

Abstract
The heat shock protein 70 kDa (Hsp70)/DnaJ/nucleotide exchange factor system assists in intracellular protein (re)folding. Using solution NMR, we obtained a three-dimensional structure for a 75-kDa Hsp70–DnaJ complex in the ADP state, loaded with substrate peptide. We establish that the J domain (residues 1–70) binds with its positively charged helix II to a negatively charged loop in the Hsp70 nucleotide-binding domain. The complex shows an unusual “tethered” binding mode which is stoichiometric and saturable, but which has a dynamic interface. The complex represents part of a triple complex of Hsp70 and DnaJ both bound to substrate protein. Mutagenesis data indicate that the interface is also of relevance for the interaction of Hsp70 and DnaJ in the ATP state. The solution complex is completely different from a crystal structure of a disulfide-linked complex of homologous proteins [Jiang, et al. (2007) Mol Cell 28:422–433].
Keywords: protein interactions, structural biology
The heat shock protein 70 kDa, heat shock protein 40 kDa, nucleotide exchange factor (Hsp70/Hsp40/NEF) system, is an essential chaperone system that facilitates the folding and refolding of proteins in healthy and stressed cells (1). The system is upregulated in tumors (2), is involved in Alzheimer’s disease (3), and is an emerging target for therapy of these diseases (3). In this work, we study the bacterial Hsp70/Hsp40/NEF system, which is called DnaK/DnaJ/GrpE. Because DnaK and DnaJ are, respectively, 68% and 54% homologous to their human counterparts Hsc70 and HDJ2, the bacterial system is generally viewed as a prototype for the human Hsp70 chaperone system.
DnaK is an allosteric protein, in which ATP binding at the nucleotide-binding domain (NBD) causes substrate release at the substrate-binding domain (SBD) with opening of the LID (residues 508–602) (1). Although structures for the individual domains and several truncations have long been known, only one structure for a near complete, not mutated Hsp70 is available to date: DnaK(1–605) of Escherichia coli in the presence of ADP, and the substrate peptide NRLLLTG (NR) (4). In this structure, the LID domain is docked to the SBD, but the SBD-LID unit moves rather unrestricted with respect to the NBD. The NBD and SBD of DnaK do interact in the ATP state (5, 6), but no structure for any Hsp70 in that state has been determined to date.
E. coli DnaJ contains an N-terminal 70-residue J domain, followed by an approximately 40-residue Gly/Phe-rich (GF) region, a Zn-Cys domain, a substrate-binding domain, and a dimerization domain. DnaJ binds to stretches of exposed hydrophobic residues in client proteins (7). The J domain alone has been shown to be sufficient to stimulate ATPase activity of DnaK (8), but cannot function in protein (re)folding assays. Mutations of a conserved 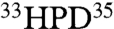 motif (His33, Pro34, Asp35), or K26 or R27 abolish DnaJ function (8), which identifies the J domain as the domain that recognizes DnaK.
motif (His33, Pro34, Asp35), or K26 or R27 abolish DnaJ function (8), which identifies the J domain as the domain that recognizes DnaK.
Residues 1–70 form an antiparallel two-helix bundle, referred to as helices II and III, with two small adjacent helical elements (9, 10). The positively charged residues referred to above are in helix II, whereas the HPD motif is located in a loop connecting helices II and III. The GF region (residues 71–108) is dynamic and disordered (10). A crystal structure (1NLT) is available for a YDJ1(110–337) dimer, a yeast protein homologous to DnaJ (11).
A common (1), but not undisputed (12) view of the DnaK–DnaJ protein refolding cycle is as follows. DnaJ binds to misfolded substrates. The J domain binds to DnaK and the substrate is transferred to the DnaK SBD. By the combined stimulation, DnaK hydrolyzes ATP, leading to a large-scale conformational change and the substrate becomes more tightly bound. An active unfolding of the substrate follows. The nucleotide exchange factor GrpE enhances the back-exchange of ATP, reversing the conformational change and reducing the affinity for the now unfolded substrate. The unfolded substrate is released and is free to refold.
Landry and coworkers (13) used NMR chemical shifts to map the binding site of DnaK on DnaJ(1–75). The chemical shift changes were centered on residues in helix II, but did not determine which residues on DnaK were involved in the interaction. Sousa and coworkers (14) covalently linked the NBD of human Hsc70 to the auxillin J domain. The apparent intermolecular interface in the crystal structure reflects the covalent linkage. As we will show, that interface does not exist in the natural complex in solution.
In the present work, we use NMR spectroscopy to determine the three-dimensional conformation of a noncovalent complex of the J domain [DnaJ(1–70)] with DnaK(1–605), both of E. coli. The intermolecular interface is composed of positive residues on helix II of DnaJ and negative residues in the segment 206–221 on DnaK. Mutations in helix II on DnaJ (15), and residues 208, 209, 217, and 218 on DnaK (ref. 16 and this work) interfere with the DnaK–DnaJ interactions, supporting the interface as determined by NMR. The complex in solution shows a very unusual tethered binding mode, which is stoichiometric, saturable, and dynamic.
Results and Discussion
Interface of DnaK on DnaJ.
Whether DnaJ’s GF region (residues 70–110) contributes to the interaction with DnaK or not (17, 18) has remained unsettled. We set out to resolve this issue by NMR. Fig. 1A shows, on the solution structure of the J domain, the chemical shift changes for the amide proton (NH) resonances of 15N-labeled DnaJ(1–108) upon titration with unlabeled DnaK(1–605) in the presence of ADP and in absence of substrate. The raw data in Fig. S1A show that the titration is in slow exchange. The binding is too tight to be quantified by NMR (KD < 1 μM). Many of the chemical shift changes are reversed (Fig. S1B) when adding excess peptide NR, which is known to bind to the DnaK substrate-binding cleft with a KD of 5 μM (19). Significantly, however, not all of the shift changes have disappeared upon adding the peptide; a subset of shifts centered on helix II remains (Fig. 1A).
Fig. 1.

Chemical shift perturbations in the 1H-15N TROSY heteronuclear single quantum correlation of DnaJ(1–70) upon titration with DnaK(1–605), in the presence of ADP. The chemical shift changes are depicted on PDB ID code 1XBL. Helix II is at the right, helix III at the left, the HPD-containing loop faces forward. Amide protons are represented as spheres. In all panels, green indicates no effect, gray, no information. (Left) In magenta and red, 1HN chemical shift changes > 0.006 ppm in the spectrum of DnaJ(1–108) upon addition of 2∶1 DnaK(1–605). In red, shifts remaining after addition of NR. (Center) DnaJ(1–70) in red, 1HN chemical shifts > 0.006 ppm and in orange, 0.004 < 1HN < 0.006; (Right) Reduction in signal height of the DnaJ(1–70) NH cross-peaks after adding 1.2∶1 DnaK(1–605)V210C-MTSL (in the presence of NR).
In Fig. 1B, we show the results of a titration of DnaJ(1–70) with DnaK(1–605). The same chemical shift change pattern occurs as for DnaJ(1–108) in the presence of NR (Fig. S2). We obtain a KD of 16 μM (Fig. S3). The data also correspond closely with the results obtained in ref. 13 for DnaJ(1–75) in the absence of substrate.
The NR competition experiment suggests that the DnaJ GF-rich region interacts with the substrate-binding cleft of DnaK. Our result reinforces and explains the results by Mayer et al. (20), who find that DnaJ fails to bind tightly to DnaK mutated in the substrate-binding cleft. We believe the GF-rich region provides a natural second site of a bivalent DnaJ–DnaK interaction in the initial encounter complex. In Fig. 2, we conceptualize the multivalency of the DnaK–DnaJ interaction. The DnaJ(1–108)–DnaK interaction is shown in Fig. 2B. Upon adding the substrate peptide, the interaction is represented by Fig. 2C.
Fig. 2.

Cartoons conceptualizing possible DnaK–DnaJ interactions. (A) DnaK (Hsp70) is on top, with NBD in red and SBD in blue. The LID domain is not shown. A DnaJ dimer is at the bottom, with the J domains in yellow, the GF regions in magenta, the SBDs in cyan, and the dimerization helices in brown. Ellipses represent constructs used in this study. (B) Bivalent interaction of a DnaJ monomer with DnaK. The area in the ellipse corresponds to the complex between DnaJ(1–108) and DnaK. (C) The trans-complex between DnaK and DnaJ with substrate peptide (NR). The area in the ellipse corresponds to the complex between DnaJ(1–108) and DnaK with NR. (D) The cis-triple complex between DnaK, DnaJ, and substrate protein. The area in the ellipse corresponds to the complex between DnaJ(1–70) and DnaK(1–605) with NR determined in this work (Fig. 4). (E) As in D, but in context of a hypothetical oligomeric complex involving DnaJ dimers.
Rudiger et al. (21) estimate that a Phe side chain binds to the DnaK cleft with a free energy of -1.17 kcal/mol (1 kcal = 4.18 kJ), which corresponds to a KD of 0.14 M. Hence a single interaction from the GF region would increase the binding affinity of DnaJ(1–108) by an order of magnitude as compared to DnaJ(1–70), bringing the interaction into the functionally relevant submicromolar affinity range (DnaK and DnaJ are in the 1-μM concentration range in E. coli (22).
However, the likely most important role of the GF-rich region is to allow flexibility in relative position of the J domain and DnaJ’s substrate-binding domain which may facilitate substrate transfer from the DnaJ SBD to the DnaK SBD (20).
There is a small, but very significant, difference between the data of Landry and coworkers (13, 23) and ours: They also observed chemical shift changes for residue Asp35 of the conserved HPD loop. However, we found that shifts for the NH signal of Asp35 can also be caused by very small changes in ionic strength. In order to exclude such effects during the titration, we used proteins that were extensively dialyzed against buffer in the same vessel. These titrations showed no chemical shift changes for residues Asp35 (and His33) of the HPD loop (Fig. 1B) (also Fig. S4).
Gross and coworkers (24) showed by surface plasmon resonance that the mutant proteins DnaJ–D35N and DnaK–R169A do not interact with their wild-type counterparts, but interact with each other almost as well as the wild-type proteins themselves. This result suggested that DnaJ–D35 and DnaK–R169 are in close proximity in the complex. Because the experiments were carried out in the presence of ATP, the conclusion formally holds only for that state. Nevertheless, such proximity has become a well-accepted paradigm for the interaction of DnaK and DnaJ in general.
We were therefore surprised to find that the NMR titration data indicate that the HPD motif is not involved in binding to DnaK in the ADP state. This observation is strongly supported by the fact that no paramagnetic relaxation enhancement (PRE) occurred in the NMR spectrum of DnaJ(1–70) complexed with DnaK-K166C-[S-(2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)methyl methanesulfonothioate] (MTSL), which contains the spin label at a position very close to R169. We conclude that the HPD loop is not within 15 Å of DnaK-R169 when DnaK is in the ADP state (see below). It appears that the HPD motif is exclusively involved in the interaction with the ATP state, although it could also have a purely structural role.
After these preliminary studies, we decided to study the interaction of the core J domain DnaJ(1–70) with full-length DnaK in the presence of NR to avoid oligomerization.
Tethered Binding.
The DnaJ(1–70) titration data saturate and yield a stoichiometry of 1∶1 and a KD of 16 μM (see Fig. S3). Remarkably, only little total 1H line broadening occurs in the process (Fig. 3), despite the fact that the nuclear spins in DnaJ(1–70) change from an 8-kDa to a 75-kDa environment upon complexation. Such a 10-fold change in molecular weight is expected to result in an approximately 10-fold increase in line width. We measured 15N spin relaxation rates for DnaJ(1–70) in the absence and presence of DnaK. 15N spin relaxation rates are widely used to obtain the rotational correlation time of proteins which, in turn, is linearly related to the molecular weight (25). Furthermore, 15N spin relaxation rates permit insight in local motions which are usually parametrized (26) by a local correlation time and an order parameter S2. For DnaJ(1–70) in the absence of DnaK, we obtain a molecular rotational correlation time of 5.2 ns (see SI Text for methods and Table S1). In the presence of DnaK, we obtain a rotational correlation time of 8.0 ns. This small value of the correlation time for bound DnaJ is surprising, knowing that the rotational correlation time for the NBD of DnaK-ADP-NR by itself is 28 ns (4). The 15N relaxation data of bound DnaJ(1–70) can be fitted in a more meaningful way with a dynamic model in which DnaJ is part of a larger complex with a 28 ns overall correlation time. In this fit, we obtain a local correlation time of 3.8 ns for the J domain and a maximum order parameter of S2 = 0.37. If the motion of DnaJ(1–70) with respect to DnaK can be described as tethered to one point and moving around in a cone with respect to that point, one calculates from S2 = 0.37 a value of 45° for the one-half opening angle of the cone. Below, we will find that the motion of DnaJ with respect to DnaK is likely more complex: DnaJ also scans the surface of part of DnaK. The translational component of this motion is not detected by NMR relaxation. We conclude that DnaJ(1–70) moves around quite freely while it is bound to DnaK. We call the phenomenon tethered binding, in which the two noncovalently bound proteins retain considerable relative mobility though a dynamic interface, likely through electrostatic interactions of the long side chains of positive Lys and Arg residues on DnaJ with negative Glu and Asp residues on DnaK (see below).
Fig. 3.

Change of the average of the amide proton line width in the 15N1H TROSY heteronuclear single quantum correlation spectrum of DnaJ(1–70) as a function of the addition of DnaK(1–605) (●). The thin line represents the function R2 = ffree × R2free + fbound × R2bound, where ffree and fbound were calculated for KD = 16 μM for the protein concentrations in the experiment. The heavy line is a fit with the same KD but allowing for chemical exchange broadening due to a koff of 14 s-1. (see SI Text)
Definition of the DnaK–DnaJ Complex.
In this work, we use DnaK(1–605), which has the same solution structure as the wild-type protein (4). Initially, we attempted to obtain the binding site of DnaJ on DnaK by chemical shift mapping. However, the chemical shift changes are too small to interpret. Hence, we mutated and spin-labeled DnaJ with MTSL to determine the PRE on the 1H-15N transverse relaxation-optimized spectroscopy (TROSY) spectrum of DnaK. The PRE is due to the dipolar interaction of the unpaired electron spin on the label with the nuclear spins on the protein (27). MTSL causes quantifiable line broadening for NMR nuclei in the range of 15–25 Å of the spin label, whereas the resonances of nuclei within 15 Å are broadened beyond detection (28).
The first choice was to spin label DnaJ residue M30. M30 is located between the functionally important residues on helix II and the 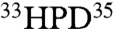 motif. M30 can be mutated to Ala without affecting functionality (15). Fig. 4A shows the effect of DnaJ(1–70)M30C-MTSL on DnaK in the presence of ADP and NRLLLTG. The structure shown in this figure is the end result of the present study and was computed from combined PRE distance constraints (see below).
motif. M30 can be mutated to Ala without affecting functionality (15). Fig. 4A shows the effect of DnaJ(1–70)M30C-MTSL on DnaK in the presence of ADP and NRLLLTG. The structure shown in this figure is the end result of the present study and was computed from combined PRE distance constraints (see below).
Fig. 4.

The average position of DnaJ(1–70) with respect to DnaK(1–605) in the presence of ADP and NR as obtained from a molecular dynamics simulation constrained by PRE distance constraints. DnaK NBD is in yellow, DnaK SBD in cyan, and DnaJ(1–70) in white. (A) Location of spin labels as discussed in the text. DnaJ M30C-MTSL (blue) affects the HN resonances on DnaK shown in red. DnaJ R19C-MTSL (green) and K41C-MTSL (green) had no effect. DnaK V210C-MTSL (red), D326C-MTSL (orange), and T417C-MTSL (orange) affect, to different extents, the resonances of the residues on DnaJ indicated in blue. D148C-MTSL, R166C-MTSL, and K421C-MTSL (all in green) had no effect. (B) A superposition of 64 MD snapshots, 0.5-ps apart, showing NBD (yellow) and DnaJ (white) only. Note that the NMR relaxation data show that DnaJ(1–70) is dynamically tethered to DnaK with S2 = 0.37, so each of the J positions is a possible dynamic average. (C) Location of functional residues, discussed in the text, in the complex (color coding as in A).
A contiguous swatch comprising DnaK residues 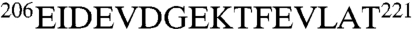 is broadened away by DnaJ(1–70)M30C-MTSL. This result precisely determines the location of the DnaJ binding site on DnaK. Next, we carried out the reciprocal experiment and spin-labeled DnaK on position 210. DnaK(1–605)V210C-MTSL strongly affects the resonances of DnaJ residues 23–33 (in the presence of ADP and NR) (see Figs. 1C and 5A). Significantly, the residues broadened on DnaJ by the spin label on DnaK correspond closely to those that show chemical shift changes without the spin label present (compare Fig. 1 B and C). Next, we spin labeled DnaJ mutations on residues R19 and K41, which are known to tolerate mutation to Ala (15). R19C-MTSL and K41C-MTSL yielded only very small effects in the spectrum of DnaK. We conclude that these DnaJ residues are on average not close to DnaK. The DnaK mutations D326C-MTSL and T417C-MTSL did affect the spectrum of DnaJ(1–70) (see Fig. 5B). The broadening pattern resembles that of V210C-MTSL, only with less intensity.
is broadened away by DnaJ(1–70)M30C-MTSL. This result precisely determines the location of the DnaJ binding site on DnaK. Next, we carried out the reciprocal experiment and spin-labeled DnaK on position 210. DnaK(1–605)V210C-MTSL strongly affects the resonances of DnaJ residues 23–33 (in the presence of ADP and NR) (see Figs. 1C and 5A). Significantly, the residues broadened on DnaJ by the spin label on DnaK correspond closely to those that show chemical shift changes without the spin label present (compare Fig. 1 B and C). Next, we spin labeled DnaJ mutations on residues R19 and K41, which are known to tolerate mutation to Ala (15). R19C-MTSL and K41C-MTSL yielded only very small effects in the spectrum of DnaK. We conclude that these DnaJ residues are on average not close to DnaK. The DnaK mutations D326C-MTSL and T417C-MTSL did affect the spectrum of DnaJ(1–70) (see Fig. 5B). The broadening pattern resembles that of V210C-MTSL, only with less intensity.
Fig. 5.

Reduction in signal height of the DnaJ(1–70) NH TROSY heteronuclear single quantum correlation cross-peaks due to the presence of spin-labeled DnaK. (Top) DnaK(1–605)V210C-MTSL and NR (DnaJ∶DnaK ratio: blue, 1∶0; green, 1∶0.5; red 1∶1.2). (Middle) As above, but with DnaK(1–605)D326C-MTSL and NR (DnaJ∶DnaK ratio: blue, 1∶0; red 1∶1.2). (Bottom) With DnaK(1–605)K421C-MTSL and NR (DnaJ∶DnaK ratio: blue, 1∶0; red 1∶1.2)
Initially we were mystified by this result because we expected that spin labels at different positions on DnaK would affect different residues on DnaJ. However, we then realized that DnaJ(1–70) is very dynamic when it is bound to DnaK, and that the paramagnetic broadening must also be dynamically averaged. The PRE results therefore indicate that DnaJ scans the surface of DnaK using residues K26 and R27 on helix II. In the scanning process, the resonances of this helix are broadened by any DnaK spin label that is located anywhere on the dynamical interface. DnaJ, when interacting with DnaK T417C-MTSL or DnaK D3626C-MTSL, still showed the same chemical shift changes on helix II as DnaJ interacting with DnaK without spin label. In addition, these (and all other spin-labeled mutants reported upon here) were unperturbed in the ATP-hydrolysis activity assays (Fig. S5). MTSL spin labels at DnaK positions K421C, N98C, D148, and K166 did not affect the NMR spectra of the bound DnaJ at all (K421C is shown in Fig. 5C). EPR control experiments verified that spin label was quantitatively present on the proteins. The proteins were still bound, given the familiar chemical shift changes in helix II. We hence conclude that K421C, N98C, D148, and K166 are not part of the DnaK–DnaJ dynamic interface.
The combined results demonstrate that the J domain dynamically scans a surface of DnaK that is centered on, but not restricted to, the sequence 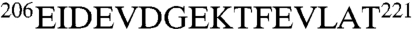 in NBD subdomain IIA. The region contains many negatively charged residues, which likely compensate the positive charges on helix II of DnaJ (also see Fig. S4). Aside from the selective broadening of resonances 206–221 by DnaJ(1–70) M30C-MTSL, there are no other major changes in the NMR spectrum of DnaK in complex with the J domain. These results strongly suggest that the J domain does not induce or mediate docking of the NBD and SBD.
in NBD subdomain IIA. The region contains many negatively charged residues, which likely compensate the positive charges on helix II of DnaJ (also see Fig. S4). Aside from the selective broadening of resonances 206–221 by DnaJ(1–70) M30C-MTSL, there are no other major changes in the NMR spectrum of DnaK in complex with the J domain. These results strongly suggest that the J domain does not induce or mediate docking of the NBD and SBD.
We encoded the positive and negative PRE data as distance constraints for a hybrid structure determination of the complex of DnaJ(1–70) with DnaK(1–605) (i.e., including the SBD-LID) using restrained molecular dynamics (MD). During all stages of the run, the local conformations of DnaK NBD, DnaK SBD-LID, and DnaJ, as obtained from available protein databank coordinates, were kept fixed with intramolecular CA–CA restraints. DnaK SBD-LID were constrained together, but not to the NBD. In the MD run, the SBD-LID domain moves around in a virtually unrestricted manner, corresponding to observations described above. Nevertheless, the presence of the SBD does restrict the DnaJ accessible space, leading to a well-defined average position of DnaJ with respect to DnaK’s NBD (see Fig. 4B).
The DnaJ binding epitope on DnaK contains several negatively charged residues that are conserved between eukaryotes and prokaryotes (see Table 1 and Table S2). The conserved electrostatic complementarity may account for the known cross-species functionality of the J domains (29) In support of this argument, it is known that DnaJ cannot stimulate ATP hydrolysis in DnaK EV(217,218)AA (16). Furthermore, DnaK DE(208,209)AA is only partially stimulated by DnaJ, whereas the ATP hydrolysis of DnaK DE(208,209)AA by itself is not affected (Fig. S6). Hence, we conclude that the acidic residues in the sequence before and after the nonconserved loop are part of the DnaJ interface on DnaK, supporting our structural findings.
Table 1.
Sequence alignment for the DnaJ–DnaK contact region of DnaK E. coli with human HspA8 (Hsc70)
| 207 | 208 | 209 | 210 | 211 | 212 | 213 | 214 | 215 | 216 | 217 | 218 | ||
| DnaK | I | D | E | — | V | D | G | E | K | T | F | E | V |
| HSPA8 | I | E | D | G | I | — | — | — | — | — | F | E | V |
E. coli count is at the top. Bold values indicate beta strands. See SI Text for a more complete allignment
The mutagenesis results also have another, important implication: Because the mutations reduce DnaJ stimulation of ATP hydrolysis, they must affect the interaction of DnaJ and DnaK in the ATP state. Hence, the negative loop 206–221 also plays a role in the DnaK–DnaJ interaction in the ATP state.
A cocrystal structure of the NBD of Hsc70 with ADP and with the J domain of auxillin has been published (14). It is not a physiologic complex because the two proteins were covalently linked by an artificial disulfide between the equivalent of Asp35 of DnaJ and the equivalent of R167 in DnaK. As expected, the crystal coordinates showed proximity for those areas of the Hsp70 and J proteins which are close to the cross-link. In the crystal structure (see Fig. 6), the positive charges of the J-domain’s helix II point away from the Hsp70, in contradiction with the mutagenesis results (15), with the NMR data of ref. 13, and with our present results.
Fig. 6.

The crystal structure of the covalent adduct (14) of Auxilin–J domain (orange) and the NBD of human Hsc70 (yellow) superposed on the solution conformation for the noncovalent complex of DnaJ (white), DnaK NBD (yellow), NBD-SBD linker (black), and DnaK SBD (cyan). Different functional residues as discussed in text are labeled. V210 is the center of the J-interaction interface on DnaK.
Relevance of the DnaJ–DnaK Complex in the ADP State.
We obtain a KD of approximately 16 µm for the binding of the isolated J(1–70) to DnaK in the ADP state. At first sight, this interaction is too weak to be of physiological relevance. However, as discussed above, DnaJ can interact in different ways with DnaK, with the J domain being only one of the different determinants (Fig. 2). Of particular physiological interest are the binding interfaces of the triple complex when DnaK and DnaJ are both bound to the same substrate (but at different sites) as illustrated in Fig. 2D. Such triple complexes have been shown to exist (12). The complex must be in the ADP state because both substrate and DnaJ stimulate DnaK’s ATP hydrolysis (1). The triple complex is expected to be tight. If DnaJ dimers are involved in the complexes with DnaK and substrate, the affinity would be higher still (Fig. 2E). We propose that the present solution complex of DnaJ(1–70) with DnaK in the ADP state, with substrate bound in trans, represents part of the triple or oligomeric complex in cis shown in Fig. 2 D or E, respectively. Indeed, our particular complex would dissociate at physiological conditions. A noncovalent complex of homologous protein domains could not be crystallized (14). Fortunately, the noncovalent complex can still be captured by NMR.
Conclusions.
The interaction of DnaJ and DnaK is multivalent. Both the J domain and GF domain interact with DnaK: the latter with the substrate-binding cleft of the SBD. In the ADP state, the J domain interacts predominantly with the DnaK NBD through a highly dynamical interface involving electrostatic interactions between conserved residues on DnaJ helix II and DnaK loop 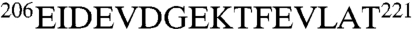 . The KD for this valency is approximately 16 µM. The interaction does not perturb the relative dynamic properties of the NBD and SBD typical of the ADP state. The dynamical interface between DnaK and DnaJ, in addition to the dynamical interface between DnaK’s NBD and SBD, may allow the Hsp70–DnaJ complex to carry out its demanding gymnastics to help unfold a client protein. The DnaJ HPD loop is not directly involved in the ADP-state interaction. This loop likely exclusively affects the ATP-state interaction and hence contributes to a third DnaJ binding valency.
. The KD for this valency is approximately 16 µM. The interaction does not perturb the relative dynamic properties of the NBD and SBD typical of the ADP state. The dynamical interface between DnaK and DnaJ, in addition to the dynamical interface between DnaK’s NBD and SBD, may allow the Hsp70–DnaJ complex to carry out its demanding gymnastics to help unfold a client protein. The DnaJ HPD loop is not directly involved in the ADP-state interaction. This loop likely exclusively affects the ATP-state interaction and hence contributes to a third DnaJ binding valency.
Materials and Methods
DnaK and DnaJ were expressed in BL21 using standard protocols (see SI Text). MTSL was procured from Toronto Research Chemicals. DnaJ or DnaK with the desired Cys mutation was loaded on a Ni-nitrilotriacetate column. After extensive washing, two- to threefold molar excess of MTSL dissolved in 3 mL DMSO was added. The column was capped and the contents were mixed. Subsequently, the column was washed with buffer to remove unbound MTSL. The MTSL-labeled protein was eluted and purified as described in the SI Text. MTSL labeling was confirmed by EPR using a Bruker ECS106 spectrometer.
NMR experiments were carried out at pH 7.4 and 30 °C using 100- to 500-μM samples in 25 mM Tris, 10 mM KCl, 20 mM KPi, 2 mM ADP, 0.02% NaN3, 3 mM EDTA, 50 μM PMSF, 5% D2O, and 2 mM NRLLLTG. The DnaJ assignments were redetermined using triple resonance and the program SAGA (30) using a Varian/Agilent NMRSYSTEM 800 MHz, equipped with a cold probe. The NMR assignments for DnaK(1–605) were taken from ref. 4. All NMR titration and PRE experiments were carried out using 15N-1H heteronuclear single quantum correlation TROSY. For each spin-labeling pair, three NMR experiments were carried out: MTSL labeled, reduced with ascorbic acid and reduced with DTT. The NMR spectra were processed with NMRPipe (31) and overlaid in Sparky (32).
Standard 15N R1 and R2 experiments were recorded at 800 MHz, using a 300-μM sample of DnaJ(1–70) in the absence or presence of 300 μM DnaK. The data were fitted using an in-house written grid-search program using a model-free spectral density function (33) (see SI Text).
In order to obtain a PRE distance calibration, we expressed the DnaJ-M30C-MTSL in 15N-labeled medium, and found that NH resonances within 15 Å of the spin label disappeared and that those between 15–20 Å were affected. The experiment was carried out at 12 °C to mimic the correlation time of DnaJ in the complex.
An average structure of the tethered complex between DnaK and DnaJ was obtained by using MD simulations and “binary” PRE restraints (see Table S3). A starting structure using the coordinates of DnaK(1–605) [Protein Data Bank (PDB) ID code 1KHO] and DnaJ(1–70) (PDB ID code 1XBL) was hand-assembled in SwissProt viewer. The MD calculation was carried out in Amber11 (34). The complex was minimized and heat ramped to 300 K in the presence of 2,353 CA–CA intradomain distance restraints with 1-Å precision based on the PDB files and a single intermolecular restraint, DnaJ Met30 CE to DnaK Val210 HN, set between 5.0 and 15.0 Å. In the 20-ps production MD run, we used 3,654 CA–CA intradomain distance restraints with 1-Å precision based on the heat-ramped structure, and 2,100 DnaK–DnaJ PRE restraints, most of which were repulsive (see Table S3). A representative snapshot selected of the MD run is shown in Fig. 4. The coordinates of 50 MD snapshots for the DnaJ–DnaK complex are given in the SI Text.
Supplementary Material
Acknowledgments.
We thank Dr. M. Berjanskii and Ms V. Semenchenko for pioneering work on the DnaK–DnaJ interaction in our lab. We thank members of the J. Gestwicki lab for assistance with the ATPase assays. This work was supported under National Institutes of Health Grants GM63027-S01 and S02.
Note.
Recently appeared work strongly suggests that DnaK residues 215–220 interact with the NBD–SBD linker in the ATP state but not in the ADP state (35). Likely, this interaction is crucial to the propagation of the allosteric signal from the NBD to the SBD (35, 36). It is significant that we find here that the J domain interacts with residues that immediately precede this site, suggesting that its presence interferes with the docking of the linker, promoting the ADP conformation. This result provides the long-awaited structural evidence for understanding how the J proteins regulate Hsp70 allostery.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1111220108/-/DCSupplemental.
References
- 1.Bukau B, Weissman J, Horwich A. Molecular chaperones and protein quality control. Cell. 2006;125:443–451. doi: 10.1016/j.cell.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 2.Rohde M, et al. Members of the heat-shock protein 70 family promote cancer cell growth by distinct mechanisms. Genes Dev. 2005;19:570–582. doi: 10.1101/gad.305405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Patury S, Miyata Y, Gestwicki JE. Pharmacological targeting of the Hsp70 chaperone. Curr Top Med Chem. 2009;9:1337–1351. doi: 10.2174/156802609789895674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bertelsen EB, Chang L, Gestwicki JE, Zuiderweg ERP. Solution conformation of wild-type E. coli Hsp70 (DnaK) chaperone complexed with ADP and substrate. Proc Natl Acad Sci USA. 2009;106:8471–8476. doi: 10.1073/pnas.0903503106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Swain JF, et al. Hsp70 chaperone ligands control domain association via an allosteric mechanism mediated by the interdomain linker. Mol Cell. 2007;26:27–39. doi: 10.1016/j.molcel.2007.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Swain JF, Schulz EG, Gierasch LM. Direct comparison of a stable isolated Hsp70 substrate-binding domain in the empty and substrate-bound states. J Biol Chem. 2006;281:1605–1611. doi: 10.1074/jbc.M509356200. [DOI] [PubMed] [Google Scholar]
- 7.Kelley WL. The J-domain family and the recruitment of chaperone power. Trends Biochem Sci. 1998;23:222–227. doi: 10.1016/s0968-0004(98)01215-8. [DOI] [PubMed] [Google Scholar]
- 8.Wall D, Zylicz M, Georgopoulos C. The NH2-terminal 108 amino acids of the Escherichia coli DnaJ protein stimulate the ATPase activity of DnaK and are sufficient for lambda replication. J Biol Chem. 1994;269:5446–5451. [PubMed] [Google Scholar]
- 9.Hill RB, Flanagan JM, Prestegard JH. 1H and 15N magnetic resonance assignments, secondary structure, and tertiary fold of Escherichia coli DnaJ(1–78) Biochemistry. 1995;34:5587–5596. doi: 10.1021/bi00016a033. [DOI] [PubMed] [Google Scholar]
- 10.Pellecchia M, Szyperski T, Wall D, Georgopoulos C, Wuthrich K. NMR structure of the J-domain and the Gly/Phe-rich region of the Escherichia coli DnaJ chaperone. J Mol Biol. 1996;260:236–250. doi: 10.1006/jmbi.1996.0395. [DOI] [PubMed] [Google Scholar]
- 11.Li JZ, Oian XG, Sha B. The crystal structure of the yeast Hsp40 Ydj1 complexed with its peptide substrate. Structure. 2003;11:1475–1483. doi: 10.1016/j.str.2003.10.012. [DOI] [PubMed] [Google Scholar]
- 12.Han WJ, Christen P. Mechanism of the targeting action of DnaJ in the DnaK molecular chaperone system. J Biol Chem. 2003;278:19038–19043. doi: 10.1074/jbc.M300756200. [DOI] [PubMed] [Google Scholar]
- 13.Greene MK, Maskos K, Landry SJ. Role of the J-domain in the cooperation of Hsp40 with Hsp70. Proc Natl Acad Sci USA. 1998;95:6108–6113. doi: 10.1073/pnas.95.11.6108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang J, et al. Structural basis of J cochaperone binding and regulation of Hsp70. Mol Cell. 2007;28:422–433. doi: 10.1016/j.molcel.2007.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Genevaux P, Fau-Schwager F, Fau K, Georgopoulos C, Kelley W. Scanning mutagenesis identifies amino acid residues essential for the in vivo activity of the Escherichia coli DnaJ (Hsp40) J-domain. Genetics. 2002;162:1045–1053. doi: 10.1093/genetics/162.3.1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gassler CS, et al. Mutations in the DnaK chaperone affecting interaction with the DnaJ cochaperone. Proc Natl Acad Sci USA. 1998;95:15229–15234. doi: 10.1073/pnas.95.26.15229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Walsh P, Bursac D, Law YC, Cyr D, Lithgow T. The J-protein family: Modulating protein assembly, disassembly and translocation. EMBO Rep. 2004;5:567–571. doi: 10.1038/sj.embor.7400172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suh WC, Lu CZ, Gross CA. Structural features required for the interaction of the Hsp70 molecular chaperone DnaK with its cochaperone DnaJ. J Biol Chem. 1999;274:30534–30539. doi: 10.1074/jbc.274.43.30534. [DOI] [PubMed] [Google Scholar]
- 19.Zhu XT, et al. Structural analysis of substrate binding by the molecular chaperone DnaK. Science. 1996;272:1606–1614. doi: 10.1126/science.272.5268.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mayer MP, Laufen T, Paal K, McCarty JS, Bukau B. Investigation of the interaction between DnaK and DnaJ by surface plasmon resonance spectroscopy. J Mol Biol. 1999;289:1131–1144. doi: 10.1006/jmbi.1999.2844. [DOI] [PubMed] [Google Scholar]
- 21.Rudiger S, Germeroth L, Schneider-Mergener J, Bukau B. Substrate specificity of the DnaK chaperone determined by screening cellulose-bound peptide libraries. EMBO J. 1997;16:1501–1507. doi: 10.1093/emboj/16.7.1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bardwell JC, et al. The nucleotide sequence of the Escherichia coli K12 dnaJ+ gene. A gene that encodes a heat shock protein. J Biol Chem. 1986;261:1782–1785. [PubMed] [Google Scholar]
- 23.Horne BE, Li TF, Genevaux P, Georgopoulos C, Landry SJ. The Hsp40 J-domain stimulates Hsp70 when tethered by the client to the ATPase domain. J Biol Chem. 2010;285:21679–21688. doi: 10.1074/jbc.M110.113118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suh WC, et al. Interaction of the Hsp70 molecular chaperone, DnaK, with its cochaperone DnaJ. Proc Natl Acad Sci USA. 1998;95:15223–15228. doi: 10.1073/pnas.95.26.15223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tanford C. Physical Chemistry of Macromolecules. New York: Wiley; 1963. [Google Scholar]
- 26.Lipari G, Szabo A. Model-free approach to the interpretation of nuclear magnetic-resonance relaxation in macromolecules. 1. Theory and range of validity. J Am Chem Soc. 1982;104:4546–4559. [Google Scholar]
- 27.Campbell ID, Dwek RA, Price NC, Radda GK. Studies on the interaction of ligands with phosphorylase b using a spin-label probe. Eur J Biochem. 1972;30:339–347. doi: 10.1111/j.1432-1033.1972.tb02103.x. [DOI] [PubMed] [Google Scholar]
- 28.Battiste JL, Wagner G. Utilization of site-directed spin labeling and high-resolution heteronuclear nuclear magnetic resonance for global fold determination of large proteins with limited nuclear overhauser effect data. Biochemistry. 2000;39:5355–5365. doi: 10.1021/bi000060h. [DOI] [PubMed] [Google Scholar]
- 29.Kampinga HH, Craig EA. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol. 2010;11:579–592. doi: 10.1038/nrm2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crippen GM, Rousaki A, Revington M, Zhang YB, Zuiderweg ERP. SAGA: Rapid automatic mainchain NMR assignment for large proteins. J Biomol NMR. 2010;46:281–298. doi: 10.1007/s10858-010-9403-2. [DOI] [PubMed] [Google Scholar]
- 31.Delaglio F, et al. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 32.Goddard TD, Kneller DG. SPARKY 3. San Francisco: Univ of California; 2000. [Google Scholar]
- 33.Lipari G, Szabo A. Model-free approach to the interpretation of nuclear magnetic-resonance relaxation in macromolecules. 2. Analysis of experimental results. J Am Chem Soc. 1982;104:4559–4570. [Google Scholar]
- 34.Case DA, et al. The amber biomolecular simulation programs. J Comput Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhuravleva A, Gierasch LM. Allosteric signal transmission in the nucleotide-binding domain of 70-kDa heat shock protein (Hsp70) molecular chaperones. Proc Natl Acad Sci USA. 2011;108:6987–6992. doi: 10.1073/pnas.1014448108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu Q, Hendrickson WA. Insights into hsp70 chaperone activity from a crystal structure of the yeast Hsp110 Sse1. Cell. 2007;131:106–120. doi: 10.1016/j.cell.2007.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.