

Abstract
Many functionally essential ionizable groups are buried in the hydrophobic interior of proteins. A systematic study of Lys, Asp, and Glu residues at 25 internal positions in staphylococcal nuclease showed that their pKa values can be highly anomalous, some shifted by as many as 5.7 pH units relative to normal pKa values in water. Here we show that, in contrast, Arg residues at the same internal positions exhibit no detectable shifts in pKa; they are all charged at pH ≤ 10. Twenty-three of these 25 variants with Arg are folded at both pH 7 and 10. The mean decrease in thermodynamic stability from substitution with Arg was 6.2 kcal/mol at this pH, comparable to that for substitution with Lys, Asp, or Glu at pH 7. The physical basis behind the remarkable ability of Arg residues to remain protonated in environments otherwise incompatible with charges is suggested by crystal structures of three variants showing how the guanidinium moiety of the Arg side chain is effectively neutralized through multiple hydrogen bonds to protein polar atoms and to site-bound water molecules. The length of the Arg side chain, and slight deformations of the protein, facilitate placement of the guanidinium moieties near polar groups or bulk water. This unique capacity of Arg side chains to retain their charge in dehydrated environments likely contributes toward the important functional roles of internal Arg residues in situations where a charge is needed in the interior of a protein, in a lipid bilayer, or in similarly hydrophobic environments.
Keywords: electrostatics continuum, hydration, self-energy, buried charges, dielectric
Ionizable groups are usually found at the protein–water interface, where their charged moieties can interact with water. However, some ionizable groups are also found buried in the relatively hydrophobic interior of proteins. These ionizable groups are usually essential for function. Arginine (Arg) is the ionizable amino acid that is found most frequently buried in the protein interior (1–3). Arg residues in relatively hydrophobic environments are essential in a variety of biological processes, such as regulation of conformation or redox potentials (4, 5), viral capsid assembly (6), electrostatic steering (7), voltage sensing across lipid bilayers (8–10), H+ transport (4, 11–13), and peptide translocation across bilayers (14, 15). Internal Arg residues also play critical roles at protein–protein interfaces (3, 16), in enzyme active sites (1, 3, 17), and in a variety of transport channels (18, 19).
To understand the functional roles of internal Arg residues, it is necessary to know their charge state throughout their functional cycle. The low polarizability and polarity of the protein interior can lead to large shifts in the pKa values of internal groups relative to their normal values in water (20–26), usually in the direction that promotes the neutral state. Arg in water has the highest pKa among naturally occurring ionizable amino acid (27), making Arg the ionizable residue least likely to exist in the neutral state near physiological pH. In the proposed functional roles for Arg residues buried in hydrophobic environments, the side chain is usually assumed to be charged, but this is seldom demonstrated experimentally (2, 8–10, 12–15, 18, 19, 28–32). Computational studies indicating that the Arg side chain is rarely deprotonated support this conclusion (2, 15, 28, 29, 33). To our knowledge, only two experimental studies have examined explicitly the charge state of an internal Arg residue at physiological pH (34, 35). Both studies investigated internal Arg in channel proteins; both studies found that the Arg was neutral.
To study the properties of Arg residues in the protein interior systematically, 25 internal positions in a highly stable form of staphylococcal nuclease (SNase) known as Δ+PHS SNase were substituted with Arg (Fig. 1). To measure the pKa values and to characterize the charge state of the internal Arg residues, the pH dependence of thermodynamic stability of the 25 variants with internal Arg was measured (20, 21, 36). Using this approach, we previously demonstrated that Lys, Asp, and Glu at these 25 internal positions can have highly anomalous pKa values (20, 22, 26, 37–39). Of the 75 Lys, Asp, and Glu studied, 64 titrates with pKa values shifted relative to their normal values in water, some by as much as 5.7 pH units. The shifts are all in the direction that favors the neutral state (elevated pKa for acidic residues and depressed pKa for basic residues). This suggests that the ionizable moieties of these residues are at least partially dehydrated and that the polarity or polarizability in the protein interior is not sufficient to compensate fully for dehydration effects. A large number of crystal structures of other variants of SNase (already available in the PDB), and and Trp fluorescence and CD and NMR spectroscopy studies show that most variants of SNase with internal ionizable groups are fully folded and native-like at pH values where the internal groups are neutral (21, 22, 26). Even upon ionization of the internal groups, most of the 75 proteins with internal Lys, Asp, and Glu remain folded; only about a third of them exhibit any evidence of conformational reorganization, minor in most cases, coupled to the ionization of the internal group (21, 22). In the current work we demonstrate that the electrostatic properties of Arg at these 25 internal positions in SNase are strikingly different from those of Lys, Glu, or Asp.
Fig. 1.

Structure of Δ+PHS nuclease showing internal positions that were substituted with Arg. Spheres represent the Cα of residues substituted with Arg. Colors identify variants that were unfolded by the substitution with Arg (red), variants that were folded but enzymatically inactive (yellow), and variants that were folded and enzymatically active (blue). Bold numbers identify the positions (72, 90, and 109) that were substituted with Arg in the three crystal structures that were solved. The circle identifies the approximate location of the active site. Structure shown is Δ+PHS (PDB ID code 3BDC) (42).
Results
Structural Consequences of Substitution with Arg.
To examine how substitution of internal residues with Arg affected the conformation of the protein globally, far-UV CD spectra were measured for each variant at pH 7 (Fig. 2A) and pH 10 (Fig. 2B). Of the 25 variants with Arg, 19 had spectra comparable to those of the parent protein (Δ+PHS nuclease), suggesting that these proteins were folded without alterations in secondary structure. The spectra of four variants (A58R, V39R, Y91R, and N100R) were slightly different from those of the parent protein. The A58R variant showed a decrease in signal intensity relative to the signal of Δ+PHS at both pH values, consistent with partial unfolding and loss of secondary structure or with the presence of some unfolded protein in equilibrium with fully folded protein. The V39R, Y91R, and N100R variants exhibited an increase in signal at 208 nm at pH 7 and a shift in spectral minimum at pH 10, consistent with a minor change in secondary structure relative to the reference protein.
Fig. 2.

Far-UV CD spectra of Arg-containing variants at pH 7 (A) and pH 10 (B). Δ+PHS nuclease (solid black line), Δ+PHS nuclease in 6 M GdnHCl (dashed black line), and the unfolded SNase variant T62P in water (dashed gray line) are shown for reference. Solid gray lines correspond to the 19 Arg variants with no apparent structural rearrangement. Variants of interest are highlighted: A58R (green); V39R, Y91R, and N100R (blue); I92R (orange); and V104R (red). GdnHCl-induced unfolding of Arg-containing variants at pH 7 (C) and pH 10 (D). Points represent the normalized fluorescence measured at 296 nm; lines represent two-state linear extrapolation model fit to each curve. Color scheme is identical to A and B. The two-state model could not be fit to the titration curves of the I92R or V104R variants.
Two other variants (I92R and V104R) showed much larger differences in both signal intensity and shape at both pH 7 and 10, relative to the reference protein. For comparison, the spectrum of an intrinsically unfolded T62P variant of SNase is included in Fig. 2. The I92R and V104R variants have deep 208-nm and shallow 222-nm minima at pH 7 and low overall signal intensity at pH 10, similar to those of the unfolded T62P variant. Although the I92R and V104R variants retain some secondary structure, they are clearly in a somewhat different conformation, more different from the native one than in any of the other variants.
Enzymatic Activity.
To determine whether the variant proteins with Arg at internal positions could achieve a folded and enzymatically active conformation, the enzymatic activity of all variants was assayed as described under Methods. Eighteen variant proteins out of 25 that were studied had detectable activity. The remaining seven variants (G20R, L36R, T41R, A58R, I92R, V104R, and A109R) were inactive. Positions 20, 36, 41, 58, and 109 are clustered around the active site (Fig. 1); it is likely that Arg at these positions interferes directly with catalysis or with substrate binding without disrupting the native fold. The lack of enzymatic activity in the I92R and V104R variants is fully consistent with CD spectra showing that these proteins are unfolded. The fact that substrate binding did not induce folding suggests that these two variants are highly destabilized relative to the reference protein.
Thermodynamic Stability.
The thermodynamic stability (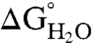 ) of all variants was determined at pH 7 and 10 by denaturation with guanidinium chloride monitored by intrinsic Trp fluorescence at 296 nm (20). The unfolding curves were analyzed using a two-state linear extrapolation model (40) (Fig. 2 C and D). The values of
) of all variants was determined at pH 7 and 10 by denaturation with guanidinium chloride monitored by intrinsic Trp fluorescence at 296 nm (20). The unfolding curves were analyzed using a two-state linear extrapolation model (40) (Fig. 2 C and D). The values of 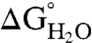 , midpoints of unfolding, and m values are listed for all proteins in Table S1. The unfolding transitions of 23 out of 25 variants exhibited apparent two-state behavior at both pH 7 and 10. Consistent with the CD spectra and with activity measurements, no unfolding transition was detected with Trp fluorescence for the I92R or the V104R variants, indicating that they are unfolded at both pH 7 and 10.
, midpoints of unfolding, and m values are listed for all proteins in Table S1. The unfolding transitions of 23 out of 25 variants exhibited apparent two-state behavior at both pH 7 and 10. Consistent with the CD spectra and with activity measurements, no unfolding transition was detected with Trp fluorescence for the I92R or the V104R variants, indicating that they are unfolded at both pH 7 and 10.
The effect of substitution with Arg on thermodynamic stability (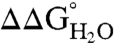 ) was measured for each variant by subtracting
) was measured for each variant by subtracting 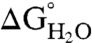 of the Arg-containing variant from
of the Arg-containing variant from 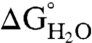 of the reference protein (Table 1). At pH 7, the energetic cost of substitution of internal groups with Arg ranged from -1.2 kcal/mol in the L38R variant to -11.8 kcal/mol or more for the I92R and V104R variants. The mean
of the reference protein (Table 1). At pH 7, the energetic cost of substitution of internal groups with Arg ranged from -1.2 kcal/mol in the L38R variant to -11.8 kcal/mol or more for the I92R and V104R variants. The mean 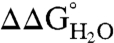 was -6.2 ± 2.7 kcal/mol at pH 7 and -5.7 ± 2.7 kcal/mol at pH 10. The mean
was -6.2 ± 2.7 kcal/mol at pH 7 and -5.7 ± 2.7 kcal/mol at pH 10. The mean 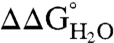 at pH 7 was comparable to the mean
at pH 7 was comparable to the mean 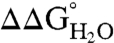 at pH 7 for substitution of the 25 internal positions with Lys (-5.7 ± 1.7 kcal/mol), Glu (-6.2 ± 1.8 kcal/mol), and Asp (-7.1 ± 2.3 kcal/mol) (21).
at pH 7 for substitution of the 25 internal positions with Lys (-5.7 ± 1.7 kcal/mol), Glu (-6.2 ± 1.8 kcal/mol), and Asp (-7.1 ± 2.3 kcal/mol) (21).
Table 1.
Thermodynamic consequences of substitutions of internal hydrophobic groups in Δ+PHS SNase with Arg
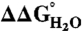 , kcal mol-1*,† , kcal mol-1*,† |
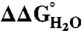 slope, kcal mol-1 pH-1‡ slope, kcal mol-1 pH-1‡
|
|||||
| Position | pH 7 | pH 10 | ||||
| G20 | −3.2 | (0.1) | −2.5 | (0.1) | 0.3 | (0.2) |
| V23 | −7.5 | (0.1) | −6.7 | (0.1) | 0.3 | (0.2) |
| L25 | −8.8 | (0.1) | −8.0 | (0.1) | 0.3 | (0.2) |
| F34 | −8.6 | (0.1) | −7.9 | (0.1) | 0.2 | (0.2) |
| L36 | −9.0 | (0.1) | −7.9 | (0.1) | 0.4 | (0.2) |
| L37 | −3.4 | (0.1) | −2.7 | (0.1) | 0.3 | (0.2) |
| L38 | −1.2 | (0.1) | −1.1 | (0.1) | 0.0 | (0.2) |
| V39 | −7.2 | (0.1) | −6.9 | (0.1) | 0.1 | (0.2) |
| T41 | −2.6 | (0.1) | −1.7 | (0.1) | 0.3 | (0.2) |
| A58 | −3.9 | (0.1) | −3.6 | (0.1) | 0.1 | (0.2) |
| T62 | −5.2 | (0.1) | −5.4 | (0.1) | −0.1 | (0.2) |
| V66 | −9.0 | (0.1) | −8.3 | (0.1) | 0.2 | (0.2) |
| I72 | −6.2 | (0.1) | −5.8 | (0.1) | 0.1 | (0.2) |
| V74 | −6.5 | (0.1) | −6.0 | (0.1) | 0.2 | (0.2) |
| A90 | −6.3 | (0.1) | −5.5 | (0.1) | 0.3 | (0.2) |
| Y91 | −6.4 | (0.1) | −5.8 | (0.1) | 0.2 | (0.2) |
| I92 | — | — | — | |||
| V99 | −10.3 | (0.1) | −9.7 | (0.1) | 0.2 | (0.2) |
| N100 | −9.5 | (0.1) | −9.1 | (0.1) | 0.1 | (0.2) |
| L103 | −6.6 | (0.1) | −6.4 | (0.1) | 0.1 | (0.2) |
| V104 | — | — | — | |||
| A109 | −3.7 | (0.1) | −3.4 | (0.1) | 0.1 | (0.2) |
| N118 | −1.8 | (0.1) | −1.2 | (0.1) | 0.2 | (0.2) |
| L125 | −9.5 | (0.1) | −9.8 | (0.2) | −0.1 | (0.2) |
| A132 | −7.0 | (0.1) | −5.9 | (0.2) | 0.4 | (0.2) |
*Values in parentheses are errors propagated from model fit.
†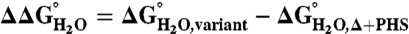 .
.
‡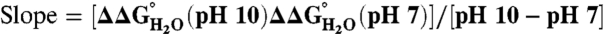 .
.
Ionization State of Arg Residues.
The pKa values of internal ionizable residues can be measured by analysis of the pH dependence of 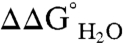 (20–22, 26, 36, 41). If the pKa values of the Arg residues were depressed relative to the normal pKa of Arg in water (pKa ∼ 12),
(20–22, 26, 36, 41). If the pKa values of the Arg residues were depressed relative to the normal pKa of Arg in water (pKa ∼ 12), 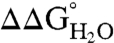 would decrease linearly with decreasing pH at pH values below 12 (21). Because the stability of SNase cannot be measured accurately above pH 10 with chemical denaturation owing to photobleaching and difficulties with pH buffering, only pKa values < 10 can be determined by analysis of
would decrease linearly with decreasing pH at pH values below 12 (21). Because the stability of SNase cannot be measured accurately above pH 10 with chemical denaturation owing to photobleaching and difficulties with pH buffering, only pKa values < 10 can be determined by analysis of 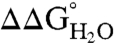 vs. pH data. Even so, the measured insensitivity of
vs. pH data. Even so, the measured insensitivity of 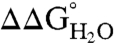 values to pH demonstrates that the 25 Arg residues at internal positions in SNase are charged at pH 10 and lower (Fig. S1). Table 1 lists
values to pH demonstrates that the 25 Arg residues at internal positions in SNase are charged at pH 10 and lower (Fig. S1). Table 1 lists 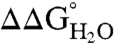 at pH 7 and 10, as well as the slopes of
at pH 7 and 10, as well as the slopes of 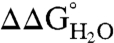 vs. pH for each variant. The slope was close to 0 kcal/mol/pH for every variant. The largest observed slope was 0.4 ± 0.2 kcal/mol/pH for Arg-36 and Arg-132. This slope could be consistent with an Arg pKa no lower than 9.2 (Fig. S1). The
vs. pH for each variant. The slope was close to 0 kcal/mol/pH for every variant. The largest observed slope was 0.4 ± 0.2 kcal/mol/pH for Arg-36 and Arg-132. This slope could be consistent with an Arg pKa no lower than 9.2 (Fig. S1). The 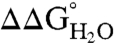 values demonstrated unequivocally that all Arg residues are ionized at pH 7 and that most if not all are also charged at pH 10.
values demonstrated unequivocally that all Arg residues are ionized at pH 7 and that most if not all are also charged at pH 10.
Microenvironments of the Arg Side Chains.
The crystal structures of the variants with I72R, A90R, and A109R were refined to final resolutions of 2.00 Å, 2.15 Å, and 1.90 Å, respectively. Crystallographic data collection parameters and refinement statistics are summarized in Table S2. These three variants had native-like CD spectra. I72R and A90R had detectable enzyme activity, but A109R was one of the variants that was enzymatically inactive. The structures suggested structural and physical origins of the ability of the Arg side chains at internal positions to retain their charge without significant structural reorganization (Fig. 3).
Fig. 3.

Crystal structures of the variants with I72R (A and D), A90R (B and E), and A109R (C and F) substitutions. (Top) The effect of the Arg substitution on the global backbone conformation of SNase. The Arg-containing variants are colored blue; the structure of the reference Δ+PHS protein is in gray. The Arg side chains are shown in stick models. (Bottom) The polar contacts made by the Arg side chains. Water molecules are shown as red spheres. Final 1.25σ 2Fo - Fc electron density is shown in blue.
Substitution of Ile-72 with Arg led to a small concerted movement of the backbone relative to the structure of the Δ+PHS nuclease (42): β-strands 1, 2, and 3 moved 3 Å in one direction and the C terminus of helix-1 moved 1 Å in the opposite direction (Fig. 3A). The overall Cα rmsd relative to Δ+PHS nuclease is 0.7 Å. In the structure of Δ+PHS nuclease, Ile-72 points directly into the β-barrel. Although the Cα and Cβ atoms of Ile-72 and Arg-72 can be superimposed, the Arg side chain does not point into the β-barrel. Instead it reaches toward bulk water, thereby allowing Arg-72 to satisfy two of its hydrogen bonds with backbone carbonyl oxygen atoms and one with a bound water molecule (Table S3 and Fig. 3D). The Arg Nϵ atom is solvent inaccessible, but both Nη atoms are approximately 50% solvent-exposed, suggesting that the guanidinium moiety satisfies its remaining two hydrogen bonds with bulk water.
The replacement of Ala-90 with Arg was tolerated with essentially no change in backbone position (Cα rmsd relative to Δ+PHS of 0.3 Å) (Fig. 3B). The Cα and Cβ of Ala-90 and Arg-90 can be superimposed well. Arg-90 is accommodated with small shifts in the positions of the side chains of Phe-76 and Arg-81 (1.1 and 0.3 Å rmsd relative to Δ+PHS). There was also a 1-Å shift in the backbone between residues 78 and 80. Only 8.5 Å2 (5.7%) of the guanidinium moiety are solvent accessible; however, despite its being almost fully buried, all five possible hydrogen bonds of the guanidine moiety are satisfied, two with backbone carbonyl oxygen atoms, two with bound water molecules, and one with the Tyr-27 OH atom (Table S3 and Fig. 3E).
Substitution of Ala-109 with Arg led to a large movement of the loop containing residues 42–53 (Fig. 3C). The change in the position of the loop prevented steric clash between the side chain of Arg-109 and the protein. 2Fo - Fc electron density contoured at 1.25σ became interpretable for the main chain in this region over several rounds of refinement; however, the density for side chains of Phe-50, Asn-51, and Glu-52 remained poor. This large structural rearrangement did not alter the global structure: The Cα rmsd relative to Δ+PHS nuclease is 0.8 Å when the loop is excluded. The average B factor of main chain atoms in this loop is higher than average in both Δ+PHS (31.3 Å2 vs. 21.4 Å2) and the Arg-109 variant (36.0 Å2 vs. 16.0 Å2), suggesting that this loop is dynamic in both proteins. Only 12.9 Å2 (8.4%) of the guanidinum moiety of Arg-109 is solvent-exposed, but it forms six possible hydrogen bonds to five atoms: the Oε1 atom of Asp-21, a backbone carbonyl oxygen atom, and three site-bound waters (Table S3 and Fig. 3F). Asp-21 is a catalytically essential residue. If the ion pair between Asp-21 and Arg-109 observed in the crystal structure is present in solution, this would explain why the variant with Arg-109 was enzymatically inactive.
At least for the three variants for which crystal structures were obtained, the substitution of internal hydrophobic groups with Arg did not trigger any substantial conformational rearrangements of the backbone. The largest change, observed in the A109R variant, involved a loop that is known to be mobile, and which was in a conformation different than in the other two variants or in the reference protein. In all three structures the Arg side chain occupied locations where the guanidinium moiety could fulfill its hydrogen bonding potential through interactions with protein oxygen atoms and site-bound water molecules. Although most of the atoms in their side chains are solvent inaccessible, the guanidinium group is sufficiently long to be able to approach the protein–water interface, allowing at least one Nη atom in each side chain to achieve limited solvent-accessibility. This is accomplished without population of unusual rotamers of the Arg side chain (43). Although the Arg side chains have a formal charge of +1, the network of hydrogen bonds is extensive and seems likely to act by solvating and effectively neutralizing the guaninidium moiety.
Discussion
The equilibrium thermodynamic data show unequivocally that the Arg side chain retains its charge even when removed from bulk water and buried, at least partially, in the relatively hydrophobic interior of a globular protein. In this respect, Arg is unique. Two-thirds of Lys, Glu, and Asp at the same 25 internal positions in SNase are neutral at pH 7, whereas all Arg retained their charge even up to pH 10. In 23 of the 25 Arg-containing variants studied, the internal Arg ionized without triggering large-scale reorganization of the protein detectable with Trp fluorescence or CD spectroscopy.
These unique properties of Arg could imply that it is less energetically costly to ionize Arg than other types of ionizable residues at internal positions. This cannot be demonstrated without knowing the pKa of Arg in water and in the protein. Unfortunately, the pKa of Arg in water is not known with certainty. It is assumed to be 12 (27), but actual experimental estimates range from 11.5 to 15.0 (44–46). If the pKa of Arg in water were 12, then the equilibrium thermodynamic experiments (Table 1) would have been able to detect shifts in pKa≥2.5. In the previous study of pKa values of internal lysines in SNase, 12 Lys exhibited shifts in this range (26); therefore, if Arg and Lys behaved similarly, approximately 12 Arg residues would have exhibited shifted pKa values. If, on the other hand, the pKa of Arg in water were closer to 15, shifts in pKa≥5.5 would have been required to be detectable. A few internal Lys residues in SNase exhibited shifts in this range; therefore, if the cost to ionize Arg and Lys was identical, a few Arg should have exhibited shifted pKa values. Although a pKa of 15 is higher than normally assumed (27), this value was measured in a study designed to eliminate sources of error in previous measurements (44). This measurement has not been validated independently or evaluated critically. Considering that the interpretation of recent studies of biological function of Arg depends on knowledge of its pKa in water, the accurate determination of this pKa should be a high priority. Owing to the uncertainty in the pKa of Arg in water, the absence of observable shifts in the pKa values of Arg residues in internal positions in SNase cannot be interpreted without ambiguity, but it is likely that at least some shifted pKa values would have been detected if they were present.
It is probably energetically less costly to ionize Arg at internal positions in a protein than it is to ionize Lys, Glu, or Asp. Because of its large size and distributed charge, the guandinium moiety is hydrated more weakly than an amino or carboxylic acid moiety; thus dehydration is energetically less costly (47). For the same reason, this moiety is likely to be less sensitive to the nature of its microenvironment than the amino group of Lys, in which the excess charge is localized on a single heavy atom. The guanidinium moiety can also participate in more types of noncovalent interactions than other ionizable groups can: ion pairs (48, 49), cation-π interactions (17, 50), five hydrogen bonds (51, 52), and even favorable interactions with other guanidiunium groups (53, 54). All these interactions promote the charged state of Arg. Owing to the length of its side chain, Arg can also sample conformational space more extensively than Lys, Glu, or Asp, thus enhancing the probability of placing the guanidinium moiety near polar protein atoms or internal or bulk water.
Many of the factors that promote the charged state of Arg were illustrated by the crystal structures (Fig. 3). The structures show that the Arg side chain reaches closer to the protein–water interface than other internal ionizable groups in SNase. In all three structures, at least one atom of the guanidinium moiety achieves some degree of exposure to bulk water, small but sufficient to partially hydrate the group (Table S3). These internal Arg side chains also fulfill all five possible hydrogen bonds with polar protein atoms or water, reminiscent of what is observed in crystal structures of proteins containing naturally occurring internal Arg residues (8–10, 12–16, 30–32). In contrast, in crystal structures of SNase variants with internal Lys, the amino moiety of the Lys side chain is often surrounded primarily by hydrophobic atoms (20, 37, 55).
The data suggest that the energy required to deform SNase slightly to keep the Arg minimally hydrated is lower than the energy required to dehydrate the group in the protein interior. If this is the case, the energy to substitute internal hydrophobic groups with ionized Arg does not only measure the energy to partially dehydrate the guanidinium group. It also measures the energy to deform the protein to accept the charged Arg side chain. This idea is supported by the high correlation between the thermodynamic cost of substitution of internal positions with charged Arg and charged Lys (Fig. 4). This indicates that, at a given site, a common process must determine the cost of substitution with both types of ionizable groups. Because the innate physicochemical properties of the amino and guanidinium moieties are radically different, the similarity in the cost of inserting Lys and Arg at pH 7 is likely dictated by a property of the protein, rather than the specific physicochemical details of the side chain. At least for the Arg-containing variants for which crystal structures are available (Fig. 3), the common process appears to involve a subtle deformation of tertiary structure to allow the hydration of the internal charged guanidinium moiety. This idea is consistent with detailed studies of the L38K, V66K, and I92K variants of SNase, all of which exhibit structural reorganization to hydrate the amino group of internal Lys residues upon ionization (55–58). An NMR study of other SNase variants with internal Lys residues (56) and computational studies on the same proteins (59) all suggest the involvement of minor conformational reorganization and water penetration in response to the ionization of internal Lys residues.
Fig. 4.

Correlation in the difference in thermodynamic stability (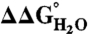 ) between variants of Δ+PHS SNase with Lys or Arg at internal positions, measured at pH 5 for variants with Lys to ensure they are all charged, and at pH 7 for variants with Arg, where they are all charged. The thermodynamic stability of Δ+PHS SNase is independent of pH between pH 5 and 7 (21). The stability of
) between variants of Δ+PHS SNase with Lys or Arg at internal positions, measured at pH 5 for variants with Lys to ensure they are all charged, and at pH 7 for variants with Arg, where they are all charged. The thermodynamic stability of Δ+PHS SNase is independent of pH between pH 5 and 7 (21). The stability of 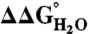 values were calculated by subtracting
values were calculated by subtracting 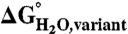 from
from 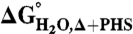 . The error bars are the estimated uncertainty of the measurement. Values for variants with Lys are from ref. 26.
. The error bars are the estimated uncertainty of the measurement. Values for variants with Lys are from ref. 26.
The deformation of SNase to allow hydration of internal charged groups suggests it would be difficult for any small globular protein to dehydrate and neutralize Arg at physiological pH. It might be possible to bury Arg in the neutral state in a larger protein with a larger hydrophobic core. Consistent with this, the only reports of Arg residues with perturbed pKa values involve large membrane proteins (34, 35). The hydrophobic interior of these proteins is much larger than for a globular protein; therefore, the Arg side chain can in principle be much farther away from bulk water than in SNase. Furthermore, minor structural deformation to allow hydration of the internal Arg side chain is less likely in a membrane protein. On the other hand, the low cost for transfer of Arg through a lipid bilayer in the interesting experiments of Freites et al. (60) has been rationalized in terms of favorable interactions between charged Arg and protein polar atoms (61), a situation comparable to what was observed with the Arg-containing variants of SNase. It also appears that specialized microenvironments do exist even in relatively small proteins that can promote the neutral form of Arg. This is the case of an internal Arg in the photoactive yellow protein, which in a neutron diffraction structure at pH 9 appears to be deprotonated (62).
Although the hydrophobic character of the interior of a protein is clearly different from that of a lipid bilayer, the ability of the Arg side chain to distort the protein to enhance its hydration and to remain charged is reminiscent of the manner in which the charged Arg side chain is thought to be able to deform a bilayer to maintain access to water and to contact polar head groups to stabilize the charged form (15, 29, 60). Our studies do not contribute definite insight into mechanisms for stabilization of charged Arg in a nonaqueous environment, but they do show that the Arg side chain has a unique capacity to retain its charge in environments that should be expected to promote the neutral form. This ability of Arg to stay charged appears to be fundamental for its functional role as a voltage sensor (8–10), as the essential filter in aquoporins (18, 30, 31), and in other proteins that perform fundamental energy transduction processes (4, 11–13).
Our data show that it is much more difficult to neutralize Arg than Lys, Asp, or Glu near physiological pH in hydrophobic protein environments. It is not clear if this occurs because the pKa of Arg in water is higher than 12, because of the unique side chain length, hydrogen bonding, and hydration properties of Arg, or some combination of both. The singular capacity of Arg to remain charged in relatively hydrophobic environments, without significantly reorganizing the protein significantly, may explain why naturally occurring internal Arg are abundant and why they seem to be the side chain of choice when biochemical function requires the presence of charge in hydrophobic environments.
Methods
Protein Engineering and Optical Spectroscopy.
All experiments were performed with variants of the highly stable Δ+PHS nuclease and were prepared and purified as described previously (57). pH titrations monitored by steady-state Trp fluorescence and CD scans from 250 to 200 nm were performed with an Aviv Automated Titration Fluorometer and an Aviv Circular Dichroims Spectrometer Model 215, as described previously (57).
Enzymatic Activity and Thermodynamic Stability.
The blue-plate assay was used to measure enzymatic activity at pH 7 as described previously (21). Thermodynamic stability was measured with GdnHCl titrations monitored with Trp fluorescence using an Aviv Automatic Titration Fluorimeter 105, as described previously.
Crystallography and Solvent-Accessibility Calculations.
Crystal structures of three variants were solved under cryogenic conditions. Proteins were crystallized at 4 °C by vapor diffusion in hanging drops over reservoir solutions that contained 25 mM potassium phosphate buffer (pH 8–9) and 24–42% MPD (wt/vol) as a precipitant. The Δ+PHS/I72R and Δ+PHS/A90R structures were cocrystallized with Ca2+ and with the inhibitor thymidine-3′,5′-diphosphate. Details of data collection and refinement statistics are provided in Table S2. Solvent-accessibility calculations were performed with NACCESS 2.1.1 using a probe radius of 1.4 Å used with the default van der Waal’s radii for each atom.
Supplementary Material
Acknowledgments.
We gratefully acknowledge Aaron C. Robinson for assistance with the activity measurements. This work was supported by National Institutes of Health Grant GM-061597 (to B.G.-M.E.), by a graduate research fellowship from the National Science Foundation (M.J.H.), and by a Research Experience for Undergraduates Fellowship (G.R.S.) and a Research Opportunity Award (J.L.S.) through the National Science Foundation Grant MCB-0743422 (to B.G-M.E.)
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 3D8G, 3DHQ, and 3D4W).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1104808108/-/DCSupplemental.
References
- 1.Bartlett GJ, Porter CT, Borkakoti N, Thornton JM. Analysis of catalytic residues in enzyme active sites. J Mol Biol. 2002;324:105–121. doi: 10.1016/s0022-2836(02)01036-7. [DOI] [PubMed] [Google Scholar]
- 2.Kim J, Mao J, Gunner MR. Are acidic and basic groups in buried proteins predicted to be ionized? J Mol Biol. 2005;348:1283–1298. doi: 10.1016/j.jmb.2005.03.051. [DOI] [PubMed] [Google Scholar]
- 3.Bogan AA, Thorn KS. Anatomy of hot spots in protein interfaces. J Mol Biol. 1998;280:1–9. doi: 10.1006/jmbi.1998.1843. [DOI] [PubMed] [Google Scholar]
- 4.Cutler RL, et al. Role of arginine-38 in regulation of the cytochrome c oxidation-reduction equilibrium. Biochemistry. 1989;28:3188–3197. doi: 10.1021/bi00434a012. [DOI] [PubMed] [Google Scholar]
- 5.Winn PJ, Lüdemann SK, Gauges R, Lounnas V, Wade RC. Comparison of the dynamics of substrate access channels in three cytochrome P450s reveals different opening mechanisms and a novel functional role for a buried arginine. Proc Natl Acad Sci USA. 2002;99:5361–5366. doi: 10.1073/pnas.082522999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hansen JL, Long AM, Schultz SC. Structure of the RNA-dependent RNA polymerase of poliovirus. Structure. 1997;5:1109–1122. doi: 10.1016/s0969-2126(97)00261-x. [DOI] [PubMed] [Google Scholar]
- 7.Wade RC, Gabdoulline RR, Ludemann SK, Lounnas V. Electrostatic steering and ionic tethering in enzyme-ligand binding: Insights from simulations. Proc Natl Acad Sci USA. 1998;95:5942–5949. doi: 10.1073/pnas.95.11.5942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiang Y, Ruta V, Chen J, Lee A, MacKinnon R. The principle of gating charge movement in a voltage-dependent K+ channel. Nature. 2003;423:42–48. doi: 10.1038/nature01581. [DOI] [PubMed] [Google Scholar]
- 9.Long SB, Campbell EB, MacKinnon R. Voltage sensor of Kv1.2: Structural basis of electromechanical coupling. Science. 2005;309:903–908. doi: 10.1126/science.1116270. [DOI] [PubMed] [Google Scholar]
- 10.Tao X, Lee A, Limapichat W, Dougherty DA, MacKinnon R. A gating charge transfer center in voltage sensors. Science. 2010;328:67–73. doi: 10.1126/science.1185954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Le NP, et al. Escherichia coli ATP synthase alpha subunit Arg-376: The catalytic site arginine does not participate in the hydrolysis/synthesis reaction but is required for promotion to the steady state. Biochemistry. 2000;39:2778–2783. doi: 10.1021/bi992530h. [DOI] [PubMed] [Google Scholar]
- 12.Rammelsberg R, Huhn G, Lubben M, Gerwert K. Bacteriorhodopsin’s intramolecular proton-release pathway consists of a hydrogen-bonded network. Biochemistry. 1998;37:5001–5009. doi: 10.1021/bi971701k. [DOI] [PubMed] [Google Scholar]
- 13.Luecke H, Richter H-T, Lanyi JK. Proton transfer pathways in bacteriorhodopsin at 2.3 angstrom resolution. Science. 1998;280:1934–1937. doi: 10.1126/science.280.5371.1934. [DOI] [PubMed] [Google Scholar]
- 14.Futaki S, et al. Arginine-rich peptides an abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J Biol Chem. 2001;276:5836–5840. doi: 10.1074/jbc.M007540200. [DOI] [PubMed] [Google Scholar]
- 15.Herce HD, Garcia AE. Molecular dynamics simulations suggest a mechanism for translocation of the HIV-1 TAT peptide across lipid membranes. Proc Natl Acad Sci USA. 2007;104:20805–20810. doi: 10.1073/pnas.0706574105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pednekar D, Tendulkar A, Durani S. Electrostatics-defying interaction between arginine termini as a thermodynamic driving force in protein-protein interaction. Proteins. 2009;74:155–163. doi: 10.1002/prot.22142. [DOI] [PubMed] [Google Scholar]
- 17.Crowley PB, Golovin A. Cation-pi interactions in protein-protein interfaces. Proteins. 2005;59:231–239. doi: 10.1002/prot.20417. [DOI] [PubMed] [Google Scholar]
- 18.Savage DF, O’Connell JD, Miercke LJW, Finer-Moore J, Stroud RM. Structural context shapes the aquaporin selectivity filter. Proc Natl Acad Sci USA. 2010;107:17164–17169. doi: 10.1073/pnas.1009864107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boudker O, Ryan RM, Yernool D, Shimamoto K, Gouaux E. Coupling substrate and ion binding to extracellular gate of a sodium-dependent aspartate transporter. Nature. 2007;445:387–393. doi: 10.1038/nature05455. [DOI] [PubMed] [Google Scholar]
- 20.Stites WE, Gittis AG, Lattman EE, Shortle D. In a staphylococcal nuclease mutant the side-chain of a lysine replacing valine 66 is fully buried in the hydrophobic core. J Mol Biol. 1991;221:7–14. doi: 10.1016/0022-2836(91)80195-z. [DOI] [PubMed] [Google Scholar]
- 21.Isom DG, Cannon BR, Castañeda CA, Robinson A, García-Moreno E. High tolerance for ionizable residues in the hydrophobic interior of proteins. Proc Natl Acad Sci USA. 2008;105:17784–17788. doi: 10.1073/pnas.0805113105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Isom DG, Castañeda CA, Cannon BR, Velu PD, García-Moreno EB. Charges in the hydrophobic interior of proteins. Proc Natl Acad Sci USA. 2010;107:16096–16100. doi: 10.1073/pnas.1004213107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li YK, Kuliopulos A, Mildvan AS, Talalay P. Environments and mechanistic roles of the tyrosine residues of delta 5-3-ketosteroid isomerase. Biochemistry. 1993;32:1816–1824. doi: 10.1021/bi00058a016. [DOI] [PubMed] [Google Scholar]
- 24.Chivers PT, et al. Microscopic pKa values of Escherichia coli thioredoxin. Biochemistry. 1997;36:14985–14991. doi: 10.1021/bi970071j. [DOI] [PubMed] [Google Scholar]
- 25.Czerwinski RM, et al. Effects of mutations of the active site arginine residues in 4-oxalocrotonate tautomerase on the pKa values of active site residues and on the pH dependence of catalysis. Biochemistry. 1999;38:12358–12366. doi: 10.1021/bi9911177. [DOI] [PubMed] [Google Scholar]
- 26.Isom DG, Castañeda CA, Cannon BR, García-Moreno EB. Large shifts in pKa values of lysine residues buried inside a protein. Proc Natl Acad Sci USA. 2011;108:5260–5265. doi: 10.1073/pnas.1010750108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tanford C. The interpretation of hydrogen ion titration curves of proteins. Adv Protein Chem. 1963;17:69–165. [Google Scholar]
- 28.Li L, Vorobyov I, MacKerell AD, Allen TW. Is arginine charged in a membrane? Biophys J. 2008;94:L11–13. doi: 10.1529/biophysj.107.121566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yoo J, Cui Q. Does arginine remain protonated in the lipid membrane? Insights from microscopic pKa calculations. Biophys J. 2008;94:L61–63. doi: 10.1529/biophysj.107.122945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Newby ZER, et al. Crystal structure of the aquaglyceroporin PfAQP from the malarial parasite Plasmodium falciparum. Nat Struct Mol Biol. 2008;15:619–625. doi: 10.1038/nsmb.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ho JD, et al. Crystal structure of human aquaporin 4 at 1.8 Å and its mechanism of conductance. Proc Natl Acad Sci USA. 2009;106:7437–7442. doi: 10.1073/pnas.0902725106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rastogi VK, Girvin ME. Structural changes linked to proton translocation by subunit c of the ATP synthase. Nature. 1999;402:263–268. doi: 10.1038/46224. [DOI] [PubMed] [Google Scholar]
- 33.Schow EV, et al. Arginine in membranes: The connection between molecular dynamics simulations and translocon-mediated insertion experiments. J Membr Biol. 2011;239:35–48. doi: 10.1007/s00232-010-9330-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Niemeyer MI, et al. Neutralization of a single arginine residue gates open a two-pore domain, alkali-activated K+ channel. Proc Natl Acad Sci USA. 2007;104:666–671. doi: 10.1073/pnas.0606173104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cymes GD, Ni Y, Grosman C. Probing the structure and electrostatics of ion-channel pores one proton at a time. Nature. 2005;438:975–980. doi: 10.1038/nature04293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fitch CA, et al. Experimental pK(a) values of buried residues: Analysis with continuum methods and role of water penetration. Biophys J. 2002;82:3289–3304. doi: 10.1016/s0006-3495(02)75670-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Karp DA, et al. High apparent dielectric constant inside a protein reflects structural reorganization coupled to the ionization of an internal Asp. Biophys J. 2007;92:2041–2053. doi: 10.1529/biophysj.106.090266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harms MJ, et al. The pKa values of acidic and basic residues buried at the same internal location in a protein are governed by different factors. J Mol Biol. 2009;389:34–47. doi: 10.1016/j.jmb.2009.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cannon BR. Baltimore, MD: Johns Hopkins University; 2008. Thermodynamic consequences of substitutions of internal positions in proteins with polar and ionizable residues. PhD thesis. [Google Scholar]
- 40.Pace CN, Shaw KL. Linear extrapolation method of analyzing solvent denaturation curves. Proteins. 2000;41:1–7. doi: 10.1002/1097-0134(2000)41:4+<1::aid-prot10>3.3.co;2-u. [DOI] [PubMed] [Google Scholar]
- 41.Dwyer JJ, et al. High apparent dielectric constants in the interior of a protein reflect water penetration. Biophys J. 2000;79:1610–1620. doi: 10.1016/S0006-3495(00)76411-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Castañeda CA, et al. Molecular determinants of the pKa values of Asp and Glu residues in staphylococcal nuclease. Proteins. 2009;77:570–588. doi: 10.1002/prot.22470. [DOI] [PubMed] [Google Scholar]
- 43.Dunbrack RL, Cohen FE. Bayesian statistical analysis of protein side-chain rotamer preferences. Protein Sci. 1997;6:1661–1681. doi: 10.1002/pro.5560060807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Noszál B, Kassai-Tánczos R. Microscopic acid—Base equilibria of arginine. Talanta. 1991;38:1439–1444. doi: 10.1016/0039-9140(91)80293-9. [DOI] [PubMed] [Google Scholar]
- 45.Clarke ER, Martell AE. Metal chelates of arginine and related ligands. J Inorg Nucl Chem. 1970;32:911–926. [Google Scholar]
- 46.Hunter A, Borsook H. The dissociation constants of arginine. Biochem J. 1924;18:883–890. doi: 10.1042/bj0180883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mason PE, Neilson GW, Dempsey CE, Barnes AC, Cruickshank JM. The hydration structure of guanidinium and thiocyanate ions: Implications for protein stability in aqueous solution. Proc Natl Acad Sci USA. 2003;100:4557–4561. doi: 10.1073/pnas.0735920100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Waldburger CD, Schildbach JF, Sauer RT. Are buried salt bridges important for protein stability and conformational specificity? Nat Struct Mol Biol. 1995;2:122–128. doi: 10.1038/nsb0295-122. [DOI] [PubMed] [Google Scholar]
- 49.Tissot AC, Vuilleumier S, Fersht AR. Importance of two buried salt bridges in the stability and folding pathway of barnase. Biochemistry. 1996;35:6786–6794. doi: 10.1021/bi952930e. [DOI] [PubMed] [Google Scholar]
- 50.Wei Y, Horng J-C, Vendel AC, Raleigh DP, Lumb KJ. Contribution to stability and folding of a buried polar residue at the CARM1 methylation site of the KIX domain of CBP. Biochemistry. 2003;42:7044–7049. doi: 10.1021/bi0343976. [DOI] [PubMed] [Google Scholar]
- 51.Mrabet NT, et al. Arginine residues as stabilizing elements in proteins. Biochemistry. 1992;31:2239–2253. doi: 10.1021/bi00123a005. [DOI] [PubMed] [Google Scholar]
- 52.Borders CL, et al. A structural role for arginine in proteins: Multiple hydrogen bonds to backbone carbonyl oxygens. Protein Sci. 1994;3:541–548. doi: 10.1002/pro.5560030402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vondrášek J, Mason PE, Heyda J, Collins KD, Jungwirth P. The molecular origin of like-charge arginine-arginine pairing in water. J Phys Chem B. 2009;113:9041–9045. doi: 10.1021/jp902377q. [DOI] [PubMed] [Google Scholar]
- 54.Kubíčková A, et al. Guanidinium cations pair with positively charged arginine side chains in water. J Phys Chem Lett. 2011;2:1387–1389. [Google Scholar]
- 55.Nguyen DM, Leila Reynald R, Gittis AG, Lattman EE. X-ray and thermodynamic studies of staphylococcal nuclease variants I92E and I92K: Insights into polarity of the protein interior. J Mol Biol. 2004;341:565–574. doi: 10.1016/j.jmb.2004.05.066. [DOI] [PubMed] [Google Scholar]
- 56.Chimenti MS, et al. Structural consequences of the ionization of internal Lys residues in a protein. Biophys J. 2009;96:587a. [Google Scholar]
- 57.Harms MJ, et al. A buried lysine that titrates with a normal pKa: Role of conformational flexibility at the protein-water interface as a determinant of pKa values. Protein Sci. 2008;17:833–845. doi: 10.1110/ps.073397708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chimenti MS, Castañeda CA, Majumdar A, García-Moreno EB. Structural origins of high apparent dielectric constants experienced by ionizable groups in the hydrophobic core of a protein. J Mol Biol. 2011;405:361–377. doi: 10.1016/j.jmb.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Damjanović A, Brooks BR, García-Moreno EB. Conformational relaxation and water penetration coupled to ionization of internal groups in proteins. J Phys Chem A. 2011;115:4042–4053. doi: 10.1021/jp110373f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Freites JA, Tobias DJ, von Heijne G, White SH. Interface connections of a transmembrane voltage sensor. Proc Natl Acad Sci USA. 2005;102:15059–15064. doi: 10.1073/pnas.0507618102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rychkova A, Vicatos S, Warshel A. On the energetics of translocon-assisted insertion of charged transmembrane helices into membranes. Proc Natl Acad Sci USA. 2010;107:17598–17603. doi: 10.1073/pnas.1012207107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yamaguchi S, et al. Low-barrier hydrogen bond in photoactive yellow protein. Proc Natl Acad Sci USA. 2009;106:440–444. doi: 10.1073/pnas.0811882106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.