

Synopsis
Aging represents a convergence of declining cardioprotective systems and increasing disease processes that is fertile ground for the development of heart failure. With 50% of all heart failure diagnoses and 90% of all heart failure deaths occurring in the segment of the population over age 70, heart failure is largely a disease of the elderly. This review discusses the microscopic and macroscopic changes in cardiovascular structure, function, protective systems, and disease associated with aging. In addition to outlining important clinical considerations and conditions in older persons, the link between normal aging and the elevated risk for development of stage B heart failure is explained and potential therapeutic pathways highlighted.
I. Introduction
While aging does not itself cause heart failure (HF), it does lower the threshold for manifestation of the disease. As the populations of most developed countries continue to become older, on average, the importance of aging as a risk factor for all cardiovascular disease increases in kind (Figure 1). In the United States alone, it is estimated that there will be 70 million people over the age of 65 by the year 2030, representing almost 25% of the population1. In many respects, HF can be thought of as the quintessential final cardiovascular aging pathway, representing the convergence of age-associated changes in cardiovascular structure and function, aging changes in other organ systems, and the progressive increase in cardiovascular diseases in the elderly.
Figure 1.

Average prevalence of heart failure according to age and sex: Framingham Heart Study, 16-year follow-up. (Modified from McKee et al “The natural history of congestive heart failure: the Framingham study.” NEJM, 1971; 285: 7796).
With the success of treatment options for ischemic and valvular diseases, there is an increasing number of older individuals with some degree of cardiac damage, type B heart failure at a minimum that are increasingly imperiled by the diminished cardiac reserve associated with normal aging. Half of all HF cases are found within the 6% of the United States population that is older than 752 (reviewed by Najjar et al3). These people often go on to develop more severe cardiac dysfunction with time. In contrast to other cardiovascular disorders, the prevalence of chronic heart failure (CHF) is increasing, with approximately 5.7 million Americans with CHF4. The incidence of heart failure doubles with each decade of life and the prevalence rises to almost 10% of those older than 80 years. CHF is a highly lethal condition, with significant mortality, morbidity, and associated costs in the older population. More than 90% of CHF deaths occur in adults older than 65 years. CHF is also the leading cause of hospitalization in Medicare beneficiaries, with those over 65 accounting for 75% of the 1.1 million heart failure discharge diagnoses4.
This review focuses on what is known about normal cardiovascular aging and the role that aging-associated changes play in reducing cardiac reserve to make the heart more susceptible to failure. There are a number of factors that link aging to HF5 and gradually reduce the amount of cardiac reserve until finally the heart is “more likely to fail” (Figure 2).
Figure 2.
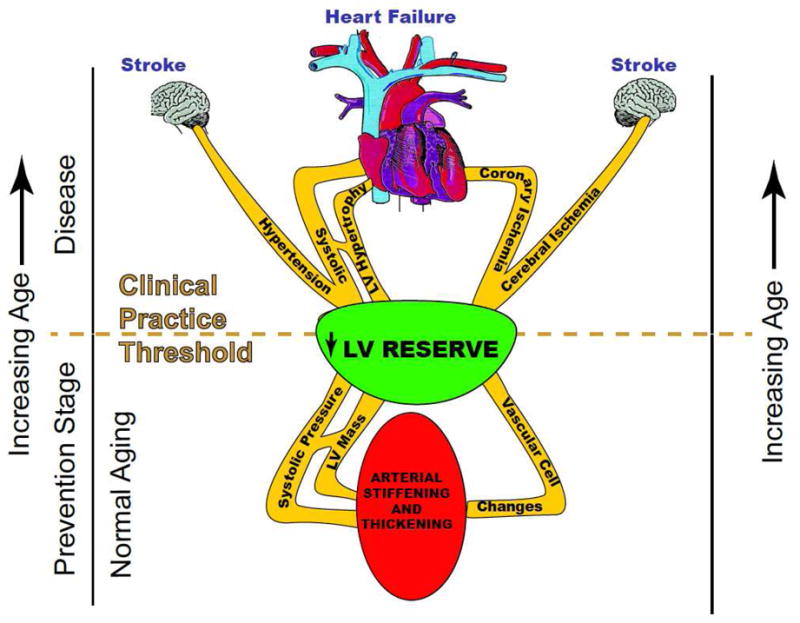
Pathways linking aging to heart failure. (Modified from Lakatta, EG. Age-associated cardiovascular changes in health: impact on cardiovascular disease in older persons. Heart Failure Rev.:2002 p1480).
Structural changes: There is significant structural change in the heart and vasculature; e.g. vascular stiffening, increased left ventricular wall thickness (within normal limits) and fibrosis with aging, leading to diastolic dysfunction, increased afterload, and HF with preserved ejection fraction (HFPEF)6,7. These are described individually in the Cardiac structural and Vascular structural sections below.
Functional changes: There are a number of functional changes and compensatory responses that the aged heart undergoes that diminish its ability to respond to increased workload and decrease its reserve capacity. Changes in maximal heart rate, end-systolic volume (ESV), end-diastolic volume (EDV), contractility, prolonged systolic contraction, prolonged diastolic relaxation, sympathetic signaling, etc. These are described individually in the Cardiac functional, Vascular functional, and Arterial-Ventricular interaction sections below.
Cardioprotection and Repair Processes: The cardiac mechanisms responsible for protection from injury and injury repair become increasingly defective with age, leading to accentuated adverse remodeling and increased dysfunction.
Increased cardiovascular disease incidence and prevalence: There is a progressive increase in the prevalence of cardiovascular disease (CVD); e.g. coronary artery disease (CAD), hypertension, and diabetes leading to the development of ischemic, hypertensive, or diabetic cardiomyopathy. The reader is referred to any number of epidemiologic studies for evidence of this fact.
Systemic disease/Other organ systems: Aging-associated changes in other organ systems may affect cardiac structure-function and thereby contribute to HF development. This facet, however, is beyond the scope of the current review.
II. Cardiovascular changes with aging
a. Cardiac changes at rest and with exercise
Cardiac structural changes
As described in Table 1 and Figure 3, there are a number of structural and functional changes in the heart with aging and each of these can have significant implications for cardiovascular disease. Structurally, there is a significant increase in myocardial thickness8 as a result of increased cardiomyocyte size9. In addition, the heart changes its overall shape from elliptical to spheroid with an asymmetric increase in the interventricular septum more than the free wall10. These changes in thickness and shape have important implications for cardiac wall stress and overall contractile efficiency.
Table 1.
Relationship of cardiovascular human aging in health to cardiovascular disease
| Age-associated changes | Plausible Mechanisms | Possible relation to human disease |
|---|---|---|
| Cardiovascular structural remodeling | ||
| ↑Vascular intimal thickness | ↑Migration of and ↑matrix production by VSMC Possible derivation of intimal cells from other sources | Early stages of atherosclerosis |
| ↑Vascular stiffness | Elastin fragmentation ↑Elastase activity ↑Collagen production by VSMC and ↑ Cross linking of collagen Altered growth factor regulation/tissue repair mechanisms |
Systolic Hypertension LV wall thickening Stroke Atherosclerosis |
| ↑LV wall thickness | ↑LV myocyte size with altered Ca2+ handling ↓Myocyte number (necrotic and apoptotic death) Altered growth factor regulation Focal matrix collagen deposition |
Retarded early diastolic cardiac filling ↑Cardiac filling pressure Lower threshold for dyspnea ↑Likelihood of heart failure with relatively normal systolic function |
| ↑Left atrial size | ↑Left atrial pressure/volume | ↑Prevalence of lone atrial fibrillation and other atrial arrhythmias |
| Cardiovascular functional changes | ||
| Altered regulation of vascular tone | ↓NO production/effects | Vascular stiffening; hypertension Early atherosclerosis |
| Reduced threshold for cell Ca2+ overload | Changes in gene expression of proteins that regulate Ca2+ handling; increased ω6ω3 PUFA ratio in cardiac membranes | Lower threshold for atrial and ventricular arrhythmia Increased myocyte death Increased fibrosis |
| ↑Cardiovascular reserve | Lower threshold for, and increased severity of heart failure | |
| Reduced physical activity | Learned lifestyle | Exaggerate age changes in some aspects of CV structure and function. Negative impact on atherosclerotic vascular disease, hypertension, and heart failure |
Abbreviations: LV, Left ventricular; PUFA, polyunsaturated fatty acids;
FIGURE 3.
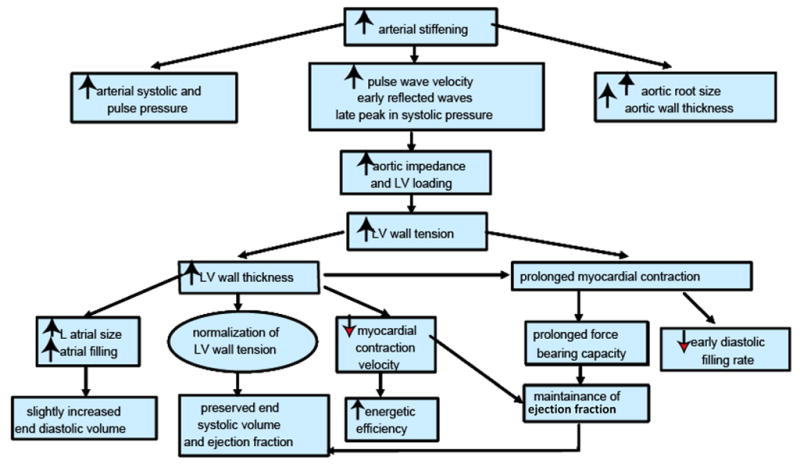
Arterial and cardiac changes that occur with aging in healthy humans. (Modified from Lakatta, EG: Cardiovascular regulatory mechanisms in advanced age. Physiol. Rev 73: 413–465, 1993).
The understanding of the changes in LV mass with aging has developed over time as researchers have made improvements in exclusion criteria, statistical correction, and technological approach. The history of this development provides a useful insight into the difficulties in conducting aging research. Initially, autopsy-based studies provided data that suggested cardiac mass increased significantly with aging11. Initial echocardiographic studies that calculated LV mass by wall thickness measurements corroborated these findings. However, measurements made from autopsies on subjects free from hypertension and CAD then corrected for body surface area, suggested that there is no actually no change in the cardiac mass of men with aging7. Similarly, autopsies of hospitalized patients free of CVD did not show an increase in cardiac mass with aging. In fact, they found a decrease in the cardiac mass of men and no change in cardiac mass for women. This finding has received support from an MRI-based study of healthy participants in the BLSA10 as well as multiple echocardiographic studies12,13. Based on these and other studies, it now appears that there is no change in LV mass in women and an actual decrease in LV mass in men with aging. It appears that the increased wall thickness represents an asymmetric increase in the interventricular septum more than the free wall redistributing cardiac muscle but not increasing total cardiac mass.
Cardiac systolic function
Despite a number of aging associated changes that may limit a person’s functional capacity and promote vascular stiffening with consequent increased afterload, the overall resting systolic function of cardiac muscle does not change (Figure 3) with healthy aging. Examination of echocardiographic LV shortening fraction14 and radionuclide ejection fraction (EF) in normotensive subjects and different aged healthy persons15 has repeatedly confirmed the stability of cardiac EF at rest (normal average EF >65%). This is not to say that there is no change in components of cardiac systole. In fact, multiple changes occur in the mean shortening velocity and the heart’s interactions with the vasculature. The combined effects of these individual changes, however, balance each other and leave the net systolic function unaltered at rest.
It is with exercise, that the effects of aging are most evident (Table 2). An overall decrease in exercise tolerance is evident in the progressive decline in VO2max, starting at age 20–30 and falling by approximately 10% per decade16 (FIGURE 4A–B). Additionally, the rate of this decline progressively increases with age. The meaning of these changes and insight into the underlying factors can be clarified through a review of the Fick Equation:
Table 2.
EXHAUSTIVE UPRIGHT EXERCISE Changes in Aerobic Capacity and Cardiac Regulation between Ages of 20 and 80 years In healthy Men and Women
| Oxygen Consumption | ↓(50%) |
|
| |
| (A-V)O2 | ↓(25%) |
|
| |
| Cardiac Index | ↓(25%) |
|
| |
| Heart Rate | ↓(25%) |
|
| |
| Stroke Volume | No Δ |
|
| |
| Preload | |
| EDV | ↓(30%) (men>women) |
|
| |
| Afterload | |
| Vascular (PVR) | ↑(30%) |
| Cardiac (ESV) | ↑(275%) |
|
| |
| LV Contractility | ↓(60%) |
|
| |
| Ejection Fraction | ↓(15%) |
|
| |
| Plasma Catecholamines | ↑ |
|
| |
| Cardiac and Vascular Responses to β-adrenergic stimulation | ↓ |
EDV, End Diastolic Volume; ESV, End Systolic Volume; LV, Left ventricular; (A-V)O2, Arterial-Venous Oxygen concentration; PVR, Peripheral Vascular Resistance.
FIGURE 4.

Longitudinal changes in peak VO2 by gender, predicted from the mixed-effects model, and separated by gender in panel A with the percent change by decade represented in panel B. A. Peak VO2 declines progressively more steeply with advancing age, with similar declines in men and women. Note that peak VO2 is only slightly higher in men than women at younger ages, converging by old age. B. Per-decade longitudinal changes in peak VO2 by gender and age decade, derived from the mixed-effects model. Note that longitudinal declines in peak VO2 steepen with age and that men decompensate at an accelerated, though similar rate after age 60. (With permission from Strait JS, Lakatta EG Cardiac Aging: Aging from human to molecules” Muscle, 2011. Modified by Ferrucci, L from Fleg JL, Morrell CH, Bos AG, et al. Accelerated longitudinal decline of aerobic capacity in healthy older adults. Circulation 2005;112:674–682).
CO=Cardiac Output, AO2=arterial O2 content, VO2=venous O2 content
Cross-sectional data reveals that VO2max falls an average of 50% from age 20–80 overall which must then be due to either/both CO and (A-V)O2. CO is known to fall 25% with aging, which by definition must be due changes in stroke volume (SV) or heart rate (hr)17. Because SV is thought to be maintained throughout life (our lab is currently completing studies to test this directly) the bulk of the CO decline is likely due to impaired heart-rate acceleration (or maximal heart rate). Although the LV-EDV (end diastolic volume) increases modestly with exercise, this results in an overall maintenance of SV (although our lab has ongoing studies to assess this assertion directly). The remaining 25% change in VO2max is due to a change in the (A-V)O2 term reflecting changes in oxygen extraction efficiency and balance (determinants include muscle mass, mitochondrial efficiency, the ability to redistribute blood flow to working muscles etc18,19) and suggests the importance of muscle mass, mitochondrial efficiency, etc. Put another way, the decline in VO2max is largely due to changes in the oxygen pulse which is the product of SV and the difference between peak VO2 and Arterial Oxygen concentration or peak VO2 /maximal hr. Overall, this reduction in cardiac reserve is a result of multiple factors including increased vascular afterload, arterial-ventricular load mismatching, reduced intrinsic myocardial contractility, impaired autonomic regulation, and physical deconditioning.
Cardiac diastolic function
Despite maintenance of systolic function at rest, there are a number of changes in the diastolic phase of the cardiac cycle that occur with aging. Normal diastolic filling can be divided into two phases, passive (represented by the “E wave” on echocardiographic study of transmitral flow) and active (represented by an A wave and produced by atrial contraction) (Figure 5A). The heart fills with blood more slowly in older vs. younger healthy individuals resulting in a lower proportion of total diastolic filling occurring during this passive, early diastolic phase8,20,21 due mainly to an increase in the isovolumic relaxation time. As the bulk of ventricular filling shifts to later in diastole and there is significant atrial enlargement with aging, the atrium contributes a greater portion of the total end diastolic volume (EDV) and a decrease in the E/A ratio (Figure 5B).
Figure 5.
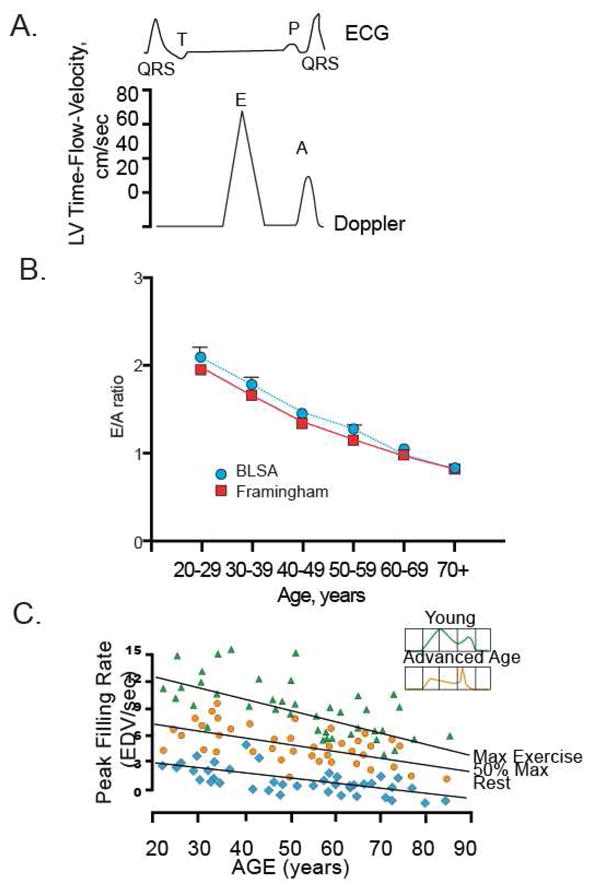
Diastolic Function at Rest and with Exercise
A. The Doppler diastolic time-flow-velocity profile, showing the E and A waves from which the indexes of diastolic filling performance are derived. Time is represented on the horizontal axis. A simultaneous electrocardiogram (ECG) is shown as a timing reference to indicate atrial and ventricular activation. LV indicates left ventricular.
B. The ratio of early left ventricular diastolic filling rate (E) to the atrial filling component (A) declines with aging, and the extent of this E/A decline with aging in healthy BLSA volunteers is identical to that in participants of the Framingham Study. (With permission from Strait JS, Lakatta EG Cardiac Aging: Aging from human to molecules” Muscle, 2011.)
C. Maximum left ventricular (LV) filling rate at rest and during vigorous cycle exercise assessed via equilibrium gated blood-pool scans in healthy volunteers from the BLSA. EDV, end-diastolic volume. (With permission from Strait JS, Lakatta EG Cardiac Aging: Aging from human to molecules” Muscle, 2011).
Normal exercise induces an increase in SV and heart rate in order to increase overall cardiac output (Table 2). This increased heart rate also increases the rate of isovolumic relaxation and produces a “suction” effect which helps fill the ventricle. However, these responses are diminished with aging as a result of slowed relaxation, reduced β-adrenergic responsiveness, and alterations in the pattern of relaxation10,22,23. Surprisingly, while the rate of filling declines and is shifted to later in diastole, the end diastolic volume actually remains unchanged or increases somewhat with rest and low-level exercise largely as a result of the slower24 heart rate which permits longer filling time (Figure 3, Table 2) and increase in the ESV. Taken together, these reductions in early diastolic filling are partially compensated for by changes in adrenergic signaling that lower the maximal heart rate. However, these compensations are not sufficient to maintain cardiac functional reserve when a subject is exposed to maximal exercise as shown in Figure 5C. In fact, a significant deficit is uncovered in the older population when the peak-filling rate at maximal exercise is compared in young vs. older individuals.
Changes in cardiac conduction system and electrocardiogram
Aging is associated with a generalized increase in elastic and collagenous tissue. Fat accumulates around the sinoatrial node, sometimes creating a partial or complete separation of the node from the atrial tissue. A pronounced decline in the number of pacemaker cells occurs after age 60; by age 75 less than 10% of the number seen in young adults remain. A variable degree of calcification on the left side of the cardiac skeleton also occurs. This can impact the atrioventricular node, atrioventricular bifurcation, and proximal left and right bundle branches leading to significant risk for atrioventricular conduction block.
While there is generally no change in the resting heart rate with aging, a number of changes do occur in the cardiac conduction system that impact its electrical properties (Table 3). A review by Fleg and Lakatta14 provides greater detail of this area. Due, likely, to a decrease in parasympathetic activity, there is a reduction in the level of phasic variation in R-R interval with respiration25 as well as the occurrence of sinus bradycardia. There can be an increase in pathological sinus bradycardia due to sick sinus syndrome and conduction pathway although the heart rate is stable in the healthy aged heart. The P-R interval, representing atrioventricular conduction, increases from 159 ms at ages 20–35 to 172 ms beyond age 6026. The QRS axis shifts leftward, possibly due to increases in LV wall thickness, with 20% of healthy subjects having a left axis deviation by age 10027. Interestingly, despite increased LV thickness, there is a decline in the R- and S-wave amplitudes with aging evident by age 4028. There is an increase in nonspecific ST-T changes with aging, although the relation of these to clinical heart disease remains in question29.
Table 3.
Normal Age-Associated Changes in Resting EKG Measurements
| Measurement | Change with Age | Effect on Mortality |
|---|---|---|
| R-R Interval | No Change | |
| P-wave duration | Minor Increase | None |
| P-R Interval | Increase | None |
| QRS duration | No change | |
| QRS axis | Leftward shift | None |
| QRS voltage | Decrease | None |
| Q-T interval | Minor increase | Probable increase |
| T-wave voltage | Decrease | None |
Modified from Lakatta EG, Sollott SJ, Pepe S. The old heart: operating on the edge. Novartis Found. Symp. 2001;235:172–96; discussion 196–201, 217–20.
In addition, there are increases in atrial arrhythmias, atria fibrillation, paroxysmal supraventricular tachycardia, and ventricular arrhythmias with aging14. The prevalence of these arrhythmias as well as their association with mortality are detailed in Tables 4 and 5. Atrial fibrillation (AF) is found in approximately 3–4% of subjects over age 60 years, a rate 10-fold higher than the general adult population. Short bursts of paroxysmal supraventricular tachycardia (PSVT) on a resting ECG are found in 1–2% of normal individuals older than 65 years. Twenty-four-hour ambulatory monitoring studies have demonstrated short runs of PSVT (usually 3 to 5 beats) in 13–50% of clinically healthy older persons30,31. The incidence increases with exercise, from 0% in the twenties to approximately 10% in the eighties. Ventricular ectopic beats (VEB) undergo an exponential increase in prevalence with advancing age. Pooled data from nearly 2500 ECGs from hospitalized patients older than 70 years revealed VEB in 8%32. The available data available in older adults without apparent heart disease support a marked age-related increase in the prevalence and complexity of exercise-related VEB, at least in men; however, the prognosis conferred by frequent or repetitive VEB induced by exercise in such individuals is unclear.
Table 4.
Relationship of Arrhythmias to Age and Mortality
| Arrhythmia | Effect of Age on Prevelance | Effect on Mortality |
|---|---|---|
| Supraventricular ectopic beats | Increased | None |
| Paroxysmal supraventricular tachycardia | Increased | Probably none |
| Atrial fibrillation (chronic) | Increased | Increased |
| Ventricular ectopic beats | Increased | Probably none |
| Ventricular tachycardia (short runs) | Increased | Probably none |
In healthy elderly
Modified from Lakatta EG, Sollott SJ, Pepe S. The old heart: operating on the edge. Novartis Found. Symp. 2001;235:172-96; discussion 196–201, 217-20.
Table 5.
Myocardial Changes with Adult Aging in Rodents
| Functional Changes | Ionic, Biophysical/Biochemical Mechanisms | Molecular Mechanisms |
|---|---|---|
| Prolonged Contraction | Prolonged cytosolic Ca2+ transient | |
| ↓SR Ca2+ pumping rate | ↓SR Ca2+ pump mRNA (no Δ calsequestrin mRNA) | |
| ↓Pump site density | ||
| Prolonged Action Potential | ↓ICa Inactivation | ↑Na/Ca exchanger mRNA |
| ↓ITo density | ||
|
| ||
| Diminished Contraction Velocity | ↓αMHC protein | ↓αMHC mRNA |
| ↑βMHC protein | ↑β MHC mRNA(no Δ Actin mRNA) | |
| ↓Myosin ATPase activity | ↓RxRβ1 and γ mRNA | |
| ↓RxRβ1 and γ mRNA | ||
| ↓RxRβ1 and γ protein | ||
| ↓Thyroid receptor protein | ||
|
| ||
| Diminished β-adrenergic Contractile Reponse | ↓Coupling BAR-ACyclase (no Δ Gi activation, no Δ BARK activity) | ↓ β1AR mRNA (no Δ BARK mRNA) |
| ↓TNI phospholambam phosphorylation | ||
| ↓ICa augmentation | ||
| ↓Cai transient augmentation | ||
| ↑Enkephalin peptides | ||
| ↑Proenkephalin peptides | ||
|
| ||
| ↑Myocardial Stiffness | ↑Hydroxyline proline content | ↑Collagen mRNA |
| ↑Activity of myocardial RAS | ↑Fibronectin mRNA | |
| ↑ AT1R mRNA | ||
|
| ||
| ↑Atrial naturetic peptide | ↑Atrial natriuretic peptide mRNA | |
|
| ||
| ↓Growth Response | ↓ Induction of immediate early genes | |
| ↓Cardiomyocyte Pro life rati ion | Δ in CyclinD1, D2, D3, pRb, p130, CDK-2 | |
| ↓Cardiomyocyte Survival | Δ in TERT, IGF-1, Caspases, AIF, Survivin | |
|
| ||
| ↓Heat Shock Response | ↓Activation of HSF | |
Abbreviations: SR, sarcoplasmic reticulum; SERCA, Sarco/endoplasmic reticulum calcium ATPase; Ca2+, Calcium ions; NaCa, Sodium Calcium; MHC, myosin heavy chain; mRNA, messenger RNA; RXR, retinoid X-receptor; BAR, Beta-adrenergic receptor; BARK, Beta-adrenergic receptor kinase; Gi, inhibitory G protein; TNI, troponin-I; Ica, calcium influx; Cai, intracellular calcium concentrations; HSF, heat shock factor; RYR2, cardiac ryanodine receptor; AT1R, Angiotensin AT-1 receptor; RAS, Renin-Angiotensin system; pRb, Retinoblastoma protein; TERT, Telomerase Reverse Transcriptase; IGF-1, Insulin-like Growth Factor-1, PI3K, phosphoinositide-3 kinase; ET-1, Endothelin-1; SIRT1, Sirtuin-1; AIF-1, Apoptosis Inducing Factor.
In summary, aging is associated with multiple ECG changes in persons without evidence of CV disease. Such changes include a blunted respiratory sinus arrhythmia, a mild P–R interval prolongation, a leftward shift of the QRS axis, and increased prevalence, density, and complexity of ectopic beats, both atrial and ventricular. Although these findings generally do not affect prognosis in clinically healthy older adults, other findings that become more prevalent with age, such as increased QRS voltage, Q waves, QT interval prolongation, and ST–T-wave abnormalities, are generally associated with increased CV risk. Abnormalities such as left BBB or AF are strongly predictive of future cardiac morbidity and mortality among older adults, even if asymptomatic.
Changes in cellular Ca2+ handling
Arrhythmias are a byproduct of aberrant Ca2+ handling. Maintenance of the calcium-electrochemical gradient is an intricate process for the individual cardiomyocyte, as depicted in Figure 11 and Table 1. With aging, a number of changes occur that slow the cellular reactions controlling the beat of the heart as a whole (more details can be found in Sollott et al33 and Janczewski et. al34). The action potential, transient increase in cytosolic Ca2+, and rate of contraction are all prolonged which consequently prolong systole and diastole of the heart 33,35 consistent with the lower maximal heart rate seen in older individuals during exercise. This is due to changes in a number of ion currents as well as a reduced rate of Ca2+ reuptake through downregulation of SERCA2 protein levels, decreased phospholambam phosphorylation, and a ~50% increase in levels of the Na+/Ca2+ exchanger. Studies in rodents that have used gene therapy or exercise conditioning to increase the level of SERCA2 have demonstrated that impaired relaxation and Ca2+ sequestration that occur with aging can be reduced by these interventions. Finally, there is a compensatory increase in L-type Ca2+ currents with an increase in the apparent number and activity of individual L-type calcium channels that prolongs the action potential, a slower inactivation of the L-type channel, and a reduction in outwardly directed K+ currents. All of which serve to prolong the action potential.
FIGURE 11.
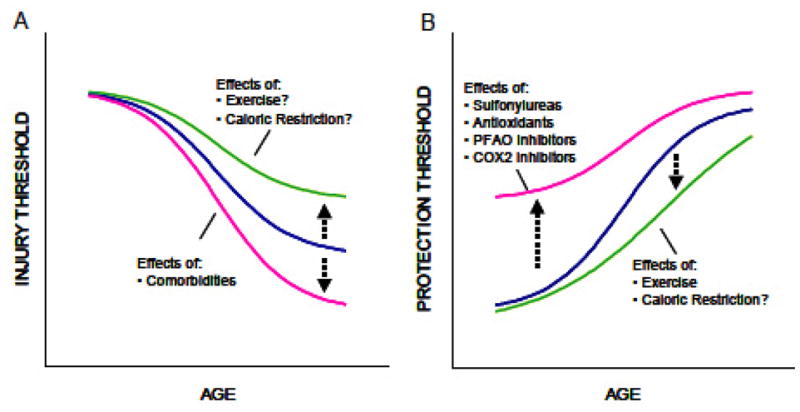
Changes in the injury and protection thresholds in aging heart. (A) Aging diminishes the hearts threshold to sustain injury (e.g., from ischemia/reperfusion, etc.). Lifestyle modifications, including exercise and possibly caloric restriction, may partially diminish the aging effect. Comorbidities (such as diabetes) have negative influence. (B) Aging increases the hearts threshold to activate protection-signaling mechanisms. Various pharmacologic agents (e.g., sulfonylureas, antioxidants, partial fatty acid oxidation (PFAO) inhibitors, and COX-2 inhibitors) that can interfere with cardioprotective signaling-pathways can exacerbate this trend and further increase the protection threshold. Exercise and caloric restriction might attenuate the age-dependent trends (Modified from Juhaszova M, Rabuel C, Zorov DB, et al: 2005. Protection in the aged heart: preventing the heart-break of old age? Cardiovasc Res 66: 233–244).
Ca2+ regulation is significantly affected by reactive oxygen species (ROS) and is therefore involved in complex pathway (Figure 11) involving membrane polyunsaturated fatty acids (PUFAs). PUFAs are an important family of dietary fats (including the ω-3 and ω-6 PUFAs) that undergo lipid peroxidation to produce aldehydes, akenals, and hydroyalkenals such as 4-hydroxy-2-nonenal (HNE). HNE reacts with protein sulfhydryl groups to induce altered protein conformations and inflict damage on the cell33. With advancing age, a number of changes occur in the lipid content of the cell membrane that reduce its capacity to tolerate and adapt to stress (e.g. ischemia-reperfusion).
Notable among these changes are a relative decrease in the ω-3: ω-6 PUFA content ratio in cellular membranes, enhanced cellular production of ROS, and alterations in proteins governing Ca2+ mobilization especially the decreased expression or function of SERCA2 and Na+- Ca2+ exchange protein33,36. These changes lead to disturbances in excitation-contraction coupling and in intracellular Ca2+ compartmentalization leading in turn to spontaneous Ca2+ oscillations and arrhythmias (Figure 11). In addition, Mitochondrial dysfunction can ensue that impairs energy metabolism (decreased ATP production) and increase ROS production. Overproduction of ROS and the inability to scavenge excess ROS lead to damaging lipid peroxidation and relatively more diffusible but still potent, reactive intermediates such as HNE which affect widespread protein targets and amplify Ca2+ dysregulation and mitochondrial abnormalities. Finally the abnormal Ca2+ handling and ROS buildup can induce mitochondrial permeability transition (MPT) which involves the release of mitochondrial contents and activation of a sequence of events that lead to cell death. Recent research has demonstrated that these effects of aging are exacerbated by poor nutritional habits but can be ameliorated by diets substituting ω-3 PUFAs for ω-6 PUFAs.
Cardiac adrenergic responsiveness
Adrenergic signaling is an important component of aging-associated cardiovascular change. Acute exercise and other stressors stimulate sympathetic modulation of the CV system, which increases heart rate, augments myocardial contractility and relaxation, reduces LV afterload, and redistributes blood to working muscles and skin to dissipate heat. However, with aging there is a diminishment of the autonomic modulation of heart rate, LV contractility, and arterial afterload37 related to a decline in the efficiency of post-synaptic β-adrenergic signaling. There is a decrease in cardiovascular responses to β-adrenergic antagonist infusion at rest with aging (Figure 7A) and a similarity in the hemodynamic profile of younger β-blocked subjects to older unblocked individuals (Figure 7B, C) that is consistent with this mechanistic explanation. A number of smaller studies37 provide further evidence for this effect. Significant β-blockade-induced LV dilation occurs only in younger subjects, the heart rate is reduced to a greater degree by acute β-adrenergic blockade in younger vs. older subjects, and the age-associated deficits in LV early diastolic filling rate, both at rest and during exercise.
FIGURE 7.
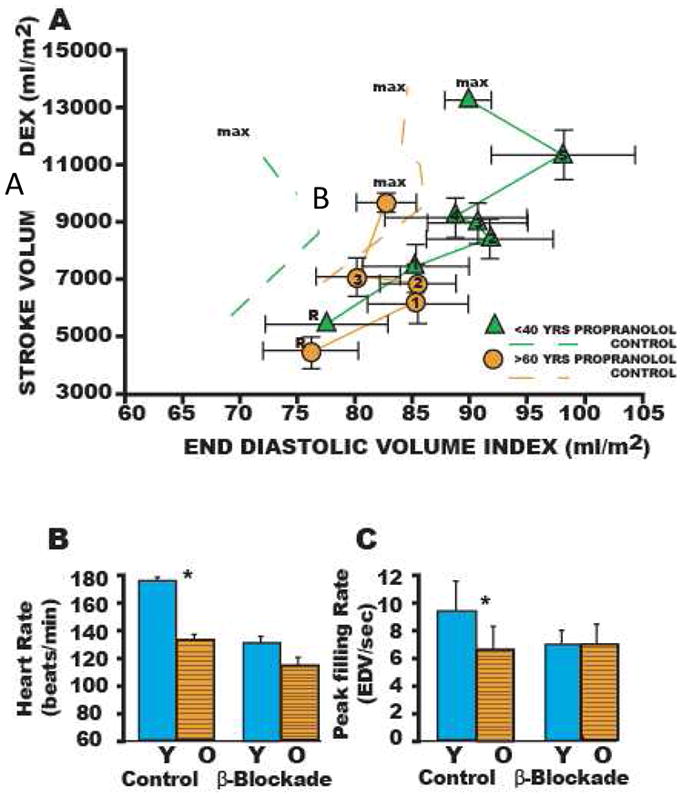
(A) Stroke volume index (SVI) as a function of end-diastolic volume index (EDVI) at rest (R) and during graded cycle workloads in the upright seated position in healthy Baltimore Longitudinal Study of Aging (BLSA ) men in the presence and absence (dashed lines) of β-adrenergic blockade. R, seated rest; 1–4 or 5, graded submaximal workloads on cycle ergometer; max, maximum effort. Stroke volume vs end-diastolic functions with symbols are those measured in the presence of propranolol; dashed line functions without symbols are the stroke volume as vs end-diastolic functions measured in the absence of propranolol. Note that in the absence of propranolol the SVI vs EDVI relation in older persons (dashed lines) is shifted rightward from that in younger ones (dashed lines with points). This indicates that the left ventricle (LV) of older persons in the sitting position compared to that of younger ones operates from a greater preload both at rest and during sub-maximal and max exercise. Propranolol markedly shifts the SV-EDVI relationship in younger persons (▲ without points) rightward, but does not markedly offset the curve in older persons (●). Thus, with respect to this assessment of ventricular function curve, β-adrenergic blockade with propranolol makes younger men appear like older ones. The abolition of the age-associated differences in the LV function curve after propranolol are accompanied by a reduction in heart rate, which at max, is shown in (B) Peak exercise heart rate in the same subjects as in A in the presence and absence of acute β-adrenergic blockade by propranolol. (C) The age associated reduction in peak LV diastolic filling rate at max exercise in healthy BLSA subjects is abolished during exercise in the presence of β-adrenergic blockade with propranolol. Y, <40 years; 0, >60 years (14). (With permission from Strait JS, Lakatta EG Cardiac Aging: Aging from human to molecules” Muscle, 2011)
The mechanism behind these changes is not entirely clear, although several alterations in the adrenergic system have been observed. It is noteworthy that the apparent deficits in sympathetic modulation of cardiac and arterial functions with aging occur in the presence of elevated sympathetic neurotransmitter levels: during any perturbation from the supine basal state, plasma levels of norepinephrine and epinephrine increase to a greater extent in older than in younger healthy individuals37,38. This increase in plasma catecholamines appears to be a compensatory response to the reduced cardiac muscarinic β-receptor density and functional decline with advancing age. The amount of circulating NE rises, in part, due to increased spillover from tissues (including the heart37–39) as a result of deficient NE re-uptake at nerve endings during acute exercise as well as reduced plasma clearance. With prolonged exercise this diminished neurotransmitter re-uptake can result in depletion of stores and reduced release and spillover in the body thereby contributing further to the blunted cardio-acceleration and LV systolic performance seen with age during such exercise.
Renin-Angiotensin system in cardiac tissue
The mechanism behind age-related cardiac remodeling is not completely understood although a number of clues have been found6,33. 1) There is a strong body of evidence for the central role for the renin-angiotensin system (RAS). 2) Increased oxidative stress has been implicated in age-related remodeling. 3) Aging has been associated with increased production of reactive oxygen species (ROS) in a number of studies. 4) RAS activation itself may stimulate ROS production. 5) NADPH oxidases appear to be important in linking these processes.
One important stimulus for RAS signaling is thought to be the stretch of cardiac myocytes and fibroblasts due to the increased load imposed by the aging vasculature40 and overall decrease in cardiomyocyte number. This initiates growth factor signaling (e.g. Angiotensin II/TGF-β), which has been shown in vitro to promote cell growth and matrix production as well as increasing apoptosis41. This ANGII does not likely come from circulating stores but is instead released from fibroblasts and cardiomycytes in an autocrine/paracrine manner in response to stretch42. ANGII binds to both Angiotensin I (AT1) and Angiotensin II (AT2) receptors to activate a complex signal transduction cascade that impact a number of transcriptionally important factors43. Either directly or through AT1, ANGII can activate the NADPH oxidase complex to generate ROS. Understanding of the relative importance of these numerous pathways will be an important component of cardiac aging research and provide an important link to similar signaling changes in the arterial wall.
b. Central arterial changes with aging
In studying the effects of aging on the CV system, it is important to recall its anatomic and functional position in series with the vascular system. In fact, many researchers now feel that the greatest risk factor for development of CV disease is “unsuccessful” age-associated arterial aging. Rather than acting as simple conduits for blood flow, blood vessels are dynamic structures that adapt, repair, remodel, and govern their structural and function properties using complex signaling pathways in response to load, stress, and age.
Central arterial macroscopic structural changes
A number of age-associated structural changes occur in the arterial system, including thickening and dilation of large arteries44 (Figure 3, Table 1). Echocardiographic studies show that the aortic root dilates modestly with age, approximating 6% between the fourth and eighth decades (Figure 8A). Similarly, serial chest x-rays over 17 years have demonstrated that the aortic knob diameter increases from 3.4 to 3.8 cm, although there is data to suggest that in hypertensives (when aortic diameter is corrected for covariates) it may represent a relative decrease in effective diameter that may contribute to increased load on the heart45. In normal aging, however, such aortic root dilation provides an additional stimulus for LV hypertrophy because the larger volume of blood in the proximal aorta leads to a greater inertial load against which the senescent heart must pump.
Figure 8.
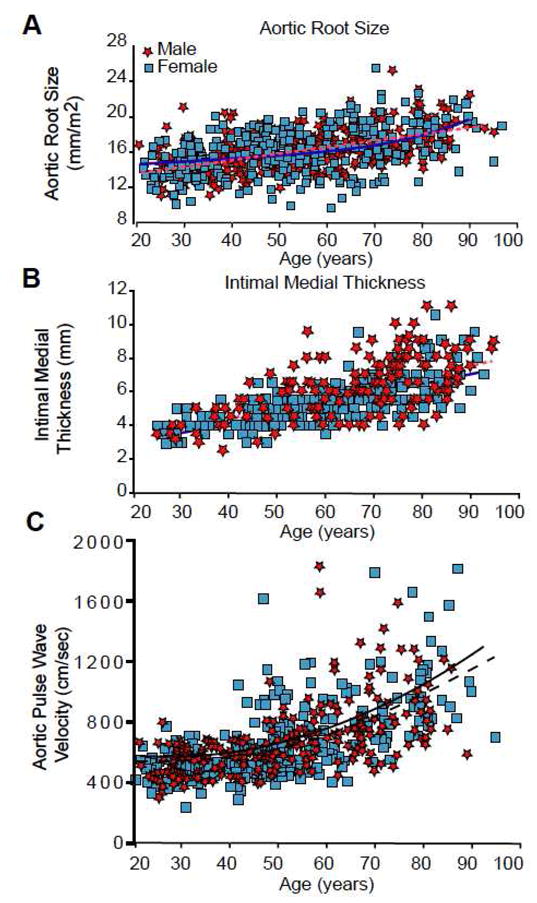
Age associated changes in arterial structure and function in men (stars) and women (squares) in the Baltimore Longitudinal Study of Aging (BLSA). Best fit regression lines (quadratic or linear) are shown for men (solid lines) and women (dotted lines). (A) Aortic root size (measured by M-mode Echocardiography) indexed to body surface area. (B) Common carotid-intima-medial thickness (measured by B-mode ultrasonography). (C) Carotid-femoral pulse wave velocity (an index of central arterial stiffness). (With permission from: Najjar SS, Lakatta EG, Gerstenblith G. Cardiovascular Aging: The Next Frontier in Cardiovascular Prevention. In Prevention of Cardiovascular Disease: Companion to Braunwald’s Heart Disease Blumenthal R, Foody J, Wong NA (editors). 2011. Saunders, Philadelphia, 2011:415–432).
Autopsy reports published as early as 1910 described age-associated aortic thickening. Cross sectional studies using ultrasound imaging have demonstrated the intimal-medial layer thickens nearly threefold (Figure 8B) between the ages of 20 and 90 years in apparently healthy individuals46. Both the average and range of intimal-medial thickness measurements is greater at higher ages, suggesting a variable response to chronological age that merits further study to identify the components of “successful aging”.
Central arterial microscopic and biochemical changes
Aging is associated with a number of structural and functional changes of the arterial wall media (hypertrophy, extracellular matrix accumulation, calcium deposits) and the vascular endothelium (decrease in the release of vasodilators and increased synthesis of vasoconstrictors) that are associated with increased vascular stiffness47,48(Table 1). Collagen and elastin provide the strength and elasticity, respectively, of the arterial wall and are normally stabilized by enzymatic cross-linking. With aging, an increase in collagen content, non-enzymatic collagen cross-linking, and fraying of elastin fibrils occur in the medial layer49, all of which reduce arterial distensibility and increase stiffness. The occurrence of irreversible non-enzymatic glycation-based crosslinking of collagen to form advanced glycation endproducts (AGEs) increases with age and is associated with increased arterial stiffness in elderly people50. Furthermore, Receptors for Advanced Glycation Endproducts (RAGE) are stimulated to produce a number of inflammatory and stress responses.
Through its secretion of nitric oxide (NO) and endothelin, the endothelium is a powerful regulator of arterial tone. Endothelial dysfunction has been identified in a number of CV disorders, including hypertension, hypercholesterolemia, and coronary and peripheral atherosclerosis51. Human and animal studies have revealed that aging is associated with a reduction in endothelial dependent vasodilatation, thought secondary to reduced NO bioavailability49. A number of animal studies have found that NO production and NO levels52 decline with aging, likely as a result of a decline in the level of endothelial nitric oxide synthase (eNOS)53.
In addition to the structural alterations (described previously), arterial function is governed by age-associated changes in several signaling cascades, most prominently the renin-angiotensin system (RAS). The classic RAS is composed of angiotensinogen, renin, angiotensin (Ang) I and II, angiotensin-converting enzyme (ACE), chymase, angiotensin, and Ang II receptor (AT1). All of these components have been found to increase within the aged arterial wall in various species54,55. The local Ang II concentration is more than 1000-fold that of circulating Ang II, is independently regulated, and plays an important role in vascular pathophysiology with aging. Ang II protein abundance increases in the aged aortic wall in rats. Studies of nonhuman primates also show that Ang II, ACE, and chymase. findings demonstrate that elements of the classic RAS are all up-regulated in the aged arterial wall55,56. The marked increase of Ang-II in the aged arterial wall appears to offset the effect of reduced plasma levels of Ang-II in the elderly. In addition, the molecules linked to the Ang II signaling cascade, including calpain-1, matrix metalloproteinase types 2 and 9 (MMP-2 and MMP-9), monocyte chemotactic protein-1 (MCP-1), and transforming growth factor-beta 1 (TGF-b1) are up-regulated within the aged arterial wall 54,57,58(Table 1 and 3). A number of studies support the concept that Ang II signaling is a central pathway that mediates the cellular and molecular mechanisms that underlie arterial aging55.
Central arterial function: Blood Pressure
Arterial pressure is determined by the interplay of peripheral vascular resistance (PVR) and arterial stiffness. Stiffness as used here, refers to time-varying cardiac elastance throughout the cardiac cycle as a result of combined effects of active contractile and “passive” structural properties as well as the interaction of the two. Systolic blood pressure (SBP), which is influenced by arterial stiffness, PVR, and cardiac function, rises with age even in normotensive cohorts. In contrast, diastolic blood pressure (DBP), rises with increased PVR but is lowered by arterial stiffness resulting in an increase in diastolic pressure till age 50, a leveling off between 50 and 60, then a decline after age 6059. Thus, hypertension in the elderly is often characterized by isolated or predominant systolic BP elevation. Pulse pressure, the difference between SBP and DBP, is a useful clinical index of arterial stiffness and the pulsatile load on the arterial tree and typically increases with aging. Some studies have suggested that pulse pressure is a more powerful predictor of future CV events than either SBP or DBP in older adults60.
Central arterial function: Pulse wave velocity and reflected pulse waves
Central arterial stiffening occurs with aging even in the absence of clinical hypertension61 as measured by pulse wave velocity (PWV). Augmentation Index (AI) illustrates an important site of cardiac-vascular interaction. When the forward pulse wave reaches an area of impedance mismatch (vessel bifurcation or movement to a higher resistance vessel), a reflected wave that travels back up the arterial tree toward the central aorta is generated. This reflected wave is identified as a small notch, inflection point, in the carotid and radial pulse waveforms, measured by arterial applanation tonometry. The situation with AI is a bit more complicated (not shown). It increases until about age 5062, even in clinically healthy volunteers. The clinical significance of these AI changes with age is that in young subjects, the reflected wave typically arrives back at the proximal aorta in diastole and may assist in coronary artery diastolic filling. However, in older individuals, the reflected waves travel faster, thus arriving at the proximal aorta during late systole, thereby creating an increased load for the ventricle, a failure to augment DBP, and a potential compromise of coronary blood flow. Several studies in cohorts with CVD have observed that higher AI is associated with adverse clinical outcomes63. A second method of arterial stiffness assessment, Pulse wave velocity (PWV), is a Doppler-based method that measures the speed with which an arterial pressure wave travels along the arterial tree, typically from the carotid region to the femoral artery. Multiple studies have shown that aortofemoral PWV increases with age, typically 2–3 fold across the adult lifespan (Figure 8C)64. Furthermore, PWV has been shown both in clinically healthy cohorts and those with CV disease, to be a predictor of future CV events, independent of blood pressure65.
c. Arterial-Ventricular Coupling
Arterial ventricular coupling background
The concept of coupling between effective arterial elastance (Ea) and ventricular elastance measures (ELV) is exceedingly important to the study of cardiovascular aging, and so merits further discussion (Figure 9)66. Oftentimes, a byproduct of the need to reduce the cardiovascular system to a manageable model system is an excessive focus on individual components rather than consideration of their function and interaction with the entire system. The ratio of effective Ea/ELV is an index of arterial ventricular coupling, related to the inverse of EF, and provides a more accurate measure of the complex cardiovascular system.
FIGURE 9.
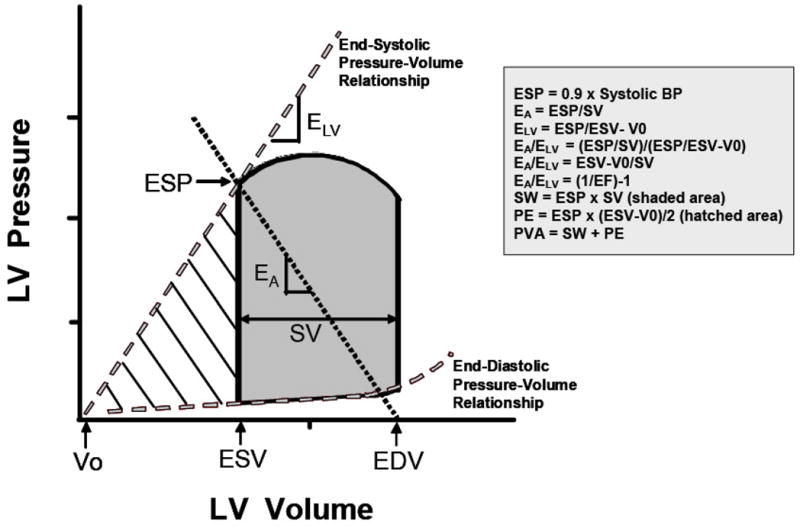
Ventricular pressure-volume diagram from which effective arterial elastance (EA) and left ventricular (LV) end-systolic elastance (ELV) are derived. EA represents the negative slope of the line joining the end-diastolic volume (EDV) and the end-systolic pressure (ESP) points. ELV represents the slope of the end-systolic pressure-volume relationship passing through the volume intercept (V0). The shaded area represents the cardiac stroke work (SW), and the hatched area represents the potential energy (PE). LV ESP is the LV pressure at the end of systole. EDV is the LV volume at the end of diastole. End-systolic volume (ESV) is the LV volume at the end of systole. Stroke volume (SV) is the volume of blood ejected by the LV with each beat and is obtained from subtracting ESV from EDV. BP, blood pressure; EF, ejection fraction; PVA, pressure-volume area. (With permission from Chantler, Lakatta, Najjar. Arterial-ventricular coupling: mechanistic insights into cardiovascular performance at rest and during exercise, J Appl Physiol 105:1342–1351, 2008, and based on Sunagawa et al. 1983. Left ventricular interaction with arterial load studied in isolated canine ventricle. The Am J of Phys, 245 (5 Pt 1), H773-80).
The effective EA is a steady state arterial parameter that characterizes the functional properties of the arterial system by incorporating peripheral vascular resistance, total lumped vascular compliance, characteristic impedance, and systolic and diastolic time intervals (characterized by the relationship of end systolic pressure and stroke volume). The ELV (left ventricular end-systolic elastance) represents myocardial performance and is defined by the relationship of end-systolic pressure to end systolic volume.
Arterial-ventricular coupling at rest
As detailed above, most of the components of EA change with age (e.g. SBP, pulse pressure) so the fact that the EA/ELV ratio is maintained at rest (Figure 10A–C) despite the known changes in the vascular system and consequent increases in afterload suggest important adaptive changes in the cardiac muscle itself67. These strategies (in part) appear to involve LV wall thickening and increased end-systolic pressure8 (moderates LV wall tension), increased end systolic volume, prolongation of systolic contraction time68 (maintains normal ejection time in the presence of late augmentation of aortic impedance), cardiac shape69, and aspects of the diastolic phase.
FIGURE 10.
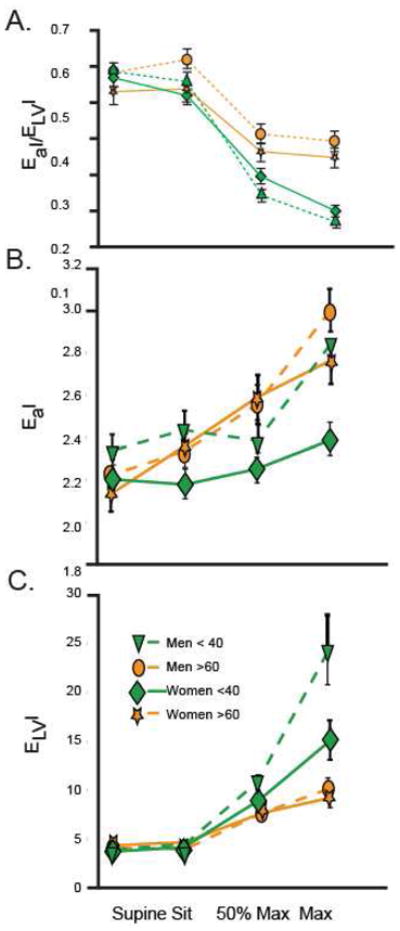
(A) Arterial ventricular coupling indexed for body surface area (EaI/ELVI), (B)Effective arterial elastance indexed to body surface area (EaI), and (C) Effective left ventricular elastance indexed to body surface area (ELVI) in men (dashed lines) <40 yr of age (triangle) and >60 yr of age (circle) as well as women (solid lines) <40 yr of age (diamond) and >60 yr of age (star) in the supine and seated positions, at 50% of maximal workload, and at peak exercise. EaI/ELVI decreases during exercise in both young and older men and women (P < 0.0001). However, older men and women have a blunted decline in EaI/ELVI (P < 0.001). EaI increases during exercise in both young and older men and women (P < 0.0001). At maximal exercise, EaI is greater in older vs. younger women (P < 0.002). In contrast, EaI does not differ between young and older men. ELVI increases during exercise in both young and older men and women (P < 0.0001). At maximal exercise, ELVI is greater in younger vs. older men (P < 0.001) and tended to be greater in younger than older women (P < 0.07). (With permission from Strait JS, Lakatta EG Cardiac Aging: Aging from human to molecules” Muscle, 2011. Modifed from Najjar SS, Schulman SP, Fleg JL, et al. Relationship of age and sex on ventricular-vascular coupling at rest and exercise. Circulation 2000(Suppl);102:II-602).
In HF, these relationships change. In systolic HF, patients have a downward and rightward shift of the end-systolic P-V relationship and thus a reduced ELVI (left ventricular end-systolic elastance index) (range 0.6–2.6 mmHgμml−1xm2)70. Systolic HF patients have an augmented EAI (arterial elastance index) (range 1.7–3.7 mmHgμml−1 × m−2) due to a decrease in stroke volume index (SVI) and to increases in HR and PVR70. The increase in EAI and decrease in ELVI result in marked, up to three- to fourfold, increases in EA/ELV (range 1.3–4.3). This suboptimal coupling reflects diminished CV performance and efficiency of the failing heart. In contrast, HF patients with preserved ejection fraction (HFpEF) have an 18 and 20% higher resting EA and ELV, respectively, compared with healthy controls71. Studies suggest a matched increase in EA and ELV and therefore no difference in their EA/ELV when compared to normotensive controls or hypertensive patients without HF.
Arterial-ventricular coupling with exercise
This coupling has significant implications for an individual’s exercise capacity as they age, however67. Otherwise normal arteries dilate and stiffen with age imposing increased load in the form of inertance, reflected pulse waves, artery compliance, and resistance (reviewed by Lakatta72). This increased load has a significant impact on the relationship of the heart to the vascular system as represented by the Ea/ELV ratio and described above. Furthermore, it increases tremendously when the body exercises (Figure 10A–C). While the resting Ea/ELV ratio is preserved by the parallel increase in both variables (described above), exercise requires an increase in ejection fraction (EF) that the aged heart may have difficulty providing. In order to increase EF, the LV end-systolic elastance (ELV) must increase to a greater extent than the EA. With increasing age, however, ELV fails to increase in proportion to the increased EA in men and there is a deficit in LVEF reserve when older subjects are exposed to acutely increased workloads (i.e. exercise). Interestingly, reduction of the afterload by an acute infusion of sodium nitroprusside in healthy, older BLSA volunteers augments LVEF at rest and with exercise and provides further evidence of the importance of vascular properties73.
The baseline alterations in EA/ELV and its components in patients with systolic HF are also evident during exercise74. Indeed, EA, ELV, and EA/ELV do not appreciably change during exercise in systolic HF patients, whereas they markedly change in healthy subjects74. Thus the limited capacity of systolic HF patients to augment their CV function during times of stress (such as exercise) involve marked deficits in both LV and arterial elastance reserves.
To date, there are no studies that have examined the change in EA/ELV or EA during exercise in HFpEF. Researchers have examined the change in ELV and EF in HFpEF during upright bicycle exercise75. Compared with HTN controls with LV hypertrophy, HFpEF patients had a threefold smaller increase in ELV and a reduced ability to lower their PVR and increase their HR during exercise. They also had a 50% smaller increase in EF during exercise. As EF is inversely related to EA/ELV, this suggests that the change in EA/ELV during exercise in HFpEF may also be severely blunted. Since female sex and systolic hypertension are risk factors for HFpEF, and as the pathophysiology of HFpEF involves a limited CV reserve76, the diminished EA/ELV reserve observed in systolic HTN women suggests that they may be exhibiting signs of subclinical (Stage B) HF. This raises the possibility that they may be on a trajectory to progressive exercise intolerance and perhaps functional limitations.
d. Cardioprotective and repair processes
Because of the many factors discussed above, the aged heart is placed under increasing levels of stress due to its diminished functional reserve. Furthermore, increasing age results in the increased occurrence of numerous disease processes e.g. diabetes, hypertension, etc). While the heart has a number of protective systems in place to deal with such insult (Figure 2) these decline with age (Figure 11A), resulting in a lower injury threshold. It is not yet clear if there are interventions (e.g. physical conditioning) that can be instituted in the elderly to delay or reverse these processes but there are a number of promising candidates.
Physical Conditioning
There is a good deal of evidence that aerobic exercise programs can provide improvements in peak oxygen consumption and its components as well as increases in ventilatory threshold and submaximal endurance for older persons with heart failure 77. A longitudinal study in older males studied in the upright position indicates that an enhanced physical conditioning status increases O2 consumption and work capacity, in part by increases in the maximum CO by increasing the maximum SV, and in part by increasing the estimated total body (A-V) O2 utilization15. The augmentation of maximum SVI is due to an augmented reduction of LVESV15 and thus, a concomitant increase in LVEF, as the effect of conditioning status to increase LVEDI exercise is minimal (recall that LVEDI during acute vigorous exercise is already appreciably increased in older, sedentary, pre-conditioned men78). This minor effect of physical conditioning on max exercise LVEDVI in older persons is in contrast to the effect of physical conditioning in younger persons, which substantially increases EDVI and SVI during vigorous exercise on the basis of the Frank Starling mechanism, as well as via an enhanced LVEF. In contrast to the improved LV ejection, the max heart rate of older persons does not vary with physical conditioning status and exercise does not appear to offset the age-associated deficiency in sympathetic modulation. It is noteworthy that the increased pulse wave velocity, carotid augmentation index or pulse pressure that occurs with aging is less in older persons who are physically conditioned than in sedentary persons59. An effect of physical conditioning to reduce these components of vascular afterload appears to be involved in the effect of conditioning to improve LV ejection61.
Stem cells and the promise of cardiac regeneration
It had traditionally been thought that cardiomyocytes terminally differentiate and withdraw from the cell cycle early in their development. However, carbon dating methods have established that cardiomyocytes continue to be synthesized and a low level of turnover in the heart persists throughout life79. New cells can arise from Endothelial Precursor Cells (EPCs) that exist in the bone marrow or precursor cells (PCs) from other locations including adipose tissue and the heart itself. There has been great interest in utilizing resident EPCs as a source of myocardial regeneration after injury or other degradatory processes and many studies of cardiac cell turnover.
The field remains contentious. Some researchers contend that the rate of cardiomyocyte turnover gradually decreases with aging from 1% per year for a 25 year old to 0.45% per year for a 75-year old79. Alternatively, Anversa et. al80 have found that cardiomyocyte regeneration from predominantly cardiac resident stem cell actually increases with aging and remains important in the heart throughout life. However, these newly generated cells appear to quickly degrade in older individuals to resemble those they have replaced. Necrotic cellular destruction progresses with aging until the heart is unable to keep pace with the level of required stem cell replacement leading to a decline in cell numbers.
The cause of this “aging memory” may be reduced telomerase activity, reactive oxygen species, or loss of telomere-related proteins80,81. The telomeres of newly generated cells in senescent human hearts quickly shorten to lengths similar to the aged cardiomyocytes they are replacing. Telomeres normally serve as protective caps for chromosomal ends, protecting them from DNA Damage Repair systems and activation of the p16INK4a pathway with eventual senescence or apoptosis82. However, telomeres lose 30–150 base pairs during each cell division due to the inability of DNA polymerase to fully replicate the 3′ end of the DNA strand. When the telomere becomes “too short” the cell can no longer divide, it becomes senescent, and eventually dies. Telomeres can be extended by Telomerase which is composed of two main components, the telomerase RNA component (TERC) and telomerase reverse transcriptase (TERT) with a third component (dyskerin) working to stabilize the complex. Agonism of telomerase activity through these components may have some benefit in stabilizing cellular DNA.
If the promise of EPC mobilization in the aged heart is to be realized, the problems of telomere shortening and cellular “memory” will have to be dealt with as well as methods to control the “internal clock” that resides in DNA-telomeres83. Insulin-like Growth Factor (IGF-1) also holds promise as a regulator of cardiac senescence. Activation of the IGF-1/IGF-1R pathway preserves telomere length and promotes cardiac progenitor cell growth and survival84. Injection of IGF-1 in damaged myocardium promotes the migration and homing of cardiac stem cells and facilitates neo-vascularization85.
Ischemic preconditioning
Ischemic preconditioning (IPC) is the hearts endogenous capacity to resist ischemic damage and its effects including suppression of ventricular arrhythmias and enhanced recovery of contractile function86. The process is dependent on an initial, brief ischemic event usually lasting less than 5 minutes. It appears to have both an early protective function lasting about 1 hour after preconditioning (PC) and also a late-delayed action that returns around 24–96 hr later. Unfortunately, aging results in decreased effectiveness of this and other protective pathways86 which leads to a decreased injury threshold in the aged heart (Figure 11A, B). This is but one example of a system that could be manipulated to enhance the cardiac protective systems in the aging heart (Figure 11B).
III. Clinical approach to heart failure in aged persons
Clinical assessment
Evaluation of the older patient presenting with failure symptoms should include a non-invasive study to determine whether the primary problem is impaired systolic function. Although systolic dysfunction is present in at least half of CHF cases, the presence of a normal, or elevated, ejection fraction is more common in older than younger heart failure patients, particularly women and those with atrial fibrillation and hypertension. The number of patients with heart failure and preserved systolic function is increasing, and is likely to continue to increase with the aging of the general population.
It is often helpful to investigate whether any reversible precipitants are present in older individuals who present with new-onset or worsening heart failure symptoms, including anemia, infection, thyroid disease, atrial fibrillation and dietary or medication non-compliance. Investigation for common co-morbidities is also useful as they are frequent and associated with increased hospitalizations and adverse clinical outcomes87. Hypertension is almost invariably present as well as increased central vascular stiffness and therefore impaired ventricular-vascular coupling. It should be noted that evidence of long standing volume overload and an S3 gallop are less likely in patients with preserved systolic function and that in the general older heart failure population exertional symptoms are less common, and those related to fatigue and mental status changes more common. The diagnosis of heart failure with preserved systolic function is primarily one of exclusion in patients with objective evidence of pulmonary vascular congestion without findings of an ischemic, hypertensive, or valvular etiology. Amyloid should also be considered in older patients presenting with heart failure symptoms in the absence of another identifiable causes.
Considerations with pharmacologic interventions
Diuretics are particularly useful in older patients with increased vascular stiffness presenting with acute congestive symptoms, since significant reductions in pressure occur with relatively small changes in intravascular volume. Heart failure with primary systolic dysfunction and with preserved systolic function are both associated with increased sympathetic and rennin-angiotensin-aldosterone activation. In those with primary systolic dysfunction, β-blockers improve survival and other important cardiovascular outcomes in the older subsets of large randomized trials88. It is unfortunate that many older individuals with heart failure and systolic dysfunction are not receiving any, or under-dosed, β-blocker therapy. In contrast to the many successful trials in those with systolic dysfunction, there are no reported randomized studies limited to the effects of beta blockers in older individuals with heart failure and preserved systolic function.
ACE-inhibitors are a cornerstone of therapy in patients with systolic dysfunction and their benefit extends to the elderly. Their benefit in patients with preserved systolic function, however, is less certain89. There is an on-going trial of spironolactone in heart failure patients with preserved systolic function, but first outcomes are not expected before 2011. There are no clear explanations for the lack of survival benefit for any medical intervention in older heart failure patients with preserved systolic function. The diagnosis is more difficult and thus patients without heart disease, for whom these therapies would not be expected to be of any benefit, may be included in some of the studies. The pathophysiology may differ, with myocardial fibrosis and impaired ventricular-vascular coupling more important responsible mechanisms, and these may not respond to the tested therapies. Treatment might therefore be focused on blood pressure control in those with hypertension, rate control in those with atrial fibrillation, and judicious use of diuretics in the setting of volume overload. It should be noted, though, that the steep volume/pressure relationship associated with increased vascular stiffness in the older individual, might result in symptomatic hypotension when diuretics result in intravascular volume depletion.
Devices (e.g. implantable cardiac defibrillators, bi-ventricular pacing devices, left ventricular assist devices, etc.) may also significantly improve outcomes in patients with persistent failure despite medical therapy. There has been significant development on this front that has made their benefits accessible to patients throughout the spectrum of disease. However, as with any treatment, there is no guarantee of benefit and it is often just as helpful to first assess more socially relevant factors. This is especially relevant in the elderly, who have a diminished cardiac reserve to begin with and may see significantly different treatment benefits when compared to the typically younger study population on which many therapeutic recommendations are based. The importance of the individual patient’s role as a partner in his/her care and of individualizing treatment and monitoring plans cannot be over-emphasized.
Although patients may carry the same heart failure diagnosis, they differ markedly in terms of disease severity and complexity, associated co-morbidities, social support, education, ingrained habits, access to medical personnel and knowledge, and understanding of health care information and directions. Noncompliance with medications or diet is often cited as a major factor contributing to hospitalization in heart failure patients. In the older patient, however, non-compliance is more likely due to social isolation, financial difficulties, limited travel and meal options, decreased tolerability for some medicines, co-morbidities, and difficult to follow complex medical regimens. In many of these patients, a multidisciplinary team approach including simplification of the medical regimen, close monitoring, and intensive patient education can decrease hospital admission and improve quality of life.
IV. Conclusions
Aging results in an increase in cardiovascular disease and a decrease in cardiac reserve at the same time that the repair processes designed to deal with these problems become less active/effective. These factors combine to set the stage for heart failure. 1) Structurally, the heart thickens and stiffens with age resulting in the increased imposition of a number of functional demands. 2) Functionally, a number of changes which assist the resting heart to deal with the effects of aging cause significant functional deficits with exercise or stress, thereby lowering the cardiac reserve that the younger heart can call on to deal with disease or insult. 3) Finally, while the increased incidence of disease, less structurally efficient heart, and decreased cardiac reserve associated with aging would be well served by an effective repair system- this too declines with age. It is hoped that improved understanding of the aged heart will enable the development of therapies which prevent the genesis of HF or at a minimum help clinicians to treat the unique properties of the failing, senescent heart.
FIGURE 6.
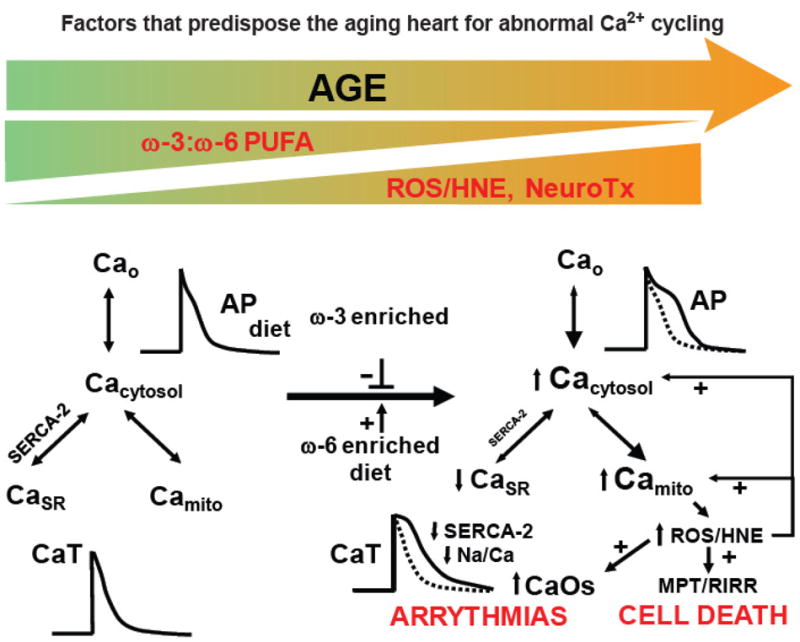
An overview of change in the aging heart that predispose to a reduced threshold for abnormal Ca2+ handling during acute stress. See text for details. (With permission from Strait JS, Lakatta EG Cardiac Aging: Aging from human to molecules” Muscle, 2011. Modified from Lakatta, E. G., Sollott, S. J., & Pepe, S. (2001). The old heart: operating on the edge Novartis Foundation symposium, 235, 172-96).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.WRITING GROUP MEMBERS. Lloyd-Jones D, Adams RJ, et al. Heart disease and stroke statistics--2010 update: a report from the American Heart Association. Circulation. 2010;121(7):e46–e215. doi: 10.1161/CIRCULATIONAHA.109.192667. [DOI] [PubMed] [Google Scholar]
- 2.THOMAS S, RICH M. Epidemiology, Pathophysiology, and Prognosis of Heart Failure in the Elderly. Heart Failure Clinics. 2007;3(4):381–387. doi: 10.1016/j.hfc.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Najjar SS, Gerstenblith G, Lakatta EG. In: Cardiovascular Medicine. Willerson JT, Wellens HJJ, Cohn JN, Holmes DR Jr, editors. London: Springer London; 2007. pp. 2439–2451. [Google Scholar]
- 4.Jessup M, Abraham WT, Casey DE, et al. 2009 focused update: ACCF/AHA Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation. 2009;119(14):1977–2016. doi: 10.1161/CIRCULATIONAHA.109.192064. [DOI] [PubMed] [Google Scholar]
- 5.Chen W, Frangogiannis NG. The role of inflammatory and fibrogenic pathways in heart failure associated with aging. Heart Fail Rev. 2010;15(5):415–422. doi: 10.1007/s10741-010-9161-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part II: the aging heart in health: links to heart disease. Circulation. 2003;107(2):346–354. doi: 10.1161/01.cir.0000048893.62841.f7. [DOI] [PubMed] [Google Scholar]
- 7.Scholz DG, Kitzman DW, Hagen PT, Ilstrup DM, Edwards WD. Age-related changes in normal human hearts during the first 10 decades of life. Part I (Growth): A quantitative anatomic study of 200 specimens from subjects from birth to 19 years old. Mayo Clin Proc. 1988;63(2):126–136. doi: 10.1016/s0025-6196(12)64945-3. [DOI] [PubMed] [Google Scholar]
- 8.Gerstenblith G, Frederiksen J, Yin F, Fortuin N. Echocardiographic assessment of a normal adult aging population. Circulation. 1977 doi: 10.1161/01.cir.56.2.273. [DOI] [PubMed] [Google Scholar]
- 9.Olivetti G, Melissari M, Capasso JM, Anversa P. Cardiomyopathy of the aging human heart. Myocyte loss and reactive cellular hypertrophy. Circ Res. 1991;68(6):1560–1568. doi: 10.1161/01.res.68.6.1560. [DOI] [PubMed] [Google Scholar]
- 10.Hees PS, Fleg JL, Lakatta EG, Shapiro EP. Left ventricular remodeling with age in normal men versus women: novel insights using three-dimensional magnetic resonance imaging. Am J Cardiol. 2002;90(11):1231–1236. doi: 10.1016/s0002-9149(02)02840-0. [DOI] [PubMed] [Google Scholar]
- 11.Linzbach AJ, Akuamoa-Boateng E. Changes in the aging human heart. I. Heart weight in the aged. Klin Wochenschr. 1973;51(4):156–163. doi: 10.1007/BF01468338. [DOI] [PubMed] [Google Scholar]
- 12.Khouri MG, Maurer MS, El-Khoury Rumbarger L. Assessment of age-related changes in left ventricular structure and function by freehand three-dimensional echocardiography. Am J Geriatr Cardiol. 2005;14(3):118–125. doi: 10.1111/j.1076-7460.2005.03845.x. [DOI] [PubMed] [Google Scholar]
- 13.Dannenberg AL, Levy D, Garrison RJ. Impact of age on echocardiographic left ventricular mass in a healthy population (the Framingham Study) Am J Cardiol. 1989;64(16):1066–1068. doi: 10.1016/0002-9149(89)90816-3. [DOI] [PubMed] [Google Scholar]
- 14.Fleg JL, Lakatta EG. Normal Aging of the Cardiovascular System. Cardiovascular Disease in the Elderly. 2007:1–46. [Google Scholar]
- 15.Schulman SP, Lakatta EG, Fleg JL, et al. Age-related decline in left ventricular filling at rest and exercise. Am J Physiol. 1992;263(6 Pt 2):H1932–8. doi: 10.1152/ajpheart.1992.263.6.H1932. [DOI] [PubMed] [Google Scholar]
- 16.Fleg JL, Lakatta EG. Role of muscle loss in the age-associated reduction in VO2 max. J Appl Physiol. 1988;65(3):1147–1151. doi: 10.1152/jappl.1988.65.3.1147. [DOI] [PubMed] [Google Scholar]
- 17.Fleg JL, Morrell CH, Bos AG, et al. Accelerated longitudinal decline of aerobic capacity in healthy older adults. Circulation. 2005;112(5):674–682. doi: 10.1161/CIRCULATIONAHA.105.545459. [DOI] [PubMed] [Google Scholar]
- 18.Le Blanc PR, Rakusan K. Effects of age and isoproterenol on the cardiac output and regional blood flow in the rat. Can J Cardiol. 1987;3(5):246–250. [PubMed] [Google Scholar]
- 19.Rakusan K, Blahitka J. Cardiac output distribution in rats measured by injection of radioactive microspheres via cardiac puncture. Can J Physiol Pharmacol. 1974;52(2):230–235. doi: 10.1139/y74-031. [DOI] [PubMed] [Google Scholar]
- 20.Miyatake K, Okamoto M, Kinoshita N, et al. Augmentation of atrial contribution to left ventricular inflow with aging as assessed by intracardiac Doppler flowmetry. Am J Cardiol. 1984;53(4):586–589. doi: 10.1016/0002-9149(84)90035-3. [DOI] [PubMed] [Google Scholar]
- 21.Bryg RJ, Williams GA, Labovitz AJ. Effect of aging on left ventricular diastolic filling in normal subjects. Am J Cardiol. 1987;59(9):971–974. doi: 10.1016/0002-9149(87)91136-2. [DOI] [PubMed] [Google Scholar]
- 22.Spina RJ, Turner MJ, Ehsani AA. Beta-adrenergic-mediated improvement in left ventricular function by exercise training in older men. Am J Physiol. 1998;274(2 Pt 2):H397–404. doi: 10.1152/ajpheart.1998.274.2.H397. [DOI] [PubMed] [Google Scholar]
- 23.Tan Y, Wenzelburger F, Lee E, et al. The pathophysiology of heart failure with normal ejection fraction: exercise echocardiography reveals complex abnormalities of both systolic and diastolic ventricular function involving torsion, untwist, and longitudinal motion. J Am Coll Cardiol. 2009;54(1):36. doi: 10.1016/j.jacc.2009.03.037. [DOI] [PubMed] [Google Scholar]
- 24.Tarasov KV, Sanna S, Scuteri A, et al. COL4A1 is associated with arterial stiffness by genome-wide association scan. Circ Cardiovasc Genet. 2009;2(2):151–158. doi: 10.1161/CIRCGENETICS.108.823245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.HISS RG, LAMB LE. Electrocardiographic findings in 122,043 individuals. Circulation. 1962;25:947–961. doi: 10.1161/01.cir.25.6.947. [DOI] [PubMed] [Google Scholar]
- 26.Fleg JL, Das DN, Wright J, Lakatta EG. Age-associated changes in the components of atrioventricular conduction in apparently healthy volunteers. J Gerontol. 1990;45(3):M95–100. doi: 10.1093/geronj/45.3.m95. [DOI] [PubMed] [Google Scholar]
- 27.Golden GS, Golden LH. The “Nona” electrocardiogram: findings in 100 patients of the 90 plus age group. Journal of the American Geriatrics Society. 1974;22(7):329–332. doi: 10.1111/j.1532-5415.1974.tb05402.x. [DOI] [PubMed] [Google Scholar]
- 28.Harlan WR, Graybiel A, Mitchell RE, Oberman A, Osborne RK. Serial electrocardiograms: their reliability and prognostic validity during a 24-yr. period. J Chronic Dis. 1967;20(11):853–867. doi: 10.1016/0021-9681(67)90023-9. [DOI] [PubMed] [Google Scholar]
- 29.Mihalick MJ, Fisch C. Electrocardiographic findings in the aged. Am Heart J. 1974;87(1):117–128. doi: 10.1016/0002-8703(74)90400-1. [DOI] [PubMed] [Google Scholar]
- 30.Manolio TA, Furberg CD, Rautaharju PM, et al. Cardiac arrhythmias on 24-h ambulatory electrocardiography in older women and men: the Cardiovascular Health Study. JAC. 1994;23(4):916–925. doi: 10.1016/0735-1097(94)90638-6. [DOI] [PubMed] [Google Scholar]
- 31.Fleg JL, Kennedy HL. Cardiac arrhythmias in a healthy elderly population: detection by 24-hour ambulatory electrocardiography. Chest. 1982;81(3):302–307. doi: 10.1378/chest.81.3.302. [DOI] [PubMed] [Google Scholar]
- 32.Fisch C. Introduction: chronic ventricular arrhythmias--a major unresolved health problem. Heart Lung. 1981;10(3):451–454. [PubMed] [Google Scholar]
- 33.Lakatta EG, Sollott SJ. Perspectives on mammalian cardiovascular aging: humans to molecules. Comp Biochem Physiol, Part A Mol Integr Physiol. 2002;132(4):699–721. doi: 10.1016/s1095-6433(02)00124-1. [DOI] [PubMed] [Google Scholar]
- 34.Janczewski AM, Spurgeon HA, Lakatta EG. Action potential prolongation in cardiac myocytes of old rats is an adaptation to sustain youthful intracellular Ca2+ regulation. J Mol Cell Cardiol. 2002;34(6):641–648. doi: 10.1006/jmcc.2002.2004. [DOI] [PubMed] [Google Scholar]
- 35.Lakatta E. Age-associated cardiovascular changes in health: impact on cardiovascular disease in older persons. Heart Fail Rev. 2002 doi: 10.1023/a:1013797722156. [DOI] [PubMed] [Google Scholar]
- 36.Lakatta EG, Sollott SJ, Pepe S. The old heart: operating on the edge. Novartis Found Symp. 2001;235:172–96. doi: 10.1002/0470868694.ch15. discussion 196–201, 217–20. [DOI] [PubMed] [Google Scholar]
- 37.Lakatta EG. Cardiovascular regulatory mechanisms in advanced age. Physiol Rev. 1993;73(2):413–467. doi: 10.1152/physrev.1993.73.2.413. [DOI] [PubMed] [Google Scholar]
- 38.Lakatta EG. Deficient neuroendocrine regulation of the cardiovascular system with advancing age in healthy humans. Circulation. 1993;87(2):631–636. doi: 10.1161/01.cir.87.2.631. [DOI] [PubMed] [Google Scholar]
- 39.Esler MD, Turner AG, Kaye DM, et al. Aging effects on human sympathetic neuronal function. Am J Physiol. 1995;268(1 Pt 2):R278–85. doi: 10.1152/ajpregu.1995.268.1.R278. [DOI] [PubMed] [Google Scholar]
- 40.Lakatta E. Cardiovascular aging research: the next horizons. Journal of the American Geriatrics Society. 1999;47(5):613–625. doi: 10.1111/j.1532-5415.1999.tb02579.x. [DOI] [PubMed] [Google Scholar]
- 41.Cigola E, Kajstura J, Li B, Meggs LG, Anversa P. Angiotensin II activates programmed myocyte cell death in vitro. Exp Cell Res. 1997;231(2):363–371. doi: 10.1006/excr.1997.3477. [DOI] [PubMed] [Google Scholar]
- 42.Sadoshima J, Xu Y, Slayter HS, Izumo S. Autocrine release of angiotensin II mediates stretch-induced hypertrophy of cardiac myocytes in vitro. Cell. 1993;75(5):977–984. doi: 10.1016/0092-8674(93)90541-w. [DOI] [PubMed] [Google Scholar]
- 43.Cave AC, Brewer AC, Narayanapanicker A, et al. NADPH oxidases in cardiovascular health and disease. Antioxid Redox Signal. 2006;8(5–6):691–728. doi: 10.1089/ars.2006.8.691. [DOI] [PubMed] [Google Scholar]
- 44.Lakatta EG, Wang M, Najjar SS. Arterial aging and subclinical arterial disease are fundamentally intertwined at macroscopic and molecular levels. Med Clin North Am. 2009;93(3):583–604. doi: 10.1016/j.mcna.2009.02.008. Table of Contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Farasat SM, Morrell CH, Scuteri A, et al. Pulse pressure is inversely related to aortic root diameter implications for the pathogenesis of systolic hypertension. Hypertension. 2008;51(2):196–202. doi: 10.1161/HYPERTENSIONAHA.107.099515. [DOI] [PubMed] [Google Scholar]
- 46.Nagai Y, Metter E, Earley C, Kemper M. Increased carotid artery intimal-medial thickness in asymptomatic older subjects with exercise-induced myocardial ischemia. Circulation. 1998 doi: 10.1161/01.cir.98.15.1504. [DOI] [PubMed] [Google Scholar]
- 47.Ungvari Z, Kaley G, de Cabo R, Sonntag WE, Csiszar A. Mechanisms of vascular aging: new perspectives. J Gerontol A Biol Sci Med Sci. 2010;65(10):1028–1041. doi: 10.1093/gerona/glq113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zieman SJ, Melenovsky V, Kass DA. Mechanisms, pathophysiology, and therapy of arterial stiffness. Arterioscler Thromb Vasc Biol. 2005;25(5):932–943. doi: 10.1161/01.ATV.0000160548.78317.29. [DOI] [PubMed] [Google Scholar]
- 49.Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part I: aging arteries: a “set up” for vascular disease. Circulation. 2003;107(1):139–146. doi: 10.1161/01.cir.0000048892.83521.58. [DOI] [PubMed] [Google Scholar]
- 50.Semba RD, Najjar SS, Sun K, Lakatta EG, Ferrucci L. Serum carboxymethyl-lysine, an advanced glycation end product, is associated with increased aortic pulse wave velocity in adults. Am J Hypertens. 2009;22(1):74–79. doi: 10.1038/ajh.2008.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Csiszar A, Wang M, Lakatta EG, Ungvari Z. Inflammation and endothelial dysfunction during aging: role of NF-kappaB. J Appl Physiol. 2008;105(4):1333–1341. doi: 10.1152/japplphysiol.90470.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tschudi MR, Barton M, Bersinger NA, et al. Effect of age on kinetics of nitric oxide release in rat aorta and pulmonary artery. J Clin Invest. 1996;98(4):899–905. doi: 10.1172/JCI118872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cernadas MR, Sánchez de Miguel L, García-Durán M, et al. Expression of constitutive and inducible nitric oxide synthases in the vascular wall of young and aging rats. Circ Res. 1998;83(3):279–286. doi: 10.1161/01.res.83.3.279. [DOI] [PubMed] [Google Scholar]
- 54.Wang M, Takagi G, Asai K, et al. Aging increases aortic MMP-2 activity and angiotensin II in nonhuman primates. Hypertension. 2003;41(6):1308–1316. doi: 10.1161/01.HYP.0000073843.56046.45. [DOI] [PubMed] [Google Scholar]
- 55.Wang M, Monticone RE, Lakatta EG. Arterial aging: a journey into subclinical arterial disease. Curr Opin Nephrol Hypertens. 2010;19(2):201–207. doi: 10.1097/MNH.0b013e3283361c0b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jiang L, Wang M, Zhang J, et al. Increased aortic calpain-1 activity mediates age-associated angiotensin II signaling of vascular smooth muscle cells. PLoS ONE. 2008;3(5):e2231. doi: 10.1371/journal.pone.0002231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang M, Zhao D, Spinetti G, et al. Matrix metalloproteinase 2 activation of transforming growth factor-beta1 (TGF-beta1) and TGF-beta1-type II receptor signaling within the aged arterial wall. Arterioscler Thromb Vasc Biol. 2006;26(7):1503–1509. doi: 10.1161/01.ATV.0000225777.58488.f2. [DOI] [PubMed] [Google Scholar]
- 58.Wang M, Zhang J, Walker SJ, et al. Involvement of NADPH oxidase in age-associated cardiac remodeling. J Mol Cell Cardiol. 2010;48(4):765–772. doi: 10.1016/j.yjmcc.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tanaka H, Dinenno FA, Monahan KD, et al. Aging, habitual exercise, and dynamic arterial compliance. Circulation. 2000;102(11):1270–1275. doi: 10.1161/01.cir.102.11.1270. [DOI] [PubMed] [Google Scholar]
- 60.Roman MJ, Devereux RB, Kizer JR, et al. High central pulse pressure is independently associated with adverse cardiovascular outcome the strong heart study. J Am Coll Cardiol. 2009;54(18):1730–1734. doi: 10.1016/j.jacc.2009.05.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vaitkevicius PV, Fleg JL, Engel JH, et al. Effects of age and aerobic capacity on arterial stiffness in healthy adults. Circulation. 1993;88(4 Pt 1):1456–1462. doi: 10.1161/01.cir.88.4.1456. [DOI] [PubMed] [Google Scholar]
- 62.Mitchell GF, Parise H, Benjamin EJ, et al. Changes in arterial stiffness and wave reflection with advancing age in healthy men and women: the Framingham Heart Study. Hypertension. 2004;43(6):1239–1245. doi: 10.1161/01.HYP.0000128420.01881.aa. [DOI] [PubMed] [Google Scholar]
- 63.Williams B, Lacy PS. Central haemodynamics and clinical outcomes: going beyond brachial blood pressure? Eur Heart J. 2010;31(15):1819–1822. doi: 10.1093/eurheartj/ehq125. [DOI] [PubMed] [Google Scholar]
- 64.Pearson T, Mensah G, Alexander R. Markers of inflammation and cardiovascular disease application to clinical and public …. Circulation. 2003 doi: 10.1161/01.CIR.0000148979.11121.6B. [DOI] [PubMed] [Google Scholar]
- 65.Najjar S, Scuteri A, Shetty V, et al. Pulse wave velocity is an independent predictor of the longitudinal increase in systolic blood pressure and of incident hypertension in the Baltimore Longitudinal Study of Aging. J Am Coll Cardiol. 2008;51(14):1377. doi: 10.1016/j.jacc.2007.10.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chantler PD, Lakatta EG, Najjar SS. Arterial-ventricular coupling: mechanistic insights into cardiovascular performance at rest and during exercise. J Appl Physiol. 2008;105(4):1342–1351. doi: 10.1152/japplphysiol.90600.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lakatta E, Najjar S. Arterial-ventricular coupling: mechanistic insights into cardiovascular performance at rest and during exercise. Journal of Applied …. 2008 doi: 10.1152/japplphysiol.90600.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lakatta EG. Cardiac muscle changes in senescence. Annu Rev Physiol. 1987;49:519–531. doi: 10.1146/annurev.ph.49.030187.002511. [DOI] [PubMed] [Google Scholar]
- 69.Hees PS, Fleg JL, Mirza ZA, et al. Effects of normal aging on left ventricular lusitropic, inotropic, and chronotropic responses to dobutamine. J Am Coll Cardiol. 2006;47(7):1440–1447. doi: 10.1016/j.jacc.2005.11.052. [DOI] [PubMed] [Google Scholar]
- 70.Asanoi H, Sasayama S, Kameyama T. Ventriculoarterial coupling in normal and failing heart in humans. Circ Res. 1989;65(2):483–493. doi: 10.1161/01.res.65.2.483. [DOI] [PubMed] [Google Scholar]
- 71.Lam CSP, Roger VL, Rodeheffer RJ, et al. Cardiac structure and ventricular-vascular function in persons with heart failure and preserved ejection fraction from Olmsted County, Minnesota. Circulation. 2007;115(15):1982–1990. doi: 10.1161/CIRCULATIONAHA.106.659763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lakatta EG. Arterial aging is risky. J Appl Physiol. 2008;105(4):1321–1322. doi: 10.1152/japplphysiol.91145.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nussbacher A, Gerstenblith G, O’Connor FC, et al. Hemodynamic effects of unloading the old heart. Am J Physiol. 1999;277(5 Pt 2):H1863–71. doi: 10.1152/ajpheart.1999.277.5.H1863. [DOI] [PubMed] [Google Scholar]
- 74.Cohen-Solal A, Faraggi M, Czitrom D, et al. Left ventricular-arterial system coupling at peak exercise in dilated nonischemic cardiomyopathy. Chest. 1998;113(4):870–877. doi: 10.1378/chest.113.4.870. [DOI] [PubMed] [Google Scholar]
- 75.Borlaug BA, Melenovsky V, Russell SD, et al. Impaired chronotropic and vasodilator reserves limit exercise capacity in patients with heart failure and a preserved ejection fraction. Circulation. 2006;114(20):2138–2147. doi: 10.1161/CIRCULATIONAHA.106.632745. [DOI] [PubMed] [Google Scholar]
- 76.Kitzman DW, Higginbotham MB, Cobb FR, Sheikh KH, Sullivan MJ. Exercise intolerance in patients with heart failure and preserved left ventricular systolic function: failure of the Frank-Starling mechanism. JAC. 1991;17(5):1065–1072. doi: 10.1016/0735-1097(91)90832-t. [DOI] [PubMed] [Google Scholar]
- 77.Fleg JL. Can exercise conditioning be effective in older heart failure patients? Heart Fail Rev. 2002;7(1):99–103. doi: 10.1023/a:1013758008044. [DOI] [PubMed] [Google Scholar]
- 78.Fleg JL, O’Connor F, Gerstenblith G, et al. Impact of age on the cardiovascular response to dynamic upright exercise in healthy men and women. J Appl Physiol. 1995;78(3):890–900. doi: 10.1152/jappl.1995.78.3.890. [DOI] [PubMed] [Google Scholar]
- 79.Bergmann O, Bhardwaj RD, Bernard S, et al. Evidence for cardiomyocyte renewal in humans. Science. 2009;324(5923):98–102. doi: 10.1126/science.1164680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kajstura J, Urbanek K, Perl S, et al. Cardiomyogenesis in the adult human heart. Circ Res. 2010;107(2):305–315. doi: 10.1161/CIRCRESAHA.110.223024. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 81.Urbanek K, Torella D, Sheikh F, et al. Myocardial regeneration by activation of multipotent cardiac stem cells in ischemic heart failure. Proc Natl Acad Sci USA. 2005;102(24):8692–8697. doi: 10.1073/pnas.0500169102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wong LSM, van der Harst P, de Boer RA, et al. Aging, telomeres and heart failure. Heart Fail Rev. 2010;15(5):479–486. doi: 10.1007/s10741-010-9173-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chen W, Frangogiannis N. The role of inflammatory and fibrogenic pathways in heart failure associated with aging. Heart Fail Rev. 2010:1–8. doi: 10.1007/s10741-010-9161-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Torella D, Rota M, Nurzynska D, et al. Cardiac stem cell and myocyte aging, heart failure, and insulin-like growth factor-1 overexpression. Circ Res. 2004;94(4):514–524. doi: 10.1161/01.RES.0000117306.10142.50. [DOI] [PubMed] [Google Scholar]
- 85.Gonzalez A, Rota M, Nurzynska D, et al. Activation of cardiac progenitor cells reverses the failing heart senescent phenotype and prolongs lifespan. Circ Res. 2008;102(5):597–606. doi: 10.1161/CIRCRESAHA.107.165464. [DOI] [PubMed] [Google Scholar]
- 86.Juhaszova M, Rabuel C, Zorov DB, Lakatta EG, Sollott SJ. Protection in the aged heart: preventing the heart-break of old age? Cardiovasc Res. 2005;66(2):233–244. doi: 10.1016/j.cardiores.2004.12.020. [DOI] [PubMed] [Google Scholar]
- 87.Najjar S. Evolving insights into the pathophysiology of heart failure with preserved ejection fraction. Current Cardiovascular Risk Reports. 2009;3(5):374–379. [Google Scholar]
- 88.Dulin BR, Haas SJ, Abraham WT, Krum H. Do elderly systolic heart failure patients benefit from beta blockers to the same extent as the non-elderly? Meta-analysis of >12,000 patients in large-scale clinical trials. Am J Cardiol. 2005;95(7):896–898. doi: 10.1016/j.amjcard.2004.11.052. [DOI] [PubMed] [Google Scholar]
- 89.Massie BM, Carson PE, McMurray JJ, et al. Irbesartan in patients with heart failure and preserved ejection fraction. N Engl J Med. 2008;359(23):2456–2467. doi: 10.1056/NEJMoa0805450. [DOI] [PubMed] [Google Scholar]