

Abstract
There is emerging evidence that people with successfully treated HIV infection age prematurely leading to progressive multi-organ disease 1, but the reasons for this are not known. Here we show that patients treated with commonly used nucleoside analog anti-retroviral drugs (NRTIs) progressively accumulate somatic mitochondrial DNA (mtDNA) mutations, mirroring that seen much later in life due to normal aging 2,3. Ultra-deep re-sequencing-by-synthesis, combined with single cell analyses, suggests that the increase in somatic mutation is not due to increased mutagenesis, but might be due to accelerated mtDNA turnover. This leads to the clonal expansion of pre-existing age-related somatic mtDNA mutations and a biochemical defect that can affect up to 10% of cells. These observations add weight to the role of somatic mtDNA mutations in the aging process, and raise the specter of progressive iatrogenic mitochondrial genetic disease emerging over the next decade.
Somatic mtDNA mutations accumulate in individual cells during normal human aging, leading to cellular bio-energetic defects of oxidative phosphorylation 2,4. Transgenic mice with a defective mtDNA polymerase (pol γ) accumulate secondary mtDNA mutations and a prematurely aged phenotype 3, but it is still not clear whether the mtDNA mutations are a cause or a consequence of aging in humans. Accelerated senescence has recently been described in humans with successfully treated HIV infection 1. These patients become frail at an early age, decline physiologically 5,6, and acquire age-associated degenerative disorders affecting the cardiovascular system and the brain leading to dementia 7,8. Several NRTIs used in the treatment of HIV inhibit the function of pol γ 9, raising the possibility that drug treatment contributes to the accelerated aging phenotype via mtDNA damage. NRTIs are well known to cause an acute, temporary and reversible reduction in the amount of mtDNA (mtDNA depletion), and one previous study detected mtDNA deletions in patients being actively treated with NRTIs 10-12. However, no previous studies have looked at the possibility of irreversible long-term effects of the drugs on mtDNA mutations after NRTI treatment has ceased.
We studied skeletal muscle from 33 HIV-infected adults, all aged 50 years or under, stratified by lifetime exposure to NRTIs previously shown to affect pol γ in vitro 9 (see Methods and Supplementary Table 1); and 10 HIV-uninfected healthy controls (HIV−) of comparable age. We initially looked for a defect of mitochondrial oxidative phosphorylation within individual cells using cytochrome c oxidase / succinate dehydrogenase (COX-SDH) histochemistry. Cellular COX defects would not be expected in this younger subject group (<0.5%) 4.The frequency of COX-deficient muscle fibers in HIV-infected non-treated (treatment-naïve, HIV+/NRTI−) subjects (n=12) was indistinguishable from that observed in HIV− controls, with the majority having no COX-deficient fibers. By contrast, NRTI-exposed (HIV+/NRTI+) subjects (n=21) had an increased frequency of COX deficient muscle fibers (maximum 9.8%, p=0.047), reaching or exceeding levels expected in healthy elderly individuals 4 (Figure 1). The severity of COX defect was strongly predicted by cumulative lifetime NRTI exposure, rather than therapy at the time of study, implicating a persistent and cumulative mitochondrial defect (r2=87%, p<0.001; Supplementary Figure 1).
Figure 1. COX (cytochrome c oxidase) deficiency in single skeletal muscle fibers.
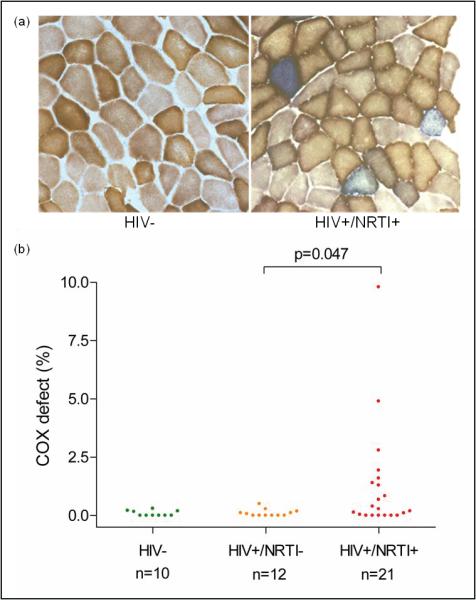
(a) COX histochemistry from a representative healthy control subject (HIV−) showing normal COX activity, whereas a nucleoside analog treated HIV-infected patient (HIV+/NRTI+) shows multiple COX-deficient fibers (counterstained blue by residual SDH (succinate dehydrogenase) activity). (b) COX defects observed in each subject group (HIV+/NRTI−, HIV-infected treatment-naïve subjects; each dot represents an individual patient biopsy; ≥500 fibers sampled per biopsy).
We then defined the molecular basis for the COX deficiency observed in NRTI-exposed subjects. We first excluded persistent mtDNA depletion. The mtDNA content in homogenized skeletal muscle did not differ between HIV+/NRTI+ and HIV+/NRTI− patients (Supplementary Figure 2). In keeping with this, the analysis of individual laser-captured single muscle fibers (n=128) showed that only a small minority of COX deficient fibers (6/70, 9%) from NRTI-treated patients had mtDNA depletion compared to adjacent fibers with normal COX activity. By contrast, the vast majority of the isolated COX deficient fibers contained markedly increased amounts of mtDNA (geometric mean 2.1 fold proliferation, maximum 21.3 fold; p<0.001 for difference in mean mtDNA content between COX deficient and normal fibers) (Figure 2a). Focal mtDNA proliferation is often seen in association with pathogenic mtDNA mutations. In keeping with this, the majority of the COX deficient fibers analyzed (40/70 fibers, from 12 HIV+/NRTI+ patients) showed high percentage levels of mtDNA molecules containing large-scale deletion mutations, exceeding the percentage level of mutation required to cause a COX defect (~60% 13). No deletion mutations were detected in adjacent skeletal muscle fibers (n=58) with normal COX activity. Analysis of the mtDNA deletion break-points (n=15 fibers, from 4 HIV+/NRTI+ patients) revealed different deletions in different fibers, all of which were clonal within individual fibers. Most of the clonally expanded deletions were unique, the only deletion observed more than once was the mt.δ4977 ‘common deletion’, the commonest age-associated somatic mtDNA mutation 14,15 (Figure 2b and 2c; Supplementary Table 2).
Figure 2. Mitochondrial DNA analysis of single skeletal muscle fibers.

(a) Mitochondrial DNA (mtDNA) content in individual COX (cytochrome c oxidase) deficient muscle fibers from nucleoside analogue treated HIV-infected (HIV+/NRTI+) subjects, expressed relative to mtDNA content in adjacent fibers of normal COX activity from the same subject. A few fibers show reduced mtDNA content, whereas the majority show increased content (geometric mean, 2.1 fold proliferation; maximum 21.3 fold; p <0.001 for difference in mean mtDNA content between COX deficient and normal fibers). (b) The majority of COX deficient fibers (COX−) contained high percentage levels of mtDNA containing a large-scale deletion of the major arc, causing the COX defect; whereas no deleted mtDNA was detected in adjacent COX positive fibers (COX+) (P <0.001). (c) Schematic representation of mtDNA large-scale deletion break-points in COX deficient fibers from HIV+/NRTI+ patients relative to the mtDNA gene positions(tRNA and rRNA not shown). Each line represents an individual deleted region. OL, origin of light chain replication; OH, origin of heavy chain replication. (n=15 fibers from 4 patients).
Although less common than large-scale deletion mutations, mtDNA point mutations are also found in COX deficient fibers from healthy aged subjects 16. In keeping with this, in the NRTI-treated patients, COX deficient fibers not containing a deletion were found to harbor non-synonymous somatic mtDNA point mutations (5/29 fibers). These mutations are predicted to alter a highly conserved amino acid, have not previously been described as inherited polymorphic variants in 5140 humans (Table 1) (one variant, 12797T>C had been observed as a somatic variant in a single human sequence) 17, and thus provide an explanation for the associated cellular COX defect. Other fibers contained high levels of non-coding control region (nt.16024 – nt.576) variants, previously described in healthy aged humans.
Table 1.
MtDNA somatic point mutations identified by whole mtDNA genome sequencing of individual COX (cytochrome c oxidase) deficient skeletal muscle fibers from nucleoside analog (NRTI) treated HIV-infected subjects where a large-scale mtDNA deletion was not detected in that fiber (n = 29 COX deficient fibers sequenced from 7 subjects; subjects identified in Supplementary Table 1, online). Likely pathogenicity (that is accounting for the cellular COX defect) was ascribed if mutations resulted in an amino acid change within a coding gene, at a position which demonstrated high inter-species conservation and is not polymorphic within human populations. Population frequencies were taken from Pereira et al (* one variant, 12797T>C had been observed only as a somatic variant in a single human sequence)17. In fibers from two subjects (7 and 20) no point mutations were identified. Syn, synonymous.
| Subject | Somatic Mutation / Variant | Population frequency (in 5140 seq.) |
|
|---|---|---|---|
| Likely Pathogenic Somatic Changes in Coding Region (mt.577-16023) | |||
| 8 | m.7818T>C | L68P (CO2) | 0 |
| 8 | m.9253G>A | D15N (CO3) | 0 |
| 8 | m.12797T>C | L154P (ND5) | 1* |
| 2 | m.6579G>A | G226Ter (CO1) | 0 |
| 11 | m.9907G>A | G234D (CO3) | 0 |
| 15 | m.6580G>A | G226E (CO1) | 0 |
| Non-Pathogenic Somatic Changes in Coding Region (mt.577-16023) | |||
| 8 | m.7906C>T | syn | 1 |
| 8 | m.11467A>G | syn | 608 |
| Somatic Changes in Control Region (mt.16024-576) | |||
| 8 | m.408T>A | non-coding | 10 |
| 8 | m.463CAC>Del | non-coding | 0 |
| 2 | m.408T>A | non-coding | 10 |
| 17 | m.408T>A | non-coding | 10 |
| 17 | m.414T>G | non-coding | 0 |
We then estimated the total burden of mtDNA deletion mutations at the whole tissue level. The proportion of mtDNA molecules containing the mt.δ4977 ‘common deletion’ was significantly higher in NRTI-treated patients compared with untreated patients (HIV+/NRTI+ (mean ±SEM), −3.45 ±0.25 log10(/mtDNA); HIV+/NRTI−, −4.56 ±0.31 log10(/mtDNA); p=0.012) (Figure 3), and were comparable with those previously reported in very elderly healthy subjects 18. Furthermore, the proportion of COX deficient muscle fibers from NRTI-treated subjects which contained the mt.δ4977 was very similar to that reported in healthy aged individuals 15. Pathogenic mutations within single fibers (of which the majority were deletions) were accompanied by proliferation of mtDNA, which occurs in an attempt to maintain adequate levels of wild-type mtDNA, as shown previously 19. As a result mutated mtDNA also proliferates within the fiber. Over time, this will lead to a detectable increase in the level of deletions at the whole tissue level.
Figure 3. Proportional level of mt.δ4977 ‘common deletion’ (CD) in homogenized skeletal muscle from HIV-infected subjects.
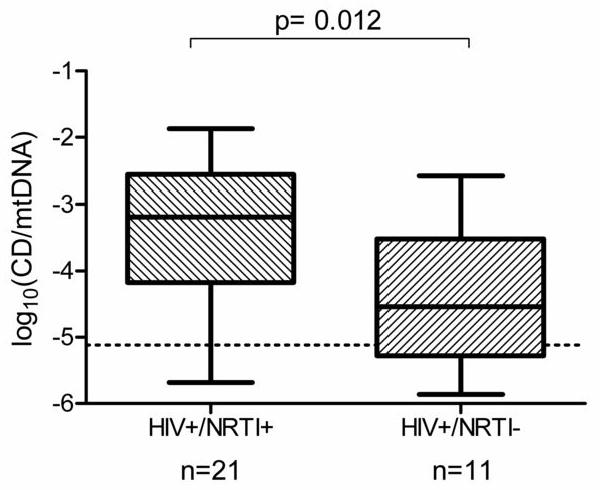
HIV+/NRTI+, HIV-infected, nucleoside analogue exposed; HIV+/NRTI−, HIV-infected, treatment-naïve. Dotted line represents lower threshold of assay. NRTI treated subjects showed significantly higher mean levels of CD than untreated subjects (HIV+/NRTI+ (mean ±SEM), −3.45 ±0.25 log10(/mtDNA); HIV+/NRTI−, −4.56 ±0.31 log10(/mtDNA); p=0.012). Box and whisker plot.
To estimate the relative burden of mtDNA point mutations between treatment groups in homogenized skeletal muscle, we designed an ultra-deep re-sequencing-by-synthesis (UDS) assay, using FLX GS technology (Roche 454). First, we carried out a series of control experiments to demonstrate the sensitivity of UDS to detect mtDNA point variants. We initially established that UDS of an mtDNA template did not generate an intrinsically different signal when compared to a nuclear DNA template by sequencing amplicons of cloned autosomal and mitochondrial DNA fragments as well as an autosomal DNA amplicon from genomic DNA (Supplementary Table 3, on-line). By this approach we confirmed a very low background noise level for the UDS assay (see Methods and Supplementary Figure 3, online). As a positive control, we then compared two mtDNA amplicons from skeletal muscle DNA of POLG patients (n=4), known to harbor high levels of somatic mtDNA point mutations 20,21. One mtDNA amplicon was in the hypervariable non-coding control region (MT-HV2), predicted from 5140 population-level sequences17 to have a high mutation rate; and one was in a highly conserved mtDNA coding region (MT-CO3). Mean coverage was 5892 sequence reads per amplicon in each direction. Consistent with an error-prone pol γ, these subjects showed an increase in mtDNA point variants detectable at >0.2% frequency in the MT-HV2 amplicon (OR 2.33, p=0.002) (Figure 4) when compared to healthy controls (n=4). No increase in variants was detected in the MT-CO3 amplicon. These findings were confirmed on replicate samples (Supplementary Figure 4, online). When we studied skeletal muscle mtDNA from the HIV+/NRTI+ subjects (n=8), the overall burden of point variants within each amplicon was indistinguishable from HIV+/NRTI− subjects (n=4) and healthy HIV− controls (n=4), all of comparable age (OR 1.08, p=0.79 for comparison of HIV+/NRTI+ and HIV− for MT-HV2). Furthermore, there was no correlation between COX defect in HIV+/NRTI+ subjects (range up to 10%) and mutation burden on UDS assay.
Figure 4. Ultra-deep re-sequencing-by-synthesis (UDS) of skeletal muscle mtDNA.

UDS (Roche 454 FLX GS) shows no difference in burden of low-level mtDNA point variants (exceeding 0.2% frequency) between HIV-infected nucleoside analog treated (HIV+/NRTI+, n=8), HIV-infected treatment-naïve (HIV+/NRTI−, n=4), and control (HIV−, n=4) subjects in two amplicons located in mtDNA hypervariable segment 2 (MT-HV2) and mtDNA COX subunit 3 (MT-CO3). In contrast, positive control subjects with inherited POLG defects (POLG, n=4) show increased burden of low-level mutations compared with healthy controls in MT-HV2 (OR 2.33, p =0.002).
Given that NRTI-treated subjects demonstrated high-level COX defects (up to 10% of fibers) which contained clonal mutated mtDNA species, one explanation for our findings is accelerated segregation of pre-existing (age-associated) mtDNA mutations due to NRTI treatment, rather than de novo somatic mutation. In contrast, the POLG subjects demonstrated a significant increase in point mutation burden in the UDS assay (although only in MT-HV2) but a low proportion of COX-deficient fibers. Although the UDS data does not exclude the possibility of a slight increase in mutagenesis in NRTI-exposed subjects, it would not be of the level predicted to be required (>100-fold increase 22) to cause the observed COX defects.
To determine whether accelerated clonal expansion was a plausible explanation for our findings, we used an established computational model, based solely on experimentally-derived parameters 22 and simulated the effects of NRTI-induced chain-termination during mtDNA replication 9. The de novo mutation rate was not altered from the original model of aging muscle. A finite NRTI exposure predicted a period of temporary mtDNA depletion which was concordant with reported mtDNA levels 12,23, and the COX-defects observed 12 in acutely treated HIV patients. This resulted in accelerated clonal expansion of pre-existing mtDNA mutations, and led to an irreversible increase in the frequency of COX-deficient muscle fibers (Figure 5a and 5b). The severity of predicted COX defect was dependent on the degree of replication failure and the duration of exposure (Figure 5b and 5c), in keeping with our observations in patient muscle which had suggested a strong dependence of on these factors (Supplementary Figure 1). In silico modeling is thus consistent with the hypothesis that accelerated clonal expansion of pre-existing (age-associated) mtDNA somatic mutations is sufficient to explain our observations in NRTI-treated subjects. Having established the model, we explored the effect of timing of NRTI exposure and showed that later periods of therapy predicted a higher frequency of COX deficiency (Figure 5d). This is due to older subjects harboring a greater number of age-related somatic mtDNA mutations than younger subjects, which rapidly clonally segregate during NRTI therapy. This is in keeping with the observation that mitochondrially-mediated clinical complications of NRTI therapy appear to be more common in older individuals 24. Finally we modeled the longer-term effects of treatment. Using this approach, an HIV-infected individual treated with NRTIs during their 3rd decade is predicted to develop ~5% COX deficient cells by age 60 (Figure 5b-d). This is similar to or exceeds that seen in the healthy very old 4.
Figure 5. Simulations of the effects of partial mitochondrial DNA (mtDNA) replication failure due to nucleoside analog (NRTI) exposure.
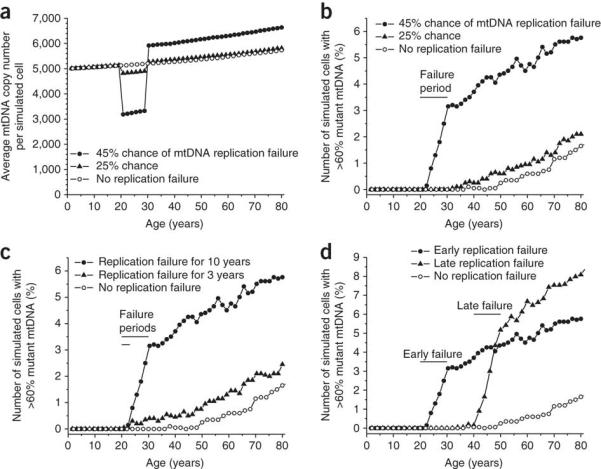
Using a validated computer model of mtDNA replication based solely on experimentally derived parameters 22, we incorporated a finite period of partial replication failure due to the mtDNA chain-terminating effects of NRTI exposure 9, assigning a probability of failure per mtDNA replication event. All other parameters remained constant, including the de novo mutation rate 22. 2000 cells were simulated for 80 years. (a) The amount of mtDNA depletion during the NRTI exposure period caused by 25% and 45% probability of replication failure between 20 and 30 years-of-age. (>50% failure led to the complete loss of mtDNA). The range of mtDNA depletion predicted is in keeping with published in vivo data12,23. (b) This led to a persistent increase in the frequency of COX (cytochrome c oxidase) deficient cells through the accelerated clonal expansion of pre-existing somatic mtDNA mutations. (c) Direct simulation of the effects of NRTI exposure within our study population (two different periods, 10 and 3 years, starting at age 20, of replication failure with 45% probability). The range of COX defects predicted closely fits our empiric data. (d) Late exposure (40 – 50 years) had a more pronounced effect than early exposure (20 – 30 years) (with 45% probability of replication failure) due to the higher number of pre-existing (age-related) somatic mtDNA mutations at the time of exposure.
Although the UDS data for mtDNA point mutations support the hypothesis of accelerated clonal expansion of pre-existing age-related mutations, rather than increased mutagenesis, it is possible that additional mechanisms may be involved for mtDNA large-scale deletions, including a replicative advantage favoring deleted molecules 25. Furthermore, although UDS provides great depth of mutational analysis, it is analogous to the PCR-cloning method of mutation rate determination, and as such will tend to exaggerate an estimate of mutation rate 26,27.
The rapid clonal expansion of somatic mtDNA mutations we observed in NRTI-treated HIV-infected patients provides a plausible mechanism for accelerated aging in treated HIV infection. This is potentially of great significance for the millions of HIV-infected patients in the developing world where these drugs remain the mainstay of therapy 28, and adds weight to a causal role for somatic mtDNA mutations in human aging.
Supplementary Material
Methods
Histochemistry
20μm frozen sections were obtained from fresh-frozen lower limb skeletal muscle biopsies and placed on PEN membrane slides (Leica) for subsequent laser micro-dissection. COX (cytochrome c oxidase) contains subunits encoded by the mitochondrial genome and stains brown (positive) in the presence of preserved respiratory chain activity. SDH (succinate dehydrogenase) provides an effective counter-stain (blue) as this respiratory chain complex is entirely encoded by the nuclear genome and will be preserved in the presence of an mtDNA defect. Thus, COX-deficient fibers are predicted to contain somatic mtDNA mutations. ATPase histochemistry was performed on adjacent frozen sections in order to determine fiber type (oxidative or glycolytic).
Molecular analyses
All primers used are listed (Supplementary Table 3). All nucleotide positions refer to the revised Cambridge Reference Sequence (rCRS, NC_012920).
Individual skeletal muscle fibers were captured by laser microdissection (Leica) and digested in 30 μl of lysis buffer (50 mM Tris-HCl pH 8.5, 0.5% Tween-20, 200 μg ml−1 proteinase K). Real-time PCR was performed as previously described 29. Briefly, mtDNA content was determined using a target template in MT-ND1. When comparing COX deficient and normal fibers these were matched for fiber type and adjusted for fiber size. We estimated the proportion of mtDNA molecules containing large-scale deletions using a target template in MT-ND4. For determination of relative mtDNA content at the whole tissue level we performed real-time PCR as above with the inclusion of the nuclear template, B2M. Proportion of mtDNA molecules in muscle homogenate containing the mt.δ4977 ‘common deletion’ were estimated by real-time PCR comparing MT-ND1 and a product (CD) specifically amplified only in the presence of the common deletion. CD/ND1 real-time PCR was performed in a 20 μl reaction comprising 1x Evagreen supermix (BioRad), 0.625 μM primers and 50 ng DNA. PCR Protocol comprised 98°C for 2 mins, followed by 40 cycles of 98°C for 5 seconds and 60°C for 20 seconds. In addition to a PCR negative, DNA extracted from whole blood of a 25 year old healthy control subject was used to define the lower limit of sensitivity for this assay, as negligible mt.δ4977 is expected to be detectable in blood by these methods 18,30.
Long-range PCR to detect mtDNA deletions in individual fibers was performed using nested PCR as previously described 31. Deletion break-points were then characterized by amplification of a ~500 bp fragment across the deletion break point. Break-point PCR reactions were performed in a 25 μl reaction containing 1x ImmoBuffer (Bioline), 2 mM MgCl2, 0.2 mM dNTPs, 1 U Immolase (Bioline) and 1 μl of long-range PCR product, diluted 1:50 with PCR-grade water. PCR conditions were 95°C for 10 min, and 25 cycles of 95°C for 15 sec, 58°C for 15 sec and 72°C for 30 sec. Cycle-sequencing was performed using BigDye Terminator v3.1 kit (Applied Biosystems) and visualized through a 3130x Genetic Analyzer (Applied Biosystems).
Whole genome sequencing from individual fibers was performed based on our previous methods 32. A nested PCR comprising a primary PCR with nine overlapping primer pairs was followed by 36 overlapping secondary PCR primer pairs. Primary PCR was performed in a 50 μl volume containing 1x PCR buffer (10 mM Tris-HCl pH 8.3, 1.5 mM MgCl2, 50 mM KCl, 0.001% w/v gelatin), 1 mM MgCl2, 0.2 mM dNTPs, 0.6 μM primers, 1.75 U AmpliTaq Gold (Applied Biosystems) and 1 μl lysate. PCR conditions were 94°C for 10 min, and 38 cycles of 94°C for 45 sec, 58°C for 45 sec and 72°C for 2 min. Final extension was 8 min. Secondary PCR was performed in a 25 μl volume containing 1x PCR buffer (as above), 0.2 mM dNTPs, 0.8 μM primers, 0.65 U AmpliTaq Gold and 1 μl of primary PCR product. PCR conditions were as above except for 1 min extension and 30 cycles. Cycle sequencing was performed as above.
Ultra-deep re-sequencing-by-synthesis (UDS; Roche 454 GS FLX) was performed by PCR amplification of two mtDNA amplicons: one in the non-coding (control region), hypervariable segment 2 (MT-HV2) (amplicon position, nt.162 – 455, 294bp), and one in the coding region, COX subunit 3 (MT-CO3) (amplicon position, nt.9307 – 9591, 285bp). Primer specificity and lack of amplification of nuclear pseudogenes was predicted by BLAST 33, and confirmed by failure of amplification of any product from rho0 cellular DNA. In addition we generated a nuclear DNA amplicon (BRCA2, NC_000013.10, 32907099-32907295). Amplicon generation was performed in a 50 μl volume containing 1x buffer for KOD Hot Start DNA Polymerase (Novagen), 1.5 mM MgSO4, 0.2 mM dNTPs, 0.3 μM primers, 1 U KOD Hot Start DNA Polymerase (Novagen) and 100 ng DNA. Cycling conditions were 95 °C for 2 minutes followed by 30 cycles of 95 °C for 20 seconds, 60 °C for 10 seconds and 70 °C for 4 seconds. EmPCR and sequencing were performed according to manufacturer’s instructions (Roche 454). Confirmatory experiments were performed by amplicon sequencing a larger amplicon in the same regions using Roche 454 GS FLX Titanium system. Amplicon positions for Titanium assay were: MT-HV2, nt.109-483; MT-CO3, nt.9304-9653; BRCA2, 32907060-32907350. Amplicon generation PCR was as for initial FLX assay with the exception of 5 seconds extension per cycle. Repeat assays were performed for HIV−, HIV+/NRTI+ and POLG subjects (n=4 each). Amplicons were additionally generated from cloned DNA fragments (MT-HV2-clone, nt.16548-771; MT-CO3-clone, nt.9127-9661; BRCA2-clone, 32906828-32907480; cloned in pGEM-T-Easy vector, Promega). An analysis pipeline of PyroBayes and Mosaik 34 was used to call and align bases from the 454 flowgram output. Subsequent analysis of variants was done within R using the custom made R library flowgram (available from the authors, IW and MSK). For comparison of samples with varying coverage depths, 5000 sub-sampled sequences were used for all samples in all analyses. Recent studies of low-level variance in mtDNA using next-generation sequencing-by-synthesis technology have employed the Illumina GA platforms 35. Experience to date suggests that this approach appears limited to a resolution of ~1-1.5% variant frequency or higher, below which true variance cannot be distinguished from noise, despite very high theoretical read depths. In order to improve on this depth of resolution, we filtered the raw FLX flowgram output for sites predicted to give poor resolution. As FLX resequencing employs pyrosequencing technology, it is prone to sequencing errors associated with mononucleotide tracts. Analysis of our outputs from cloned DNA confirmed this observation and such sites were excluded from further analysis. Such an approach enabled resolution to variants with measured frequency of ≥0.2%, whereby there was negligible variance detected in any cloned DNA amplicon or the nuclear (BRCA2) amplicon from genomic DNA at this level (~0.5% of base positions). Comparison with mtDNA amplicon sequence variants from patient samples, thus indicated that almost all low frequency variants (>0.2%) reflect true sequence variation rather than noise. Power calculations indicate that this assay will have 80% power to detect an absolute increase in mutation burden of 2.7% at P<0.05.
Modeling of NRTI effects on mtDNA replication
Modeling was performed by development of a validated simulation model of mtDNA replication and age-associated clonal expansion of mtDNA mutations, based solely on experimentally derived parameters 22. The effect of NRTIs on mtDNA replication was modeled by including a probability of failure for each replication event. In the case of a replication failure the mtDNA molecule being copied was assumed to be destroyed. With this assumption, any failure rate of 50% or greater results in the complete loss of the mtDNA from the simulation. De novo mutations were modeled by including a probability of mutation formation at each replication event, with a probability of 5 × 10−5 per replication, which was kept constant across all simulated exposure groups. The de novo mutation rate was set at this value in order to keep the probability of forming clonal expansions below 1% before age 70 in the control case. Other relevant parameter values were the optimal mtDNA copy number (Nopt = 5000), the mtDNA half life (10 days), and the maximum proliferation factor (alpha = 15). 2000 cells were simulated to measure the probability of developing clonal expansions of mtDNA mutations. Simulated cells which fixed on the mutant (a very rare occurrence) were removed from the model. The simulation was written in FORTRAN and is available from the author (DCS).
Statistical analyses
Percentage levels of COX defect were compared between groups (≥500 fibers per subject) by Mann Whitney test. Comparison of proportions of COX-deficient and normal fibers showing mtDNA deletions was made by Chi-squared test. Comparison of mean log10(mtDNA) content and log10(CD/mtDNA) levels in skeletal muscle homogenates was made using t-test. Statistical comparisons were performed using R. The multiple regression models were run in Origin 7 (OriginLab).
ACKNOWLEDGEMENTS
This work was support by grants from the Medical Research Council (B.I.A.P.), British Infection Society (B.I.A.P.), the Newcastle Healthcare Charity (D.A.P.), the UK NIHR Biomedical Research Centre for Aging and Age-related disease award to the Newcastle upon Tyne Foundation Hospitals NHS Trust (P.F.C), and the Wellcome Trust (P.F.C.). The authors thank G.L. Toms for the use of his Containment Level 3 facility, and E.L.C. Ong, M.L. Schmid, U. Schwab and R. Pattman for access to their clinic cohorts; J. Coxhead (NewGene, Newcastle-upon-Tyne, UK) for assistance with Roche 454 FLX and D. Deehan for assistance with obtaining control muscle samples.
Footnotes
AUTHOR CONTIBUTIONS P.F.C. designed the study and wrote the paper. B.A.I.P. designed the study, carried out the molecular analyses and wrote the paper. C.A.H. designed and carried out the mt.δ4977 assay. R.H. and D.A.P. designed the study. I.J.W. and M.S-K. designed and performed the UDS analysis. D.C.S. designed, programmed and carried out the in silico modeling.
COMPETING INTERESTS STATEMENT The authors declare that they have no competing financial interests.
ETHICS This study was approved by the Newcastle and North Tyneside Local Research Ethics Committee. Informed consent was obtained from all subjects.
References
- 1.Effros RB, et al. Aging and Infectious Diseases: Workshop on HIV Infection and Aging: What Is Known and Future Research Directions. Clinical Infectious Diseases. 2008;47:542–553. doi: 10.1086/590150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bua E, et al. Mitochondrial DNA-deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am J Hum Genet. 2006;79:469–80. doi: 10.1086/507132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Trifunovic A, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–23. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 4.Brierley EJ, Johnson MA, James OF, Turnbull DM. Effects of physical activity and age on mitochondrial function. Qjm. 1996;89:251–8. doi: 10.1093/qjmed/89.4.251. [DOI] [PubMed] [Google Scholar]
- 5.Desquilbet L, et al. HIV-1 infection is associated with an earlier occurrence of a phenotype related to frailty. J Gerontol A Biol Sci Med Sci. 2007;62:1279–86. doi: 10.1093/gerona/62.11.1279. [DOI] [PubMed] [Google Scholar]
- 6.Oursler KK, Sorkin JD, Smith BA, Katzel LI. Reduced aerobic capacity and physical functioning in older HIV-infected men. AIDS Res Hum Retroviruses. 2006;22:1113–21. doi: 10.1089/aid.2006.22.1113. [DOI] [PubMed] [Google Scholar]
- 7.Guaraldi G, et al. Coronary Aging in HIV-Infected Patients. Clin Infect Dis. 2009 doi: 10.1086/648080. [DOI] [PubMed] [Google Scholar]
- 8.Valcour V, et al. Higher frequency of dementia in older HIV-1 individuals: the Hawaii Aging with HIV-1 Cohort. Neurology. 2004;63:822–7. doi: 10.1212/01.wnl.0000134665.58343.8d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lim SE, Copeland WC. Differential incorporation and removal of antiviral deoxynucleotides by human DNA polymerase gamma. J Biol Chem. 2001;276:23616–23. doi: 10.1074/jbc.M101114200. [DOI] [PubMed] [Google Scholar]
- 10.McComsey GA, et al. Improvements in lipoatrophy, mitochondrial DNA levels and fat apoptosis after replacing stavudine with abacavir or zidovudine. Aids. 2005;19:15–23. doi: 10.1097/00002030-200501030-00002. [DOI] [PubMed] [Google Scholar]
- 11.Cote HC, et al. Changes in mitochondrial DNA as a marker of nucleoside toxicity in HIV-infected patients. N Engl J Med. 2002;346:811–20. doi: 10.1056/NEJMoa012035. [DOI] [PubMed] [Google Scholar]
- 12.Maagaard A, et al. Mitochondrial (mt)DNA changes in tissue may not be reflected by depletion of mtDNA in peripheral blood mononuclear cells in HIV-infected patients. Antivir Ther. 2006;11:601–8. [PubMed] [Google Scholar]
- 13.Hayashi J, et al. Introduction of disease-related mitochondrial DNA deletions into HeLa cells lacking mitochondrial DNA results in mitochondrial dysfunction. Proc Natl Acad Sci U S A. 1991;88:10614–8. doi: 10.1073/pnas.88.23.10614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Corral-Debrinski M, et al. Mitochondrial DNA deletions in human brain: regional variability and increase with advanced age. Nat Genet. 1992;2:324–9. doi: 10.1038/ng1292-324. [DOI] [PubMed] [Google Scholar]
- 15.Brierley EJ, Johnson MA, Lightowlers RN, James OF, Turnbull DM. Role of mitochondrial DNA mutations in human aging: implications for the central nervous system and muscle. Ann Neurol. 1998;43:217–23. doi: 10.1002/ana.410430212. [DOI] [PubMed] [Google Scholar]
- 16.Fayet G, et al. Ageing muscle: clonal expansions of mitochondrial DNA point mutations and deletions cause focal impairment of mitochondrial function. Neuromuscul Disord. 2002;12:484–93. doi: 10.1016/s0960-8966(01)00332-7. [DOI] [PubMed] [Google Scholar]
- 17.Pereira L, et al. The diversity present in 5140 human mitochondrial genomes. Am J Hum Genet. 2009;84:628–40. doi: 10.1016/j.ajhg.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee HC, Pang CY, Hsu HS, Wei YH. Differential accumulations of 4,977 bp deletion in mitochondrial DNA of various tissues in human ageing. Biochim Biophys Acta. 1994;1226:37–43. doi: 10.1016/0925-4439(94)90056-6. [DOI] [PubMed] [Google Scholar]
- 19.Chinnery PF, Samuels DC. Relaxed replication of mtDNA: A model with implications for the expression of disease. Am J Hum Genet. 1999;64:1158–65. doi: 10.1086/302311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wanrooij S, et al. Twinkle and POLG defects enhance age-dependent accumulation of mutations in the control region of mtDNA. Nucleic Acids Res. 2004;32:3053–64. doi: 10.1093/nar/gkh634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Del Bo R, et al. Remarkable infidelity of polymerase gammaA associated with mutations in POLG1 exonuclease domain. Neurology. 2003;61:903–8. doi: 10.1212/01.wnl.0000092303.13864.be. [DOI] [PubMed] [Google Scholar]
- 22.Elson JL, Samuels DC, Turnbull DM, Chinnery PF. Random intracellular drift explains the clonal expansion of mitochondrial DNA mutations with age. Am J Hum Genet. 2001;68:802–6. doi: 10.1086/318801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cherry CL, et al. Tissue-specific associations between mitochondrial DNA levels and current treatment status in HIV-infected individuals. J Acquir Immune Defic Syndr. 2006;42:435–40. doi: 10.1097/01.qai.0000224974.67962.ce. [DOI] [PubMed] [Google Scholar]
- 24.Smyth K, et al. Prevalence of and risk factors for HIV-associated neuropathy in Melbourne, Australia 1993-2006. HIV Med. 2007;8:367–73. doi: 10.1111/j.1468-1293.2007.00478.x. [DOI] [PubMed] [Google Scholar]
- 25.Diaz F, et al. Human mitochondrial DNA with large deletions repopulates organelles faster than full-length genomes under relaxed copy number control. Nucleic Acids Res. 2002;30:4626–33. doi: 10.1093/nar/gkf602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Greaves LC, et al. Quantification of mitochondrial DNA mutation load. Aging Cell. 2009;8:566–72. doi: 10.1111/j.1474-9726.2009.00505.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kollberg G, et al. Low frequency of mtDNA point mutations in patients with PEO associated with POLG1 mutations. Eur J Hum Genet. 2005;13:463–9. doi: 10.1038/sj.ejhg.5201341. [DOI] [PubMed] [Google Scholar]
- 28.WHO Antiretroviral therapy for HIV infection in adults and adolescents: A public health approach. 2006. [PubMed]
- 29.Durham SE, Samuels DC, Cree LM, Chinnery PF. Normal levels of wild-type mitochondrial DNA maintain cytochrome c oxidase activity for two pathogenic mitochondrial DNA mutations but not for m.3243A-->G. Am J Hum Genet. 2007;81:189–95. doi: 10.1086/518901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shieh DB, et al. Mitochondrial DNA alterations in blood of the humans exposed to N,N-dimethylformamide. Chem Biol Interact. 2007;165:211–9. doi: 10.1016/j.cbi.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 31.Bender A, et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet. 2006;38:515–7. doi: 10.1038/ng1769. [DOI] [PubMed] [Google Scholar]
- 32.Durham SE, Samuels DC, Chinnery PF. Is selection required for the accumulation of somatic mitochondrial DNA mutations in post-mitotic cells? Neuromuscul Disord. 2006;16:381–6. doi: 10.1016/j.nmd.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 33.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 34.Quinlan AR, Stewart DA, Stromberg MP, Marth GT. Pyrobayes: an improved base caller for SNP discovery in pyrosequences. Nat Methods. 2008;5:179–81. doi: 10.1038/nmeth.1172. [DOI] [PubMed] [Google Scholar]
- 35.He Y, et al. Heteroplasmic mitochondrial DNA mutations in normal and tumour cells. Nature. 2010;464:610–4. doi: 10.1038/nature08802. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.