

Abstract
Hypovolemic shock (Dengue shock syndrome (DSS)), is the commonest life-threatening complication of dengue. We conducted a genome-wide association study of 2,008 pediatric cases treated for DSS and 2,018 controls from Vietnam. Replication of the most significantly associated markers was carried out in an independent Vietnamese follow-up sample of 1,737 cases and 2,934 controls. Polymorphisms within two genes showed genome-wide significant association with DSS (Pmeta = 4.41 × 10−11, per-allele odds ratio (OR) = 1.34 for MICB rs3132468 located within the broad MHC region and Pmeta = 3.08 × 10−10, per-allele OR = 0.80 for PLCE1 rs3765524). Our data implicates MICB is an important determinant in early immune control of dengue virus infection and PLCE1 a factor in vascular endothelial dysfunction and circulatory hypovolemia.
Introduction
Dengue is an acute systemic viral infection caused by one of four serotypes of dengue (DEN) virus and is globally the commonest mosquito-borne infection after malaria 1. The burden of dengue is growing, with an estimated 100 million infections now occurring annually and with 2.5 billion people living in areas at risk of transmission. A wide spectrum of disease manifestations is seen, ranging from subclinical infection to severe and fatal disease. Severe dengue in children is characterised by an increase in vascular permeability that leads to life-threatening hypovolemic shock (dengue shock syndrome-DSS). This is often accompanied by thrombocytopenia and haemostatic dysfunction, which may result in severe bleeding. Children are at greatest risk of developing DSS but with careful supportive care the case fatality rate is less than 1% 2. In southern Vietnam, serological studies have estimated the population based exposure to dengue virus infection to reach 85% by the end of childhood (15 years old) 3, while the incidence of DSS is estimated to occur at less than 1% of exposed individuals 2 (see “The use of population controls” in the Methods section). A host genetic basis to susceptibility to severe dengue has been alluded to in epidemiological studies, and various candidate gene studies of modest sample sizes have been performed 4-8.
To estimate the genetic contribution underlying severe dengue, we genotyped 2,118 DNA samples from Vietnamese children with established or incipient DSS and 2,089 cord blood controls in a genome-wide association study (GWAS). After exclusion of samples for discrepancies between clinical and genetically inferred gender, relatedness or for per-sample call rates of less than 95 percent (Supplementary Figure 1), there were 2,008 DSS cases and 2,018 controls available for analysis. The clinical and virological characteristics of the case population are described in Supplementary Table 1. A total of 657,366 SNPs were initially included within the Illumina 660W Beadchip used for genome-wide genotyping. After various stringent QC exclusions (Supplementary Figure 2), a total of 481,342 SNPs were retained for downstream association analysis.
Upon conducting the routine GWAS statistical tests (see Statistical findings in the Methods section), detailed examination of the overall scan results revealed strong evidence of disease association at two distinct loci; (Figure 1) MICB on Chromosome 6 and PLCE1 on Chromosome 10, both represented by SNPs which were close to the formal threshold for genome-wide significance (P = 5.38 × 10−8 for MICB rs3132468 and P = 5.84 × 10−8 for PLCE1 rs3740360) (Table 1). Together with the SNPs at MICB and PLCE1, a total of 85 SNPs exceeded P < 10−4 on single SNP analysis (Supplementary Table 2). We were able to design assays for 72 out of these 85 SNPs using the Sequenom Mass-Array platform. The remaining 13 SNPs in the broad MHC region were refractory to assay design, thus necessitating ABI Taqman assays to be designed for the sentinel SNP at MICB (rs3132468) and rs3134899 (also within MICB; GWAS P = 1.03 × 10−4, OR = 1.31). We then genotyped these 74 SNPs (72 non-MHC SNPs and two SNPs within MICB) in a replication sample of 1,824 DSS cases and 3,019 controls. We applied the same GWAS QC filters for the replication set: five SNPs had poor genotyping clusters and were excluded from analysis (Supplementary Table 2), and 132 samples (87 cases and 85 controls) had per-sample call-rates of less than 95 percent; these were excluded from further analysis. This left 69 SNPs to be analyzed in 1,737 cases and 2,934 controls for the replication stage. In keeping with the GWAS observations, the strongest evidence of association was observed with SNPs at MICB (rs3132468, Prepl = 9.32 × 10−5 and rs3134899, Prepl = 0.0082) and PLCE1 (3 SNPs with Prepl ranging from 5.23 × 10−4 to 1.6 × 10−4, Table 1). Using inverse-variance weights, data from both the GWAS and replication cohorts (N = 3,745 DSS cases and N = 4,952 controls) were combined in formal meta-analysis, and this revealed strong evidence of association with rs3132468 at MICB (P = 4.41 × 10−11; per-allele odds ratio (OR) = 1.34, [1.23 - 1.46]) and 7 SNPs at PLCE1 (4.18 × 10−9 ≤ P ≤ 3.08 × 10−10; 0.75 ≤ OR ≤ 0.87, Table 1). To aid in refining the original signal of association, we performed imputation analysis at regions flanking both loci (Chr. 6: 30 - 32 Mb, and Chr. 10: 95.5 - 96.5 Mb). This did not reveal signals of association over and above that of the directly genotyped SNPs. The associations observed at MICB and PLCE1 were not specific to any Dengue virus serotype on subgroup analysis of viral serotype, nor were they associated with the degree of thrombocytopenia or the degree of clinical shock (data not shown).
Figure 1.
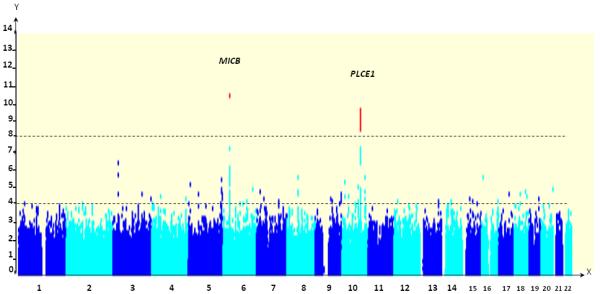
Manhattan plot showing directly genotyped SNPs plotted according to chromosomal location (X-axis, with −Log10P-values (Y-axis) derived from the trend test. The lower horizontal dotted line indicates the threshold for bringing SNPs forward to the replication stage (P < 10−4). SNPs surpassing P < 10−8 (upper horizontal dotted line) on combined analysis of both GWAS and replication data are reflected by red dots, and gene names are given for these loci. SNPs in MICB and PLCE1 have significant associations.
Table 1.
Association analysis between Dengue shock syndrome and SNP genotypes at MICB and PLCE1.
| Gene/Marker (Alleles) |
Chromosome (Position) |
Stage | MAF Cases |
MAF Controls |
OR | P | ORmeta (95% CI) |
P meta |
|---|---|---|---|---|---|---|---|---|
| MICB/rs3132468 | 6 | GWAS | 0.176 | 0.132 | 1.41 | 5.39 × 10−8 | ||
| (C/T) | 31583465 | Replication | 0.163 | 0.134 | 1.27 | 9.32 × 10−5 | 1.34 (1.23 - 1.46) |
4.41 × 10−11 |
| MICB/rs3134899 | 6 | GWAS | 0.130 | 0.102 | 1.31 | 1.09 × 10−4 | ||
| (G/A) | 31581265 | Replication | 0.114 | 0.096 | 1.20 | 0.0082 | 1.26 (1.14 - 1.38) |
4.08 × 10−6 |
|
PLCE1/rs37655 24 |
10 | GWAS | 0.249 | 0.300 | 0.77 | 2.68 × 10−7 | ||
| (T/C) | 96048288 | Replication | 0.265 | 0.302 | 0.83 | 1.60 × 10−4 | 0.80 (0.75 - 0.86) |
3.08 × 10−10 |
|
PLCE1/rs22742 23 |
10 | GWAS | 0.250 | 0.303 | 0.77 | 1.19 × 10−7 | ||
| (G/A) | 96056331 | Replication | 0.267 | 0.300 | 0.85 | 5.23 × 10−4 | 0.81 (0.75 - 0.86) |
6.89 × 10−10 |
|
PLCE1/rs37403 60 |
10 | GWAS | 0.219 | 0.271 | 0.75 | 5.84 × 10−8 | ||
| (C/A) | 96015481 | Replication | 0.242 | 0.273 | 0.85 | 0.0012 | 0.80 (0.75 - 0.86) |
1.15 × 10−9 |
|
PLCE1/rs12263 737 |
10 | GWAS | 0.250 | 0.301 | 0.77 | 3.73 × 10−7 | ||
| (A/G) | 96034903 | Replication | 0.266 | 0.300 | 0.84 | 3.95 × 10−4 | 0.81 (0.75 - 0.87) |
1.22 × 10−9 |
|
PLCE1/rs11187 842 |
10 | GWAS | 0.219 | 0.269 | 0.76 | 1.19 × 10−7 | ||
| (T/C) | 96042501 | Replication | 0.240 | 0.271 | 0.85 | 0.0011 | 0.80 (0.75 - 0.86) |
1.78 × 10−9 |
|
PLCE1/rs75372 4 |
10 | GWAS | 0.219 | 0.269 | 0.76 | 1.28 × 10−7 | ||
| (T/G) | 96041407 | Replication | 0.242 | 0.272 | 0.85 | 0.0012 | 0.81 (0.75 - 0.86) |
2.27 × 10−9 |
|
PLCE1/rs37812 64 |
10 | GWAS | 0.229 | 0.278 | 0.77 | 3.43 × 10−7 | ||
| (G/A) | 96060365 | Replication | 0.250 | 0.280 | 0.85 | 0.0011 | 0.81 (0.76 - 0.87) |
4.18 × 10−9 |
MAF cases: Minor allele frequency in DSS cases
MAF controls: Minor allele frequency in the controls
OR: Odds of DSS per-copy of the minor allele
P: P-value using the 1 degree of freedom score-test.
ORmeta : Odds ratio for the combined GWAS and replication cohorts.
Pmeta: P-value for the combined GWAS and replication cohorts.
95% CI: 95% confidence interval for the OR.
Phet: Heterogeneity P-value.
GWAS: Sample size of 2,008 DSS cases and 2,018 cord blood controls.
Replication: Sample size of 1,737 DSS cases and 2,934 cord blood controls.
Found within the broad Major Histo-Compatability (MHC) locus, MICB lies just outside both the type I and type II HLA regions, ~140,000 base-pairs centromeric to the nearest Class I gene (HLA-B) and more than 1 million base-pairs away from the nearest Class II gene (HLA-DR). Apart from the peak signal at rs3132468 which was observed directly within MICB, twelve other SNPs in this region also showed association signals exceeding P < 10−4 on single-SNP analysis. We thus performed conditional analysis to assess the independence of the association observed at MICB rs3132468 from that of the nearby genes. Although the most significant SNP from the GWAS (rs3132468) could account for the majority of the association signal across the locus, we observed residual signals of association (0.0003 < P < 0.05) with SNPs near the vicinity of HLA-B and HLA-C as well as other neighboring genes (Supplementary Figure 3). These residual associations indicate that definitive identification of MICB as a gene associated with DSS could be complicated by its location within the broad MHC region, which is known for its extensive linkage disequilibrium (LD) spanning multiple genes (Supplementary Figure 4). This precludes definitive identification of the causative gene without extensive further fine-mapping and re-sequencing. With regards to PLCE1 on Chromosome 10, association analysis conditioning for the lead SNP (rs3743060, directly genotyped) did not reveal any secondary signals of association (Supplementary Figure 5), which suggests that the lead SNP -or any of its close correlates in complete LD with it and confined within their distinct genomic region (Supplementary Figure 6)- best explains the association signal at the locus. We did not observe any evidence of epistasis between SNPs at MICB and PLCE1 (P = 0.11).
MICB appears to be a promising candidate based on the present strength of the statistical associations observed in the Chromosome 6 hit region. MICB encodes for MHC class I polypeptide-related sequence B, an inducible activating ligand for the NKG2D type II receptor on natural killer (NK) and CD8+ T cells 9-10 Ligation of NKG2D by MICB stimulates anti-viral effector functions in NK cells including cytokine expression and the cytolytic response 11. We have previously reported that MICB, together with other genes associated with NK cell activation, are highly expressed in the leukocytes of acute dengue patients 12. We therefore propose the association between the MICB rs3132468 genotype and susceptibility to severe dengue might reflect altered or dysfunctional NK and/or CD8+ T cell activation early in infection that results in a higher viral burden in vivo, a recognized factor in clinical outcome 13-14. The recent finding that a SNP near the closely related MICA gene (rs2596542) is associated with Hepatitis C virus induced hepatocellular carcinoma is suggestive of a pivotal role for MIC proteins in the pathogenesis of these Flaviviridae infections 15.
Mutations within PLCE1 are associated with nephrotic syndrome 16. Nephrotic syndrome is a kidney disorder in which dysfunction of the glomeruli basement membrane results in proteinuria and hypoproteinemia that when severe leads to reduced vascular oncotic pressure and edema. These elements of nephrotic syndrome have striking similarities with severe dengue and suggest an important role for PLCE1 in maintaining normal vascular endothelial cell barrier function. In summary, our study identifies genetic variants in MICB and PLCE1 as being associated with severe dengue.
Supplementary Material
Acknowledgements
This work was supported by the Wellcome Trust, United Kingdom (grant 088791/A/09/Z and 084368/Z/07/Z) and the Agency for Science, Technology, and Research, Singapore.
Appendix
Methods
Patient enrolment and diagnosis
Blood samples for genotyping were collected from patients enrolled into one of two research studies of children with dengue. In both studies, children were eligible if they were ≤15 years of age and had clinical signs, symptoms and hematological findings that led to a clinical diagnosis of incipient or established DSS as defined by WHO criteria (WHO, see URLs). All patients were resuscitated with bolus intravenous fluid therapy (≥15ml/kg in the first hour). Summary laboratory and clinical findings were recorded into case record forms during the inpatient period until the patient was either discharged from hospital, died, or was transferred to another hospital. Blood samples for research and diagnostic tests were collected at the time of enrolment and again prior to patient discharge from Hospital. The first study enrolled patients in the pediatric intensive care unit of the Hospital for Tropical Diseases between 2001-2009. The second study enrolled patients in high dependency rooms or the intensive care departments of Children’s Hospital No. 1, Children’s Hospital No. 2, Tien Giang Provincial Hospital, Dong Thap Provincial Hospital and Sa Dec Hospital between 2008-2010. The parent or guardian of each participant gave written informed consent to participate. The Scientific and Ethical Committees of each study site approved the study protocols, as did the Oxford University Tropical Research Ethical Committee.
The GWAS was performed on DNA samples (n=1039) from patients enrolled between 2001-2009 at the Hospital for Tropical Diseases and from patients (n=969) enrolled at the other five participating hospitals during 2008 only. The replication study was performed in patients (n=1737) enrolled between 2009-2010 at Children’s Hospital No. 1, Children’s Hospital No. 2, Tien Giang Provincial Hospital, Dong Thap Provincial Hospital and Sa Dec Hospital. All patients represented in the GWAS and replication phases had laboratory evidence of dengue as shown by RT-PCR detection of viral RNA in plasma collected at the time of enrolment and/or by serological detection of DEN-virus reactive IgM or IgG in single or paired plasma specimens.
Cord blood DNA samples
Blood from the cord of newborn infants was collected in one of two prospective studies. The first study was conducted at Hung Vuong Hospital, Ho Chi Minh City, between 2004-2006. The second study was conducted at Hung Vuong Hospital, Ho Chi Minh City and Dong Thap Hospital, Dong Thap Province, between 2009-2010. All participants gave written informed consent to participate. The Scientific and Ethical Committees of each study site approved the study protocols, as did the Oxford University Tropical Research Ethical Committee. DNA was extracted from cord blood using Nucleon BACC Genomic DNA Extraction Kits (GE Healthcare, USA).
The use of population controls
The number of potentially misclassified cord blood controls in the GWAS and replication stages was estimated to be 11 (out of 2,018) in the GWAS stage, and 15 (out of 2,394) in the replication stage, based on the following three assumptions that- a) all individuals in a given birth cohort will experience two sequential infections by different serotypes during their lifetime,17 b) that only up to 25% of these infections are clinically apparent,18-23 c) 2% of clinically apparent secondary infections develop DSS. These assumptions estimate a life-time population risk of DSS to be 0.5%. This is consistent with estimates of the prevalence of DSS cases expected over the first 15 years in a given birth cohort under the assumption that the incidence of DSS is constant (DSS incidence in Southern Viet Nam in 2009 was 26.59/100,000; based on statistics obtained from the Dengue Control program, Ministry of Health Viet Nam, 2010). Under this assumption we would expect 0.4% of a birth cohort to experience DSS before the age of 15yrs.
Genotyping
Cases and controls were randomized on plates and were genotyped with the Illumina Human 660W Quad BeadChips following manufacturer instructions. This successful use of this chip has been previously documented 24. For the replication stage, 72 of the selected SNPs which were not on the broad MHC region were genotyped with the Sequenom MassArray (Sequenom, see URLs) primer extension iPLEX system. MICB rs3132468 and rs3134899 were genotyped using the ABI Taqman platform (Applied Biosystems, see URLs).
Statistical analysis
Stringent QC filters were applied to remove poorly performing SNPs and samples using tools implemented in PLINK version 1.7 25. The QC criteria were as follows: SNPs that had > 5% of missing genotypes, gross departure from Hardy-Weinberg equilibrium (test for HWE showing P < 10−7) or were of minor allele frequency below 1% were excluded from downstream analysis. For sample QC, samples with an overall genotyping call rate of < 95% were excluded from analysis. The remaining samples were then subjected to biological relationship verification by using the principle of variability in allele sharing according to the degree of relationship. Identity-by-state (IBS) information was derived using PLINK.25 For those pairs of individuals who showed evidence of cryptic relatedness (possibly either due to duplicated or biologically related samples), we removed the sample with the lower call rate before performing principal component (PC) analysis. PC analysis was undertaken to account for spurious associations resulting from ancestral differences of individual SNPs 26, and PC plots were performed using the R statistical program package (R-project, see URLs).
For both the GWAS and replication stages, analysis of association with DSS was carried out using a 1 degree of freedom (d.f.) score-based test. This test models for a trend-per-copy of the minor allele on disease risk and has been extensively described 27-28. It has the best statistical power to detect association for complex traits across a wide range of alternative hypotheses, with the exception of those involving rare recessive variants 29. The threshold for significant independent replication was set at P < 0.05 in the combined replication data sets 30.
Meta-analysis was conducted using inverse variance weights for each cohort, which calculates an overall Z-statistic, corresponding P-value and accompanying odds ratios for each SNP analyzed 30-31. Genotyping clusters were directly visualized for the 85 SNPs exceeding P < 10−4 and confirmed to be of good quality before inclusion for statistical analysis. Supplementary Figure 7 shows the cluster plots for MICB rs3132468 and PLCE1 rs3740360, the two SNPs with showing P < 10−7 in the GWAS stage. Analysis of linkage disequilibrium was performed using Haploview 32.
Statistical findings after routine GWAS analysis
Analysis of genetic ancestry using principal components (PC) revealed no significant population substructure between the DSS cases and controls (Supplementary Figure 8). As the individual PCs were non-significant (Bonferroni corrected P > 0.05, Supplementary Table 3) when tested as continuous covariates using logistic regression, we did not adjust for them in subsequent association analysis.
Single SNP analysis was performed using logistic regression assuming additive genetic effects relating genotype dosage (scoring for 0, 1, or 2 copies of the minor allele) to DSS. A quantile-quantile plot of the single SNP analysis showed a clear excess of extreme P-values compared to the null distribution (Supplementary Figure 9). As this excess was observed against a background of minimal genome-wide inflation of test statistics (λgc = 1.024), it excludes the possibility of substantial population substructure and differential genotyping call rate between cases and controls as a reason for the excess. Instead this suggests that at least some of these extreme P-values (P < 10−4) may represent true genetic associations with DSS;
Footnotes
URLs:: WHO, http://whqlibdoc.who.int/publications/2009/9789241547871_eng.pdf; Sequenom, www.sequenom.com; Applied Biosystems, www.appliedbiosystems.com; R project,www.r-project.org/.
Accession codes: MICB (MHC class I polypeptide-related sequence B) mRNA, NM_005931; PLCE1 (phospholipase C, epsilon 1) mRNA, NM_001165979, NM_016341.
Author Contributions: M.L.H. and C.P.S are the study principal investigators who conceived and obtained funding for the project. C.C.K. organized and supervised the GWAS and replication genotyping pipeline, devised the overall analysis plan and wrote the first draft of the manuscript with input from M.L.H., C.P.S., and S.D. C.T.N.B. is the lead coordinator of clinical samples and phenotypes for both the discovery and replication stages. J.P. and D.E.K.T. performed genotyping and quality checks on all samples. C.C.K., S.D., R.T.H.O., and Y.Y.T. analyzed the data. H.T.L., S.J.D., B.W., J.F., T.V.T., T.T.G., N.T.N.B., L.T.T., L.B.L., N.M.T., N.T.H.T., M.N.L., N.M.N., N.T.H., N.V.V.C., T.T.T., and A.S. coordinated and contributed patient and database phenotype collections as lead investigators for their respective sample collections. D.E.K.T. and J.P. performed genotyping and DNA quality checks. All authors critically reviewed manuscript revisions and contributed intellectual input to the final submission.
References
- 1.Gubler DJ. Dengue and dengue hemorrhagic fever. Clin Microbiol Rev. 1998;11:480–496. doi: 10.1128/cmr.11.3.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anders KL, et al. Epidemiological factors associated with dengue shock syndrome and mortality in hospitalized dengue patients in Ho Chi Minh City, Vietnam. Am J Trop Med Hyg. 84:127–134. doi: 10.4269/ajtmh.2011.10-0476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thai KT, et al. Seroprevalence of dengue antibodies, annual incidence and risk factors among children in southern Vietnam. Trop Med Int Health. 2005;10:379–386. doi: 10.1111/j.1365-3156.2005.01388.x. [DOI] [PubMed] [Google Scholar]
- 4.Loke H, et al. Susceptibility to dengue hemorrhagic fever in vietnam: evidence of an association with variation in the vitamin d receptor and Fc gamma receptor IIa genes. Am J Trop Med Hyg. 2002;67:102–106. doi: 10.4269/ajtmh.2002.67.102. [DOI] [PubMed] [Google Scholar]
- 5.Loke H, et al. Strong HLA class I--restricted T cell responses in dengue hemorrhagic fever: a double-edged sword? J Infect Dis. 2001;184:1369–1373. doi: 10.1086/324320. [DOI] [PubMed] [Google Scholar]
- 6.Stephens HA, et al. HLA-A and -B allele associations with secondary dengue virus infections correlate with disease severity and the infecting viral serotype in ethnic Thais. Tissue Antigens. 2002;60:309–318. doi: 10.1034/j.1399-0039.2002.600405.x. [DOI] [PubMed] [Google Scholar]
- 7.Sakuntabhai A, et al. A variant in the CD209 promoter is associated with severity of dengue disease. Nat Genet. 2005;37:507–513. doi: 10.1038/ng1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vejbaesya S, et al. TNF and LTA gene, allele, and extended HLA haplotype associations with severe dengue virus infection in ethnic Thais. J Infect Dis. 2009;199:1442–1448. doi: 10.1086/597422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Steinle A, et al. Interactions of human NKG2D with its ligands MICA, MICB, and homologs of the mouse RAE-1 protein family. Immunogenetics. 2001;53:279–287. doi: 10.1007/s002510100325. [DOI] [PubMed] [Google Scholar]
- 10.Gonzalez S, Lopez-Soto A, Suarez-Alvarez B, Lopez-Vazquez A, Lopez-Larrea C. NKG2D ligands: key targets of the immune response. Trends Immunol. 2008;29:397–403. doi: 10.1016/j.it.2008.04.007. [DOI] [PubMed] [Google Scholar]
- 11.Garrity D, Call ME, Feng J, Wucherpfennig KW. The activating NKG2D receptor assembles in the membrane with two signaling dimers into a hexameric structure. Proc Natl Acad Sci U S A. 2005;102:7641–7646. doi: 10.1073/pnas.0502439102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoang LT, et al. The early whole-blood transcriptional signature of dengue virus and features associated with progression to dengue shock syndrome in Vietnamese children and young adults. J Virol. 84:12982–12994. doi: 10.1128/JVI.01224-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Libraty DH, et al. Differing influences of virus burden and immune activation on disease severity in secondary dengue-3 virus infections. J Infect Dis. 2002;185:1213–1221. doi: 10.1086/340365. [DOI] [PubMed] [Google Scholar]
- 14.Vaughn DW, et al. Dengue viremia titer, antibody response pattern, and virus serotype correlate with disease severity. J Infect Dis. 2000;181:2–9. doi: 10.1086/315215. [DOI] [PubMed] [Google Scholar]
- 15.Kumar V, et al. Genome-wide association study identifies a susceptibility locus for HCV-induced hepatocellular carcinoma. Nat Genet. doi: 10.1038/ng.809. [DOI] [PubMed] [Google Scholar]
- 16.Hinkes B, et al. Positional cloning uncovers mutations in PLCE1 responsible for a nephrotic syndrome variant that may be reversible. Nat Genet. 2006;38:1397–1405. doi: 10.1038/ng1918. [DOI] [PubMed] [Google Scholar]
- 17.Chau TN, et al. Dengue virus infections and maternal antibody decay in a prospective birth cohort study of Vietnamese infants. J Infect Dis. 2009;200:1893–1900. doi: 10.1086/648407. doi:10.1086/648407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tien NT, et al. A prospective cohort study of dengue infection in schoolchildren in Long Xuyen, Viet Nam. Trans R Soc Trop Med Hyg. 2010;104:592–600. doi: 10.1016/j.trstmh.2010.06.003. doi:S0035-9203(10)00132-X [pii] 10.1016/j.trstmh.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 19.Balmaseda A, et al. Trends in patterns of dengue transmission over 4 years in a pediatric cohort study in Nicaragua. J Infect Dis. 2010;201:5–14. doi: 10.1086/648592. doi:10.1086/648592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Porter KR, et al. Epidemiology of dengue and dengue hemorrhagic fever in a cohort of adults living in Bandung, West Java, Indonesia. Am J Trop Med Hyg. 2005;72:60–66. doi:72/1/60 [pii] [PubMed] [Google Scholar]
- 21.Endy TP, et al. Epidemiology of inapparent and symptomatic acute dengue virus infection: a prospective study of primary school children in Kamphaeng Phet, Thailand. Am J Epidemiol. 2002;156:40–51. doi: 10.1093/aje/kwf005. [DOI] [PubMed] [Google Scholar]
- 22.Mammen MP, et al. Spatial and temporal clustering of dengue virus transmission in Thai villages. PLoS Med. 2008;5:e205. doi: 10.1371/journal.pmed.0050205. doi:08-PLME-RA-0417 [pii] 10.1371/journal.pmed.0050205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Endy TP, et al. Determinants of inapparent and symptomatic dengue infection in a prospective study of primary school children in Kamphaeng Phet, Thailand. PLoS Negl Trop Dis. 2011;5:e975. doi: 10.1371/journal.pntd.0000975. doi:10.1371/journal.pntd.0000975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mells GF, et al. Genome-wide association study identifies 12 new susceptibility loci for primary biliary cirrhosis. Nat Genet. doi: 10.1038/ng.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Price AL, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 27.Purdue MP, et al. Genome-wide association study of renal cell carcinoma identifies two susceptibility loci on 2p21 and 11q13.3. Nat Genet. 43:60–65. doi: 10.1038/ng.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thomas G, et al. A multistage genome-wide association study in breast cancer identifies two new risk alleles at 1p11.2 and 14q24.1 (RAD51L1) Nat Genet. 2009;41:579–584. doi: 10.1038/ng.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lettre G, Lange C, Hirschhorn JN. Genetic model testing and statistical power in population-based association studies of quantitative traits. Genet Epidemiol. 2007;31:358–362. doi: 10.1002/gepi.20217. [DOI] [PubMed] [Google Scholar]
- 30.McGovern DP, et al. Genome-wide association identifies multiple ulcerative colitis susceptibility loci. Nat Genet. 42:332–337. doi: 10.1038/ng.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Asano K, et al. A genome-wide association study identifies three new susceptibility loci for ulcerative colitis in the Japanese population. Nat Genet. 2009;41:1325–1329. doi: 10.1038/ng.482. [DOI] [PubMed] [Google Scholar]
- 32.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.