, and fosamprenavir and lopinavir as components of alternative regimens [http://www.aidsinfo.nih.gov/guidelines]. All protease inhibitors are used in combination with a low dose of ritonavir to boost plasma concentrations. To investigate the impact of ART on AD, we performed this study to examine the effect of ART drugs on Aβ clearance in human monocyte-derived macrophages (MDM) and on Aβ production in primary human cortical neurons. Results showed that select ART drugs had a modest effect on Aβ clearance in MDM, whereas lopinavir, nelfinavir, ritonavir, and saquinavir potently inhibited the β and γ-secretase activities and reduced Aβ production from human neurons. On the other hand, the nelfinavir and lopinavir/ritonavir (Kaletra®) had no effect on Aβ accumulation in an AD mouse model brain, suggesting the limitation of the biological activity of ART drugs on beta-amyloidosis due to their poor CNS penetration in vivo.
Materials and methods
ART drugs
Amprenavir, atazanavir, darunavir, indinavir, lopinavir, nelfinavir, ritonavir, saquinavir, and tipranavir were obtained from NIH AIDS Research and Reference Reagent Program, and were resolved in dimethyl sulfoxide (DMSO, Sigma-Aldrich, St. Louis, MO) to make 20 mM stocking solutions which were stored at −20°C. Before being dissolved in BACE1 reaction buffer or neuron medium, all various concentrations of ART drugs were first prepared so that all the assay groups contained the same amount of DMSO. For the animal studies, co-formulated lopinavir/ritonavir (Kaletra®, Abbott Laboratories, Abbott Park, IL), and nelfinavir (AG1343, Pfizer Inc, Groton, CT) were used.
Cell culture
Isolation and cultivation of monocytes were performed as previously reported (Yamamoto et al. 2008). Briefly, human monocytes were recovered from peripheral blood mononuclear cells (PBMC) of HIV-1 and hepatitis B seronegative donors after leukopheresis and purified by countercurrent centrifugal elutriation (Gendelman et al. 1988). Monocytes were cultured in Medium A containing Dulbecco’s minimal essential media (DMEM, Invitrogen, Carlsbad, CA, USA) supplemented with 10% heat-inactivated human serum, 2 mM L-glutamine, 50 μg/mL gentamicin, 10 μg/mL ciprofloxacin, and 1000 U/mL macrophage-colony stimulating factor (M-CSF, R&D Systems, Minneapolis, MN, USA). Monocytes were cultivated in Medium A for 7 days allowing their differentiation into macrophages, which were then referred to as MDM and maintained in Medium A without M-CSF (Medium B).
Human fetal brain tissue (12–16 weeks postconception) was obtained from the Birth Defects Laboratory, University of Washington, Seattle, in full compliance with the ethical guidelines of the NIH and the Institutional Review Board of the University of Nebraska Medical Center. Human neurons were isolated from fetal brain tissue cortices as previously described (Peng et al. 2004). Cells were plated onto poly-D-lysine-coated 6-well plates at a density of 1×106 cells/well for extraction protein; in 48-well plates at 1×105 cells/well for ELISA or MTT assays. Cells were cultured in Neurobasal Media supplemented with B27, 1 mM L-glutamine, and 2% heat-inactivated human serum (all from Invitrogen). The medium was half-changed every 4 days, and the cultivation lasted for 2–3 weeks allowing the cells to differentiate into neurons. Well-differentiated human neurons were then treated with 25 or 50 μM lopinavir, nelfinavir, ritonavir, and saquinavir for 48 h. The media were collected for the Aβ40 ELISA assay (Invitrogen), while the neurons were used for the MTT assay or protein extraction.
125I-Aβ degradation study in MDM
Iodinated Aβ42 peptides were prepared using IODO-beads (Pierce, Rockford, IL), synthetic human Aβ42 peptide (Invitrogen), and iodine-125 (125I, Amersham Bioscience, Piscataway, NJ) according to the manufacturer’s instructions. 125I-Aβ42 peptides were aggregated by stirring at 37°C for 3 days, followed by centrifugation at 40,000 × g for 20 min at 4°C. The precipitated fraction was used as fibrillar Aβ. Human MDM (500,000 cells/well) were pulse-labeled with fibrillar 125I-Aβ (200,000 cpm/well) at the final concentration of 1 μM in tissue culture media for 1 h at 37°C, washed three times with PBS, and chased with fresh tissue culture media for 72 h in the presence or absence of the following compounds: DMSO, ART drugs, and thiorphan at various concentrations (1-100 μM). At each time point, media were collected and cells lysed in lysis buffer (1 M NaOH) for counting intracellular Aβ. 99% of this fraction was undigested Aβ as determined by trichloroacetic acid (TCA) precipitation (final concentration of 10%). The collected media were subsequently mixed with 10% TCA for polypeptide precipitation, and centrifuged at 3,000 × g for 15 min at 4°C. The radioactivity level of both the TCA-soluble (degraded Aβ) and the precipitated (non-degraded Aβ) fractions were determined for calculating the fractions of intracellular Aβ, extracellular intact Aβ, and extracellular degraded Aβ.
Measurement of Aβ in neuron culture
The amounts of Aβ40 and Aβ42 in neuron media were quantitatively determined with Aβ40/42 Human ELISA Kits (Invitrogen) following the manufacturer’s instructions. MTT assay values were used to normalize the amounts of Aβ in each well.
MTT assay
The viability of human neurons in the presence of ART drugs was estimated by determining the activity of mitochondrial dehydrogenases with 3(4,5-dimethylthiazol-2-yl)-2.5-diphenyltetrazolium bromide (MTT) assay (Sigma-Aldrich). ART drug-treated neurons were supplemented with neuron differentiation medium containing 0.5 mg/mL MTT (Sigma-Aldrich), and incubated at 37°C for 4 h. After the incubation, the media were carefully removed, and 300 μl DMSO was added to the cells, followed with incubation at room temperature in the dark for 10 min with shaking. Then, the resulting solution was transferred into a 96-well plate, and the absorbance of the samples was measured at a wavelength of 490 nm.
Measurement of BACE1 activity in solution
The activity assay was carried out on recombinant human BACE1 protein (R & D systems) following the manufacturer’s instructions with some modifications. Briefly, 1 μg BACE1 protein was mixed with ART drugs (25, 50, 100, or 300 μM) in 80 μL of the reaction buffer (100 mM sodium acetate, pH 4.5, 100 ng heparin, Sigma-Aldrich) at room temperature for 30 min with gentle shaking, and the resulting solution was designated as Solution A. Then, 50 μM fluorogenic substrate Mac-SEVNLDAEFRK(Dnp)-R-R-NH2 (Substrate IV) (R & D systems) in 20 μL reaction solution was added into Solution A, and incubated in the dark for 1 h at 37°C for enzymatic reaction. Fluorescent intensity of the cleaved fragments was measured by fluorometry with excitation at 320 nm and emission at 405 nm.
Immunoblotting
The protein (20 μg) from the neuron lysate was electrophoresed on 16% SDS-PAGE and transferred to Immuno-Blot PVDF membranes (Bio-Rad Laboratory, Hercules, CA), followed by incubation in blocking buffer (5% skim fat milk in TBST) for 1 h at room temperature. Membranes were then incubated overnight at 4°C with primary antibodies against APP-CT (1:5000 dilution; rabbit polyclonal, Cat#171610, EMD Chemicals, Gibbstown, NJ), and β–actin (1:10,000 dilution; mouse monoclonal, AC-15, Sigma-Aldrich). Subsequently, membranes were incubated with horseradish peroxidase-conjugated secondary anti-rabbit secondary antibodies (1:10,000 dilution; Jackson ImmunoResearch Laboratories, West Grove, PA) for 1 h at room temperature. Immunoreactive bands were visualized by enhanced chemiluminescene (Amersham Bioscience) and captured with CL-XPosure Film (Pierce).
Animal study
To evaluate the impact of HIV-1 protease inhibitors on Aβ production in mice we produced immunodeficient mice expressing a human APP695 transgene with the KM670/671NL and V717F familial Alzheimer’s disease mutations (Chishti et al. 2001). Crosses were performed between BALB/cBy-Prkdcscid mice (obtained from The Jackson Laboratory, Bar Harbor, ME) and 129.Tg(APPNLI)CRND8 mice selecting for the presence of the transgene and for homozygosity for the scid allele of Prkdc as previously described (Chishti et al. 2001; Tyor et al. 1993). The shorthand “APP SCID” is used for APP transgene heterozygous, Prkdcscid/ Prkdcscid homozygous mice. Immunodeficiency caused by homozygosity for the scid mutation did not prevent or delay amyloid plaque formation or microgliosis. Plaque numbers, plaque size, and amyloid burden did not differ between APP SCID mice and immunocompetent Tg(APPNLI)CRND8 transgenic mice (data not shown). All the mice had non-viral disease, and were treated from 2 months old for 30 days.
The APP-SCID mice received oral administration of nelfinavir (AG1343, 30 mg/kg/day, b.i.d.), co-formulated lopinavir/ritonavir (Kaletra® containing 200 mg lopinavir and 50 mg ritonavir in one tablet, ground to administer 200 mg/kg/day, b.i.d.), or vehicle (n=3 per group). The mice were sacrificed 4 weeks after the treatment, and cortexes and hippocampi were collected. The collected tissues were homogenized in 10 volumes of ice-cold guanidine buffer (5.0 M guanidine-HCl, 50 mM Tris-HCl, pH8.0). The homogenates were mixed at room temperature for 3 to 4 h, then either assayed or stored at −80°C. The protein concentrations were determined by standard BCA, and the Aβ concentrations were measured with Aβ 40/42 Human ELISA Kits (Invitrogen) following the manufacturer’s instructions.
Immunohistochemistry was performed as described previously (Kiyota et al. 2010; Kiyota et al. 2009a, b). Briefly, mice were anesthetized with isoflurane and transcardially perfused with 25 ml of ice-cold PBS. The brains were rapidly removed, immersed in freshly depolymerized 4% paraformaldehyde for 48 h at 4°C, and cryoprotected by successive 24-h immersions in 15% and 30% sucrose in 1× PBS. Fixed, cryoprotected brains were frozen and sectioned coronally using a Cryostat (Leica, Bannockburn, IL, USA) and stored at −80°C. Immunohistochemistry was performed using anti-Aβ rabbit polyclonal antibody (1:100, Invitrogen). Immunodetection was visualized using Envision Plus (DAKO, Carpenteria, CA, USA) with 3, 3′-diaminobenzidine (Vector Laboratories, Burlingame, CA, USA).
Drug measurement
The concentration of nelfinavir in mouse brain was performed using an Agilent 1200 reverser-phase high performance liquid chromatography (RP-HPLC) system and diode array detector (Agilent Technologies, Santa Clara, CA, USA). The drug was extracted from the brain tissue by mixing with the equal volume of chloroform and then vortexing, followed by centrifugation at 12,000 rpm for 1 min. The organic phase was collected, and the extraction was repeated three times. The pooled organic phase was lyophilized and the sample was dissolved in 100 μl of the mobile phase buffer. Each 5 μl sample and standard samples (nelfinavir with known concentrations) were injected into the RP-HPLC column (4.6 150 mm, 3.5 m ZORBAX Eclipse AAA analytical columns, Agilent), and the drug peak was monitored using a diode array detector with wavelengths of 230 nm. A gradient elution program was optimized for nelfinavir measurement, which peaked at 2.12 min, with a flow rate 1.2 ml/min. This program began at 100% mobile phase A, containing 40 mM sodium dihydrogen phosphate (pH 7.8). Gradient elution started at 2 min with mobile phase B containing 45% acetonitrile and 45% methanol from 0 to 60% in 7 min and 100% thereafter for an additional 18 min. Total analysis time was 26 min. The drug concentration was determined by the peak area of the eluted time point of injected drug standards and samples and by normalization of the original concentration based on the chloroform extraction efficiency and the protein concentration of the samples.
Statistical tests
All the data are presented as means ± standard error (SEM) unless otherwise noted. All experiments were repeated at least three times with different donors, and all data were evaluated statistically by the analysis of variance (ANOVA), followed by Turkey multiple comparison tests using software (Prism 4.0, GraphPad Software, Seattle, WA). In the case of single mean comparison, data were analyzed by t test. P values of <0.05 were regarded as statistically significant.
Results
Effect of ART drugs on Aβ clearance in MDM
Using primary cultured MDM, the effect of ART drugs on the clearance of Aβ42 fibrils were tested using pulse-chase analysis of 125I-Aβ42 peptides, which were pulse-labeled for 1 h, followed by chasing for 72 h. The 125I-Aβ42 peptides were fractionated as intracellular (mostly undigested fibril Aβ42), TCA-soluble extracellular (digested Aβ42), and TCA-insoluble extracellular fractions (undigested Aβ42) (Table 1). Among the nine ART drugs tested, none of them showed statistically significant changes in the intracellular fraction of Aβ42, although select drugs (atazanavir, darunavir, indinavir, ritonavir, saquinavir, and tipranavir) retained more intracellular Aβ42. However, atazanavir, ritonavir, and saquinavir showed significant reduction in the extracellular TCA-soluble fraction, indicating that Aβ degradation was partly inhibited by the drugs. Interestingly we saw more apparent increase in the TCA-insoluble fraction when treated with thiorphan, a neprilysin inhibitor, lopinavir, nelfinavir, or ritonavir, suggesting that phagocytized Aβ fibrils were more prone to exocytosis without digestion in the presence of select ART drugs. The drugs that showed significant changes in Aβ clearance were further tested in different doses in MDM. As shown in Fig. 1, none of the doses showed significant changes in the intracellular fraction of Aβ42 (Fig. 1a), whereas atazanavir, ritonavir, and saquinavir showed a significant reduction in the TCA-soluble extracellular fraction at 50 or 100 μM but not 1 or 10 μM doses (Fig. 1b). The TCA-insoluble extracellular fraction was significantly increased by thiorphan, lopinavir, nelfinavir, and ritonavir at 50 or 100 μM but not 1 or 10 μM doses, except nelfinavir at 10 μM (Fig. 1c). These data indicated that ART drugs modestly change Aβ clearance in MDM at higher concentrations only in the extracellular fractions, ruling out the possibility that ART drugs potently altered Aβ clearance in peripheral macrophages.
Table 1.
125I-Aβ42 clearance in MDM
| Intracellular |
Extracellular TCA-soluble |
Extracellular TCA-insoluble |
||||
|---|---|---|---|---|---|---|
| Mean | Std. Error | Mean | Std. Error | Mean | Std. Error | |
| DMSO | 31.99 | 4.012 | 57.23 | 3.844 | 10.78 | 0.4983 |
| Thiorphan | 39.35 | 3.086 | 39.76 | 5.02 | 20.89* | 3.566 |
| Amprenavir | 43.7 | 4.328 | 42.29 | 4.476 | 14.01 | 1.173 |
| Atazanavir | 53.11 | 5.857 | 33.59* | 4.478 | 13.3 | 1.58 |
| Darunavir | 42.59 | 3.58 | 41.6 | 3.384 | 15.81 | 0.8098 |
| Indinavir | 44.24 | 5.395 | 41.13 | 5.408 | 14.62 | 0.6964 |
| Lopinavir | 33.27 | 5.08 | 35.36 | 5.587 | 31.37** | 8.235 |
| Nelfinavir | 37.98 | 4.389 | 38.48 | 2.374 | 23.54** | 3.018 |
| Ritonavir | 41.79 | 4.063 | 34.33* | 3.843 | 23.88** | 2.883 |
| Saquinavir | 52.06 | 4.439 | 34.34* | 4.159 | 13.6 | 1.934 |
| Tipranavir | 45.7 | 6.775 | 39.56 | 5.496 | 14.74 | 1.525 |
denote p<0.05 and 0.01 vs. DMSO-treated group as determined by ANOVA and Turkey post hoc
denote p<0.05 and 0.01 vs. DMSO-treated group as determined by ANOVA and Turkey post hoc
Fig. 1.
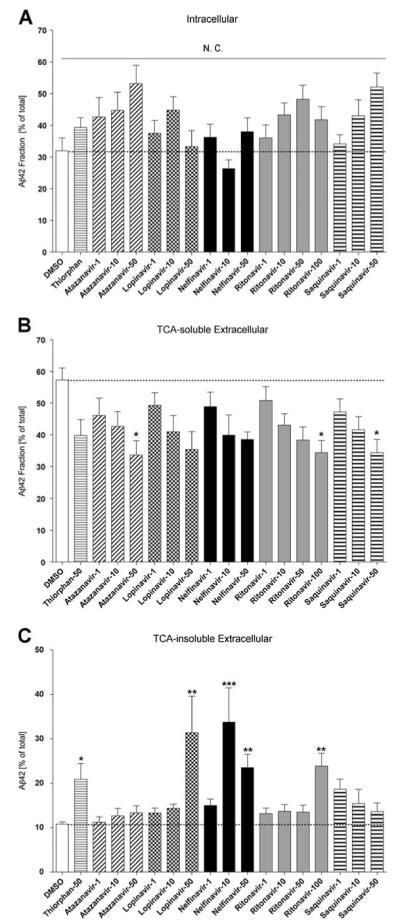
The effect of ART drugs on Aβ42 fibril clearance in MDM. a–c, MDM were pulse-labeled with aggregated 125I-Aβ42 and chased with fresh tissue culture media for 72 h in the presence or absence of ART drugs (1, 10, 50, and 100 μM) and thiorphan (neprilysin inhibitor). After chasing, the total cell lysate was collected and subjected to γ-counting, which represented the intracellular 125I-Aβ retention (a), extracellular TCA-soluble 125I-Aβ (b) and insoluble 125I-Aβ fractions (c). Each fraction was presented as % total 125I-Aβ42 (a sum of each fraction for each group). The dotted line shows the %fraction value of DMSO-treated MDM as a control group. *, **, or *** denotes p<0.05, 0.01, or 0.001 vs. DMSO-treated control MDM (open column) as determined by ANOVA and Turkey post hoc, respectively. N.C.; no statistically significant change
ART drugs decreased Aβ production in neurons
Second, we examined the effect of ART drugs on Aβ production in human neurons. Differentiated neurons were cultured with medium containing 25 or 50 μM ART drugs for 48 h. Media were collected for Aβ detection with an ELISA kit, while the cells underwent the MTT assay. As shown in Fig. 2a, lopinavir, nelfinavir, ritonavir, and saquinavir decreased Aβ production in human neurons, among which nelfinavir showed the strongest inhibition of Aβ production, reaching 90% when compared with the control group. MTT assay results showed that nelfinavir showed significantly reduced mitochondrial activity, while all other drugs had no such toxicity (Fig. 2b).
Fig. 2.
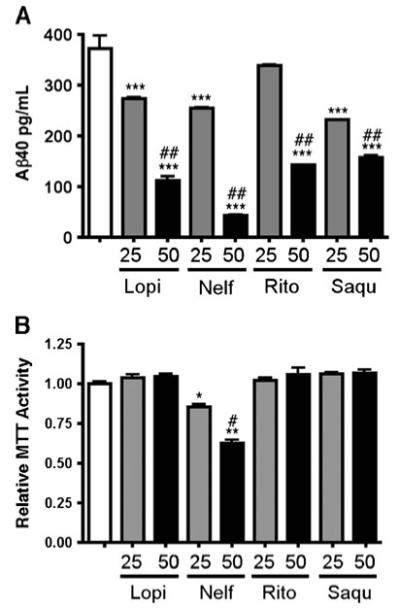
ART drugs decreased Aβ40 production in human neurons a-b, Primary human neurons were treated with 25 or 50 uM lopinavir (Lopi), nelfinavir (Nelf), ritonavir (Rito), or saquinavir (Saqu) for 48 h, and subjected to quantification of Aβ40 in the media (a) and MTT assay (b). *, **, and *** denotes p<0.05, 0.01, or 0.001 vs. vehicle-treated group (blank column), and # or ## denotes p<0.05 or 0.01 vs. 25-μM group of the same drug as determined by one-way ANOVA and Turkey post-hoc, respectively
The effect of ART drugs on purified BACE1 enzyme activity
Since BACE1 is regarded as a key enzyme to produce Aβ peptides (Vassar et al. 1999), we performed an experiment to examine if the ART drugs directly inhibited BACE1 enzyme activity in vitro. Drugs with serial concentrations were added to the recombinant BACE1 protein and the enzyme-specific fluorogenic substrate. Lopinavir, nelfinavir, ritonavir, and saquinavir at 300-μM concentration showed as high as 60, 30, 32, and 48% inhibition as compared to control (Fig. 3). Although these were statistically significant, the potency and efficiency of inhibition was modest as compared to the ART-induced reduction of Aβ production in primary human neurons. In particular, neither nelfinavir nor ritonavir had a significant effect on purified BACE1 enzyme activity at 25 or 50 μM concentrations. These data suggest that BACE1 is not a direct target of ART drugs in neurons.
Fig. 3.
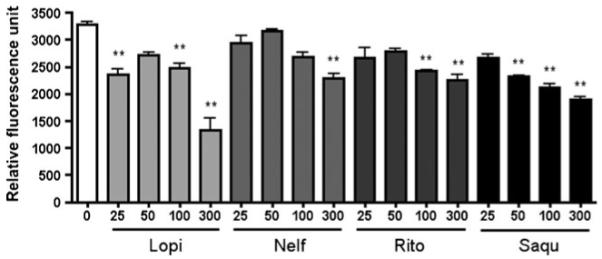
ART drugs decreased recombinant human BACE1 activity. One μg purified BACE1 protein was incubated together with lopinavir (Lopi), nelfinavir (Nelf), ritonavir (Rito), or saquinavir (Saqu) at different concentrations at room temperature for 30 min and fluorogenic Substrate IV was added for BACE1 activity measurement. * or ** denotes p<0.05 or 0.01 vs. vehicle-treated group (blank column) as determined by one-way ANOVA and Turkey post-hoc, respectively
ART drugs decreased β- and γ-secretase activities in human neurons
To understand the effect of ART drugs on β- and γ-secretase activity, we measured the BACE1 activity in the human neurons after ART drug treatment, and subjected the cell lysate for immunoblotting against the C-terminal stub of APP. After the treatment with 50 and 25 μM ART drugs for 48 h, neuron lysates were collected, and their BACE1 activities were determined with the fluorogenic substrate. As shown in Fig. 4a, ritonavir and saquinavir strongly decreased the BACE1 activity (69 and 70% inhibition at 50 μM concentration, respectively). Nelfinavir also significantly decreased BACE1 activity in human neurons (41% at 50 μM), although the effect was not as strong as ritonavir and saquinavir. Lopinavir, on the other hand, showed no effect on the BACE activity in human neurons. The neuronal lysate was subjected to immunoblot analysis of the C-terminal stub of endogenous APP (APPct), which is the processing product of α/β-processing and is accumulated when γ-secretase activity is inhibited. These results showed that all the 4 drugs tested here enhanced the production of APPct, indicating their inhibition of γ-secretase activity (Fig. 4b).
Fig. 4.
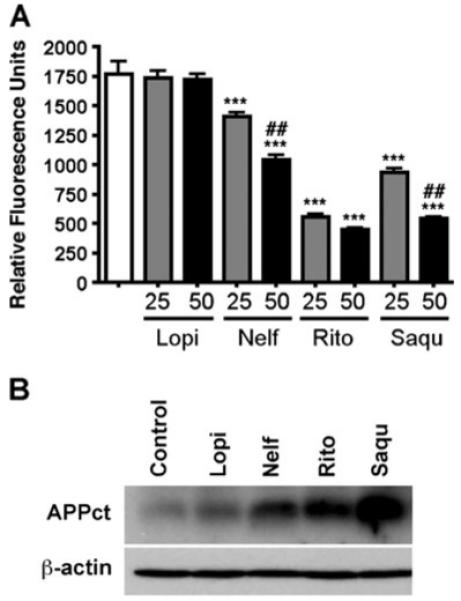
ART drugs decreased BACE1 and γ-secretase activity in human neuron lysate. a. Human neurons were planted on a 6-well plate for 2 weeks, and treated with ART (50 or 25 uM) drugs for 48 h. Then the cell lysates were collected, and 5 μg of each lysate was subjected to BACE1 activity assays. b. Immunobotting of 20 μg of protein lysate using anti-APP c-terminal region specific antibody (CT-15) and anti-β-actin antibody as a reference protein. *** or ## denotes p<0.01 or 0.001 vs. vehicle control (open column) or 25μM of the same compound as determined by ANOVA and Newman-Keuls post-test
Nelfinavir had no effect on Aβ accumulation in APP SCID mice
Finally, to understand the effect of ART drug administration on Aβ accumulation in the immunocompromised brain, APP SCID mice were orally administered pellets containing ground nelfinavir (30 mg/kg/day) for 30 days starting at 2 months of age, and sacrificed for biochemical analysis of both hippocampi and cortexes. We chose nelfinavir since this drug showed a significant effect on Aβ synthesis in neurons and Aβ clearance in MDM, with a CSF/IC50 ratio of 4.3 at the commonly prescribed dose of 1250 mg b.i.d. indicating drug distribution to the CNS (Madariaga and Swindells 2008). There were no changes in accumulation of total Aβ40 or Aβ42 as determined by human Aβ40- or Aβ42- specific ELISA of guanidine buffer-solubilized samples from hippocampi and cortexes (Fig. 5a-b). This mouse model expressed a double mutant form of APP 695 (KM670/671NL + V717F). Aβ accumulation was mainly intraneuronal in the pyramidal layer of the hippocampus at this age, without significant difference between the control vehicle and nelfinavir-administered groups (Fig. 5c-d). In addition, we performed the same experiment with co-formulated lopinavir/ritonavir (Kaletra®, 200 mg/kg/day, b.i.d) on APP SCID mice for 30 days from 2 months of age. Lopinavir with ritonavir has a CSF/IC50 ratio of 3.3 and is a commonly prescribed ART drug (Madariaga and Swindells 2008). However, we found no difference in Aβ42 accumulation in the hippocampus or cortex between drug and vehicle-treated groups (189.8±78.4 vs. 139.2±64.9 pg/μg in cortex, and 56.0±9.6 vs. 81.0±9.2 pg/μg in hippocampus, respectively). These data suggested that orally-administered nelfinavir or lopinavir/ritonavir has no effect on Aβ accumulation, at least with the tested doses.
Fig. 5.
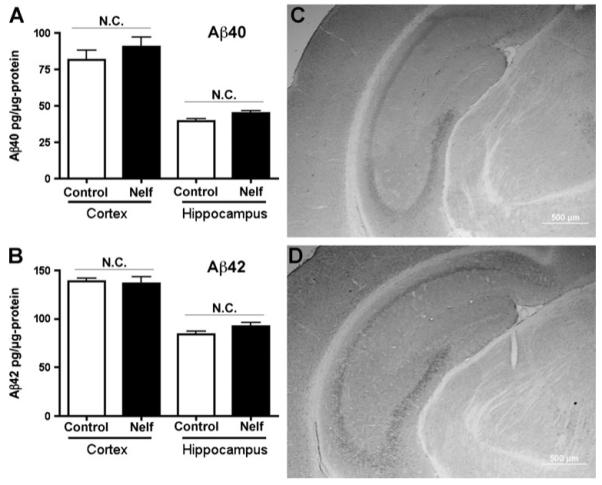
Nelfinavir had no effect on Aβ accumulation in the APP SCID mouse brain. a-b. APP mice at 2 months of age received oral administration nelfinavir (30 mg/kg/day) or vehicle for 30 days, then euthanized (n=3 per group). The cortexes and hippocampi were lysed in guanidine buffer and subjected to Aβ40/42 ELISA. “N.C.” denotes no statistically significant change. c-d. Immunohiostochemistry of coronal hippocampal sections stained by anti-human Aβ polyclonal antibody after control vehicle (c) or nelfinavir treatment (d). The scale bar represents 500 μm. Original magnification: ×100
To determine the delivery of ART drug in the brain, the cortical tissues of ART drug-administered APP mice were subjected to chloroform extraction and reverse-phase HPLC-based quantification of nelfinavir. We determined that the concentration was 6.1 μmol/kg in nelfinavir-administered APP SCID mice and was absent in vehicle-administered APP SCID mice. Although the exact concentration of nelfinavir in neuron was unknown, these concentrations were well above the therapeutic dose of nelifinavir (IC50=13–563 ng/ml, which is equivalent to 0.022-0.99 μM, dependent on the tested viral strain) (Zhang et al. 1999).
Discussion
The introduction of potent, combination ART had a significant impact on the natural history of HIV (Palella et al. 1998). Morbidity and mortality have declined dramatically, and HIV has evolved from a fatal disease to a chronic, manageable condition. Immune reconstitution achieved with ART can be sufficient and sustained enough that primary and secondary prophylaxis against opportunistic infections can be safely discontinued.
In this study, we screened nine HIV protease inhibitors for their effect on the clearance of fibrillar Aβ42 in primary human MDM. This was based on the prior study that found that nelfinavir inhibited the activity of IDE, one of two major Aβ-degrading enzymes, in HepG2 cell line and in semi-purified conditions (Hamel et al. 2006). However, we did not observe a significant reduction in Aβ degradation except with atazanavir, ritonavir, and saquinavir as determined by the amount of TCA-soluble extracellular fraction of degraded 125I-Aβ. Select ART drugs (lopinavir, nelfinavir, and ritonavir) rather enhanced abortive secretion of phagocytized Aβ in the TCA-insoluble extracellular fraction, although these were minor fractions as compared to the other two. These relatively modest changes in Aβ clearance by ART could be due to the relative abundance of neprilysin activity over IDE in differentiated MDM, minimizing the effect of ART drugs, including nelfinavir, on Aβ degradation. On the other hand, select ART drugs potently reduced Aβ40 production from primary cultured human neurons. Aβ42 production was consistently undetectable in the tested experimental design. However, considering the known Aβ40/Aβ42 ratio of around 9 when generated from wild type APP, we assumed Aβ42 generation would be similarly affected by ART treatment. Among the tested ART drugs, only nelfinavir showed modest neurotoxicity at both 25 and 50 μM concentrations when applied to primary neurons. This suggests that nelfinavir is potentially neurotoxic if CNS penetration of the drug is enhanced in disease conditions. However, in vivo application of nelfinavir to APP SCID mice showed no obvious neuronal cell loss in hippocampal or cortical regions, indicating that the neurotoxic effect of nelfinavir is not significant when the BBB is intact.
The effect of ART drugs on purified BACE1 enzyme activity was consistent with the Aβ synthesis data obtained from the neuron culture for lopinavir and saquinavir, but inconsistent for nelfinavir and ritonavir, since there was no difference at 25 or 50-μM concentrations. However, these ART drugs, with the exception of lopinavir, significantly inhibited BACE1 activity in neuron culture lysate. These data indicate that the effect of ART drugs on purified BACE1 enzyme is artificial and does not reflect their effect on intact BACE1 activity in neurons, or it indicates that the drug effects on BACE1 are largely indirect.
The major difference between the purified enzyme and intact BACE1 in neurons is that the former is a mixture of pro- and mature-forms, whereas ART drugs can interrupt the maturation step of BACE1 when applied to the neuron culture. pro-BACE1is cleaved by Furin and other members of the Furin family of convertases to remove the 24-amino acid N-terminal region of the pro-peptide within the trans-Golgi network. BACE1 then undergoes core glycosylation in the endoplasmic reticulum (ER) co-translationally, and is produced as pro-BACE1. pro-BACE1 is transported from the ER to the Golgi apparatus, and undergoes rapid maturation by the proteolytic removal of the prodomain followed by complex N-glycosylation in the Golgi apparatus (Bennett et al. 2000, 2001; Capell et al. 2000; Haniu et al. 2000; Huse et al. 2000). HIV protease inhibitors may interfere with any of the maturation steps of BACE1, likely the first Furin-mediated cleavage of pro-BACE1.
Our observation of enhanced APPct in all the tested ART drugs is striking, since it indicates that γ-secretase activity is inhibited by antiretroviral treatment regardless of their effect on BACE1-mediated β-processing. The data interpretation is complicated, since inhibition of BACE1 will reduce the amount of APPct (99 amino acids). However, APPct can also be generated by enhanced processing at α-cite, which generates a shorter version of APPct (83 amino acids). Our data does not explicitly separate the two APPct bands, but both of them will be accumulated if γ-processing is inhibited. This also explains the robust reduction of Aβ40 generation by ART drug treatment of neurons, which is difficult to explain by their effect on BACE1 activity. The molecular mechanism of γ-secretase complex assembly and maturation is highly complicated (for review, see (Dries and Yu 2008)), and involves assembly of presenlin-1/2, Aph-1, Pen-2, and Nicastrin, post-translational modification and trafficking of the proteins including N-glycosylation and endoproteolysis, where ART drugs may interfere.
Finally, we tested the effect of select ART drugs on CNS Aβ accumulation in APP SCID mice. We chose this mouse model because it over-expresses an FAD-linked double mutant form of human APP695 in a CNS-specific manner, and mimics the immunodeficient condition of human HIV-infected patients. Although ART drugs will have a direct effect on the peripheral clearance of Aβ, the SCID mouse model has an intact BBB, unless an additional modification is installed (Persidsky and Gendelman 1997), preventing the robust CNS penetration of ART drugs. Nonetheless, we detected nelfinavir in the drug-administered APP SCID mouse brain, which was higher than the reported concentration of nelfinavir in the mouse brain after acute iv injection of the drug (10 mg/kg) in wild type or P-glycoprotein (Mdr)−/− mice (0.22 or 3.6 μmol/kg, respectively) (Choo et al. 2000; Kim et al. 1998; Salama et al. 2005). However, the animals in this study were tested at the endpoint of 30-day consecutive administration and the daily dose (30 mg/kg/day or 52.8 μmol/kg/day) was 3-fold higher than the acute injection protocol, which may explain the higher retention of the drug in the brain. Our data indicate that ART drugs had no effect on Aβ accumulation in cortical and hippocampal regions of APP SCID mice after 30-day administration at 2–3 months of age. Although the exact concentration of the drug in neurons was unknown, this indicated that the penetration of the drug was well above the therapeutic dose but not sufficient to alter Aβ clearance or production in the brain.
Penetration of ART drugs into the CNS depends on many factors. Protein binding is particularly important because only free drug is able to diffuse across the BBB. Other factors that help with penetration into the CNS include small molecular size, a higher degree of acidity, higher lipophilicity, and the presence of inflammation, which may favor the accumulation of drugs in the CNS. The CSF level of an ART drug is used as a surrogate for drug penetration into the brain parenchyma, although this is not necessarily the optimal approach. Protease inhibitors have poor penetration because they are highly protein-bound in the plasma. Improvement in immune status from ART may also reconstitute the defective BBB, which is seen in later stages of HIV infection (Evers et al. 2004). The importance of CNS penetration in the treatment of HAND is therefore controversial, and drugs which do not have adequate CSF levels may still be effective in human but not in animal models. These points are important for evaluating the potential side effect of ART drugs on beta-amyloidosis in brain, which is a growing concern (Xu and Ikezu 2009).
In conclusion, we have evaluated the effect of 10 HIV protease inhibitors on Aβ clearance and production in human primary MDM, primary neurons, and the APP SCID mouse model. Select ART drugs showed modest inhibition of Aβ clearance in human MDM, and significant inhibition of Aβ production from human neurons. This was accompanied by inhibition of neuronal BACE1 and γ-secretase activity in neurons. On the other hand, 30-day administration of nelfinavir or lopinavir/ritonavir had no effect on Aβ accumulation in the APP SCID mouse brain. Although a longer-time treatment study should be considered to understand the chronic effect of ART treatment, these data are highly relevant for our understanding of the effect of ART drugs on the prevalence of AD in HIV-affected populations.
Acknowledgement
We would like to thank Katherine A. Estes for preparation of drugs for administration to mice, and Seiko Ikezu, Kaitlin Ingraham, Megan Varnum, and Robert Freilich for manuscript editing.
This work is supported in part by NIH R01 MH083523, R01MH072539, and P01 NS043985 (TI). Drug supports: Pfizer, Inc. for nelfinavir (AG1343), Abbott Laboratory for Kaletra®, and NIH AIDS Research and Reference Reagent Program for amprenavir, atazanavir, darunavir, indinavir, lopinavir, nelfinavir, ritonavir, saquinavir, and tipranavir.
Footnotes
Disclaimers Susan Swindells reports receiving research grants or contracts from or was a consultant for Abbott Pharmaceuticals, Bavarian Nordic, GlaxoSmithKline, Novartis Pharmaceuticals Corporation, Pfizer, and Tibotec Therapeutics. The other authors have no potential conflict of interest with this article.
Contributor Information
Xiqian Lan, Department of Pharmacology and Experimental Neuroscience, University of Nebraska Medical Center, Omaha, NE 68198-5880, USA.
Tomomi Kiyota, Department of Pharmacology and Experimental Neuroscience, University of Nebraska Medical Center, Omaha, NE 68198-5880, USA.
Richa Hanamsagar, Department of Pharmacology and Experimental Neuroscience, University of Nebraska Medical Center, Omaha, NE 68198-5880, USA.
Yunlong Huang, Department of Pharmacology and Experimental Neuroscience, University of Nebraska Medical Center, Omaha, NE 68198-5880, USA.
Scott Andrews, Department of Pharmacology and Experimental Neuroscience, University of Nebraska Medical Center, Omaha, NE 68198-5880, USA.
Hui Peng, Department of Pharmacology and Experimental Neuroscience, University of Nebraska Medical Center, Omaha, NE 68198-5880, USA.
Jialin C. Zheng, Department of Pharmacology and Experimental Neuroscience, University of Nebraska Medical Center, Omaha, NE 68198-5880, USA
Susan Swindells, Department of Internal Medicine, University of Nebraska Medical Center, Omaha, NE 68198-5880, USA.
George A. Carlson, McLaughlin Research Institute, Great Falls, MT 59405, USA
Tsuneya Ikezu, Department of Pharmacology and Experimental Neuroscience, University of Nebraska Medical Center, Omaha, NE 68198-5880, USA; Department of Pharmacology and Experimental Therapeutics, Boston University School of Medicine, Boston, MA 02118, USA; Department of Neurology, Boston University School of Medicine, Boston, MA 02118, USA; Alzheimer’s Disease Center, Boston University School of Medicine, Boston, MA 02118, USA.
References
- Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, Seubert P, Schenk D, Yednock T. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6(8):916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- Benjannet S, Elagoz A, Wickham L, Mamarbachi M, Munzer JS, Basak A, Lazure C, Cromlish JA, Sisodia S, Checler F, Chretien M, Seidah NG. Post-translational processing of beta-secretase (beta-amyloid-converting enzyme) and its ectodomain shedding. The pro- and transmembrane/cytosolic domains affect its cellular activity and amyloid-beta production. J Biol Chem. 2001;276(14):10879–10887. doi: 10.1074/jbc.M009899200. [DOI] [PubMed] [Google Scholar]
- Bennett BD, Denis P, Haniu M, Teplow DB, Kahn S, Louis JC, Citron M, Vassar R. A furin-like convertase mediates propeptide cleavage of BACE, the Alzheimer’s beta -secretase. J Biol Chem. 2000;275(48):37712–37717. doi: 10.1074/jbc.M005339200. [DOI] [PubMed] [Google Scholar]
- Besson C, Goubar A, Gabarre J, Rozenbaum W, Pialoux G, Chatelet FP, Katlama C, Charlotte F, Dupont B, Brousse N, Huerre M, Mikol J, Camparo P, Mokhtari K, Tulliez M, Salmon-Ceron D, Boue F, Costagliola D, Raphael M. Changes in AIDS-related lymphoma since the era of highly active antiretroviral therapy. Blood. 2001;98(8):2339–2344. doi: 10.1182/blood.v98.8.2339. [DOI] [PubMed] [Google Scholar]
- Brew BJ, Crowe SM, Landay A, Cysique LA, Guillemin G. Neurodegeneration and ageing in the HAART era. J Neuroimmune Pharmacol. 2009;4(2):163–174. doi: 10.1007/s11481-008-9143-1. [DOI] [PubMed] [Google Scholar]
- Capell A, Steiner H, Willem M, Kaiser H, Meyer C, Walter J, Lammich S, Multhaup G, Haass C. Maturation and propeptide cleavage of beta-secretase. J Biol Chem. 2000;275(40):30849–30854. doi: 10.1074/jbc.M003202200. [DOI] [PubMed] [Google Scholar]
- Chishti MA, Yang DS, Janus C, Phinney AL, Horne P, Pearson J, Strome R, Zuker N, Loukides J, French J, Turner S, Lozza G, Grilli M, Kunicki S, Morissette C, Paquette J, Gervais F, Bergeron C, Fraser PE, Carlson GA, George-Hyslop PS, Westa-way D. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem. 2001;276(24):21562–21570. doi: 10.1074/jbc.M100710200. [DOI] [PubMed] [Google Scholar]
- Choo EF, Leake B, Wandel C, Imamura H, Wood AJ, Wilkinson GR, Kim RB. Pharmacological inhibition of P-glycoprotein transport enhances the distribution of HIV-1 protease inhibitors into brain and testes. Drug Metab Dispos. 2000;28(6):655–660. [PubMed] [Google Scholar]
- Dries DR, Yu G. Assembly, maturation, and trafficking of the gamma-secretase complex in Alzheimer’s disease. Curr Alzheimer Res. 2008;5(2):132–146. doi: 10.2174/156720508783954695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evers S, Rahmann A, Schwaag S, Frese A, Reichelt D, Husstedt IW. Prevention of AIDS dementia by HAART does not depend on cerebrospinal fluid drug penetrance. AIDS Res Hum Retroviruses. 2004;20(5):483–491. doi: 10.1089/088922204323087723. [DOI] [PubMed] [Google Scholar]
- Forman MS, Trojanowski JQ, Lee VM. Neurodegenerative diseases: a decade of discoveries paves the way for therapeutic breakthroughs. Nat Med. 2004;10(10):1055–1063. doi: 10.1038/nm1113. [DOI] [PubMed] [Google Scholar]
- Gendelman HE, Orenstein JM, Martin MA, Ferrua C, Mitra R, Phipps T, Wahl LA, Lane HC, Fauci AS, Burke DS, et al. Efficient isolation and propagation of human immunodeficiency virus on recombinant colony-stimulating factor 1-treated monocytes. J Exp Med. 1988;167(4):1428–1441. doi: 10.1084/jem.167.4.1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamel FG, Fawcett J, Tsui BT, Bennett RG, Duckworth WC. Effect of nelfinavir on insulin metabolism, proteasome activity and protein degradation in HepG2 cells. Diabetes Obes Metab. 2006;8(6):661–668. doi: 10.1111/j.1463-1326.2005.00546.x. [DOI] [PubMed] [Google Scholar]
- Haniu M, Denis P, Young Y, Mendiaz EA, Fuller J, Hui JO, Bennett BD, Kahn S, Ross S, Burgess T, Katta V, Rogers G, Vassar R, Citron M. Characterization of Alzheimer’s beta -secretase protein BACE. A pepsin family member with unusual properties. J Biol Chem. 2000;275(28):21099–21106. doi: 10.1074/jbc.M002095200. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Heaton RK, Franklin DR, Ellis RJ, McCutchan JA, Letendre SL, Leblanc S, Corkran SH, Duarte NA, Clifford DB, Woods SP, Collier AC, Marra CM, Morgello S, Mindt MR, Taylor MJ, Marcotte TD, Atkinson JH, Wolfson T, Gelman BB, McArthur JC, Simpson DM, Abramson I, Gamst A, Fennema-Notestine C, Jernigan TL, Wong J, Grant I. HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: differences in rates, nature, and predictors. J Neurovirol. 2011;17(1):3–16. doi: 10.1007/s13365-010-0006-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hisatomi T, Nakazawa T, Noda K, Almulki L, Miyahara S, Nakao S, Ito Y, She H, Kohno R, Michaud N, Ishibashi T, Hafezi-Moghadam A, Badley AD, Kroemer G, Miller JW. HIV protease inhibitors provide neuroprotection through inhibition of mitochondrial apoptosis in mice. J Clin Invest. 2008;118:2025–2038. doi: 10.1172/JCI34267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse JT, Pijak DS, Leslie GJ, Lee VM, Doms RW. Maturation and endosomal targeting of beta-site amyloid precursor protein-cleaving enzyme. The Alzheimer’s disease beta-secretase. J Biol Chem. 2000;275(43):33729–33737. doi: 10.1074/jbc.M004175200. [DOI] [PubMed] [Google Scholar]
- Kim RB, Fromm MF, Wandel C, Leake B, Wood AJ, Roden DM, Wilkinson GR. The drug transporter P-glycoprotein limits oral absorption and brain entry of HIV-1 protease inhibitors. J Clin Invest. 1998;101(2):289–294. doi: 10.1172/JCI1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyota T, Yamamoto M, Schroder B, Jacobsen MT, Swan RJ, Lambert MP, Klein WL, Gendelman HE, Ransohoff RM, Ikezu T. AAV1/2-mediated CNS gene delivery of dominant-negative CCL2 mutant suppresses gliosis, beta-amyloidosis, and learning impairment of APP/PS1 mice. Mol Ther. 2009a;17(5):803–809. doi: 10.1038/mt.2009.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyota T, Yamamoto M, Xiong H, Lambert MP, Klein WL, Gendelman HE, Ransohoff RM, Ikezu T. CCL2 accelerates microglia-mediated Abeta oligomer formation and progression of neurocognitive dysfunction. PLoS One. 2009b;4(7):e6197. doi: 10.1371/journal.pone.0006197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyota T, Okuyama S, Swan RJ, Jacobsen MT, Gendelman HE, Ikezu T. CNS expression of anti-inflammatory cytokine interleukin-4 attenuates Alzheimer’s disease-like pathogenesis in APP + PS1 bigenic mice. FASEB J. 2010;24(8):3093–3102. doi: 10.1096/fj.10-155317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojro E, Fahrenholz F. The non-amyloidogenic pathway: structure and function of alpha-secretases. Subcell Biochem. 2005;38:105–127. doi: 10.1007/0-387-23226-5_5. [DOI] [PubMed] [Google Scholar]
- Lan X, Xu J, Kiyota T, Peng H, Zheng JC, Ikezu T. HIV-1 reduces Abeta-degrading enzymatic activities in primary human mononuclear phagocytes. J Immunol. 2011;186(12):6925–6932. doi: 10.4049/jimmunol.1100211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llibre JM, Falco V, Tural C, Negredo E, Pineda JA, Munoz J, Ortega E, Videla S, Sirera G, Martinez E, Miralles C, Iribarren J, Galindo MJ, Domingo P, d’Arminio-Monforte A, Miro JM, Clotet B. The changing face of HIV/AIDS in treated patients. Curr HIV Res. 2009;7(4):365–377. doi: 10.2174/157016209788680633. [DOI] [PubMed] [Google Scholar]
- Madariaga M, Swindells S. HIV-1 associated dementia. In: Ikezu T, Gendelman HE, editors. Neuroimmune pharmacology. Springer; New York: 2008. pp. 605–619. [Google Scholar]
- Palella FJ, Jr, Delaney KM, Moorman AC, Loveless MO, Fuhrer J, Satten GA, Aschman DJ, Holmberg SD, HIV Outpatient Study Investigators Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. N Engl J Med. 1998;338(13):853–860. doi: 10.1056/NEJM199803263381301. [DOI] [PubMed] [Google Scholar]
- Peng H, Huang Y, Rose J, Erichsen D, Herek S, Fujii N, Tamamura H, Zheng J. Stromal cell-derived factor 1-mediated CXCR4 signaling in rat and human cortical neural progenitor cells. J Neurosci Res. 2004;76(1):35–50. doi: 10.1002/jnr.20045. [DOI] [PubMed] [Google Scholar]
- Persidsky Y, Gendelman HE. Development of laboratory and animal model systems for HIV-1 encephalitis and its associated dementia. J Leukoc Biol. 1997;62:100–106. doi: 10.1002/jlb.62.1.100. [DOI] [PubMed] [Google Scholar]
- Salama NN, Kelly EJ, Bui T, Ho RJ. The impact of pharmacologic and genetic knockout of P-glycoprotein on nelfinavir levels in the brain and other tissues in mice. J Pharm Sci. 2005;94(6):1216–1225. doi: 10.1002/jps.20344. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81(2):741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Tyor WR, Power C, Gendelman HE, Markham RB. A model of human immunodeficiency virus encephalitis in scid mice. Proc Natl Acad Sci U S A. 1993;90(18):8658–8662. doi: 10.1073/pnas.90.18.8658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varatharajan L, Thomas SA. The transport of anti-HIV drugs across blood-CNS interfaces: summary of current knowledge and recommendations for further research. Antiviral Res. 2009;82(2):A99–109. doi: 10.1016/j.antiviral.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, Collins F, Treanor J, Rogers G, Citron M. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286(5440):735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- Xu J, Ikezu T. The comorbidity of HIV-associated neurocognitive disorders and Alzheimer’s disease: a foreseeable medical challenge in post-HAART era. J Neuroimmune Pharmacol. 2009;4(2):200–212. doi: 10.1007/s11481-008-9136-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Kiyota T, Horiba M, Buescher JL, Walsh SM, Gendelman HE, Ikezu T. Interferon-gamma and tumor necrosis factor-alpha regulate amyloid-beta plaque deposition and beta-secretase expression in Swedish mutant APP transgenic mice. Am J Pathol. 2007;170(2):680–692. doi: 10.2353/ajpath.2007.060378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Kiyota T, Walsh SM, Liu J, Kipnis J, Ikezu T. Cytokine-mediated inhibition of fibrillar amyloid-beta peptide degradation by human mononuclear phagocytes. J Immunol. 2008;181(6):3877–3886. doi: 10.4049/jimmunol.181.6.3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XQ, Schooley RT, Gerber JG. The effect of increasing alpha1-acid glycoprotein concentration on the antiviral efficacy of human immunodeficiency virus protease inhibitors. J Infect Dis. 1999;180(6):1833–1837. doi: 10.1086/315123. [DOI] [PubMed] [Google Scholar]