

Abstract
Vascularization plays a key role in processes such as wound healing and tissue engineering. Non-thermal plasma, which primarily produces reactive oxygen species (ROS), has recently emerged as an efficient tool in medical applications including blood coagulation, sterilization and malignant cell apoptosis. Liquids and porcine aortic endothelial cells were treated with a non-thermal dielectric barrier discharge plasma in vitro. Plasma treatment of phosphate-buffered saline (PBS) and serum-free medium increased ROS concentration in a dose-dependent manner, with a higher concentration observed in serum-free medium compared with PBS. Species concentration inside cells peaked 1 h after treatment, followed by a decrease 3 h post treatment. Endothelial cells treated with a plasma dose of 4.2 J cm–2 had 1.7 times more cells than untreated samples 5 days after plasma treatment. The 4.2 J cm–2 plasma dose increased two-dimensional migration distance by 40 per cent compared with untreated control, while the number of cells that migrated through a three-dimensional collagen gel increased by 15 per cent. Tube formation was also enhanced by plasma treatment, with tube lengths in plasma-treated samples measuring 2.6 times longer than control samples. A fibroblast growth factor-2 (FGF-2) neutralizing antibody and ROS scavengers abrogated these angiogenic effects. These data indicate that plasma enhanced proliferation, migration and tube formation is due to FGF-2 release induced by plasma-produced ROS. Non-thermal plasma may be used as a potential tool for applying ROS in precise doses to enhance vascularization.
Keywords: vascularization, wound healing, angiogenesis, non-thermal plasma, reactive oxygen species, fibroblast growth factor-2 release
1. Introduction
Angiogenesis, the growth of new blood vessels from existing vessels, plays a key role in physiological processes including development, growth and wound healing [1]. Insufficient vascularization contributes to impaired wound healing in patients with diabetes [2] and systemic sclerosis [3], the elderly [4], and patients treated with immunosuppressive drugs [5], chemotherapy [6] and radiotherapy [7]. In vitro, vascularization is essential for engineering complex, large tissues and organs such as bone, muscle, liver and heart. Although successful tissue fabrication of a few hundred micrometres has been achieved, tissue engineering is limited by the inability to vascularize constructs to provide nutrients to the tissue core [8–11].
Over the past few decades, many growth factors that stimulate angiogenesis have been discovered, including vascular endothelial growth factor (VEGF) [12,13] and fibroblast growth factor-2 (FGF-2) [14,15]. These growth factors are the standard by which other angiogenic therapies are measured in either in vivo assays (e.g. Matrigel plug, corneal implant) [16–19] or in vitro assays (e.g. cell proliferation, Boyden chamber migration, collagen gel tube formation) [20–24]. While these growth factors are critical to the angiogenic process, their success depends on timing, dose and gradients [25–28]. Efforts to apply growth factors in wounds or in tissue-engineered constructs to induce angiogenesis have met with limited success [28–30].
FGF-2, a pro-angiogenic molecule that binds heparan sulphate proteoglycans as well as cell surface receptors, is associated with cell survival, proliferation and migration [31–33]. FGF-2 is particularly interesting because it does not have a recognized signal sequence for secretion. FGF-2 is known to be released by injured cells, whether the injury is by ionizing radiation, pulsed electromagnetic field, mechanical forces or elevated glucose [34–38]. FGF-2 released during cell injury promotes cell survival and also increases FGF-2 expression in endothelial cells, smooth muscle cells and cardiac myocytes [39–43]. Thus, cells that have suffered an injury prepare themselves against subsequent injuries by synthesizing additional FGF-2. FGF-2 may also induce expression of other angiogenic growth factors, such as VEGF, in endothelial cells [44].
Plasma medicine is a rapidly expanding interdisciplinary field combining engineering, physics, biochemistry and life sciences [45]. Plasma, the fourth state of matter, is an ionized gas composed of charged particles (electrons, ions), excited atoms and molecules, radicals, and UV photons. Man-made plasma is created with an electrical discharge and a large electric field. In non-thermal plasma, which is far from thermal equilibrium, electron temperature is much higher than heavy particle temperature. While high-temperature electrons interact with gas molecules to create reactive species, overall gas temperature remains close to room temperature and hence plasma can be applied directly to cells and tissue without measurable damage [46]. Non-thermal plasma has recently emerged as a novel technology for various medical applications such as blood coagulation [47–50], wound healing [51], tissue sterilization [48], medical equipment sterilization [52], dental cavity treatment [53], malignant cell apoptosis [54,55] and tissue-engineering scaffold treatment [56].
Non-thermal plasma devices, specifically dielectric barrier discharge (DBD) plasma, are used extensively in medicine because of their selectivity, portability, scalability, ease of operation, and low manufacturing and maintenance costs. DBD plasma is generated at atmospheric pressure in air when short duration, high-voltage pulses are applied between two electrodes, with one electrode being insulated to prevent a current increase [57,58]. DBD plasma characteristics in air, including electron density, reduced electric field, gas temperature and active species concentration, vary depending on applied voltage, dielectric material and interelectrode distance [58]. Non-thermal DBD plasma produces a variety of biologically active reactive species, in particular, reactive oxygen species (ROS) [58].
ROS, including hydrogen peroxide (H2O2), superoxide (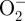 ), hydroxyl radicals (OH·) and singlet oxygen (O2(1Δg)), are highly reactive ions or molecules that contribute to angiogenic signalling [59]. While in an early wound high ROS levels mediate inflammation [60] and may inhibit angiogenesis, later low ROS levels initiate wound healing partially through angiogenesis [61–63]. Low ROS doses may increase endothelial cell migration, proliferation and tube formation in vitro and in vivo through growth factor-related mechanisms. ROS can enhance production of pro-angiogenic factors such as VEGF and FGF-2 [62,64–66], as well as other mitogens such as insulin-like growth factor-1 (IGF-1) [67] and transforming growth factor-α [68]. Low ROS doses may enhance growth factor binding to receptors, induce receptor tyrosine kinase phosphorylation, or act as messengers in downstream signalling along growth factor pathways [59,69–72]. Finally, ROS can damage the cell membrane leading to FGF-2 release, which then has proliferative and survival effects on adjacent cells [37,73].
), hydroxyl radicals (OH·) and singlet oxygen (O2(1Δg)), are highly reactive ions or molecules that contribute to angiogenic signalling [59]. While in an early wound high ROS levels mediate inflammation [60] and may inhibit angiogenesis, later low ROS levels initiate wound healing partially through angiogenesis [61–63]. Low ROS doses may increase endothelial cell migration, proliferation and tube formation in vitro and in vivo through growth factor-related mechanisms. ROS can enhance production of pro-angiogenic factors such as VEGF and FGF-2 [62,64–66], as well as other mitogens such as insulin-like growth factor-1 (IGF-1) [67] and transforming growth factor-α [68]. Low ROS doses may enhance growth factor binding to receptors, induce receptor tyrosine kinase phosphorylation, or act as messengers in downstream signalling along growth factor pathways [59,69–72]. Finally, ROS can damage the cell membrane leading to FGF-2 release, which then has proliferative and survival effects on adjacent cells [37,73].
Our group previously demonstrated that a non-thermal DBD plasma promoted endothelial cell proliferation in vitro through FGF-2 release [74]. However, the mechanism for this effect was unknown. We hypothesized that FGF-2 release is related to plasma-induced ROS, and that the combination of ROS and FGF-2 further induce endothelial cells to create new blood vessels. In the current work, we measured plasma-produced ROS in liquid and cells over time, and then quantified plasma ROS and cell-released FGF-2 effects on endothelial cell proliferation, migration and tube formation. This study will help identify how plasma can be used to promote vascularization for wound healing and tissue engineering.
2. Experimental procedure
2.1. Cell culture and materials
Porcine aortic endothelial cells (PAEC) were isolated from swine aorta by the collagenase dispersion method [75]. PAEC were selected because they do not require growth factor supplementation for survival, and there is high cross reactivity between human and porcine growth factors [37,76]. Cells were cultured in low glucose Dulbecco's modified Eagle's medium (DMEM, Mediatech) supplemented with 5 per cent foetal bovine serum (FBS, Hyclone), 1 per cent penicillin–streptomycin and 1 per cent l-glutamine (Invitrogen). Cells between passages 4 and 9 were maintained in a humidified incubator at 37°C and 5 per cent CO2 with medium change every 2 days.
Recombinant human FGF-2 was from Peprotech, and the neutralizing FGF-2 antibody was from Upstate Biotechnology. An intracellular ROS scavenger, 10 mM N-acetyl cysteine (NAC, Sigma) and an extracellular ROS scavenger, 50 mM sodium pyruvate (SP, Sigma), were used to block plasma-produced ROS.
2.2. Plasma treatment
Non-thermal DBD plasma was generated by applying alternating polarity pulsed voltage (1.5 kHz, 20 kV peak-to-peak) between an insulated high-voltage electrode and the grounded base holding the cell sample (figure 1a,b) [74,77]. A variable voltage and current power supply custom made by Quinta Ltd. (Moscow, Russia) was used for treating liquids and cells. The power supply was connected to a 25.4 mm diameter copper electrode with a 1 mm thick quartz insulating dielectric covering its bottom surface. The insulating dielectric prevented current flow between electrodes, creating plasma with high reactive species concentrations but minimal gas heating. The discharge gap between the quartz dielectric and the cell sample was fixed at 2 mm. Voltage and current were measured using a voltage (North Star PVM-4, 100 MHz bandwidth) and current probe (Pearsons Model 2878), respectively (figure 1c). Signals were recorded with an oscilloscope (TDS 5052B, Tektronix Inc.). The pulse waveform had a 1.65 µs width and a 5 V ns–1 rise time. Discharge power density was calculated to be 0.84 W cm–2 (1.5 kHz) using Matlab (Mathworks). The plasma treatment dose in Joules per square centimetre was calculated by multiplying plasma discharge power density with plasma treatment duration.
Figure 1.

Non-thermal DBD plasma (a) schematic, 1: high-voltage wire, 2: ultem insulation, 3: copper electrode core, 4: quartz (dielectric), 5: cell culture media, 6: grounded base, 7: endothelial cells, 8: plasma; (b) device; and (c) voltage and current waveforms. (Online version in colour.)
Cells in glass bottom dishes (14 mm, MatTek) or on 18 mm coverslips (VWR) were directly treated with plasma in the presence of 100 µl serum-free medium to prevent sample drying. Plasma was produced between the insulated high-voltage electrode and the grounded base holding the cell sample, which acted as the other electrode. Similar DBD plasmas have gas and surface temperatures of around 47°C [78,79]. To ensure that plasma-induced heating did not damage cells, we measured the temperature change in 200 µl phosphate-buffered saline (PBS) or serum-free medium post-plasma treatment using a thermistor probe and meter (YSI 427 and 43TA, Yellow Springs Instruments). The 4.2 J cm–2 plasma dose increased liquid temperature by less than 1°C. In PBS and serum-free medium that were warmed to 37°C before the experiment, liquid temperature never rose above 28.5°C even for a high plasma dose of 134.4 J cm–2, since a rapid drop in liquid temperature was observed when it was exposed to room temperature air. Additionally, no significant change in PBS and serum-free medium pH was observed up to 9.24 J cm–2 plasma.
2.3. Reactive oxygen species detection in liquid and cells
Since DBD plasma produces a variety of ROS [80], plasma ROS formed in liquid were quantified with 2′, 7′-dichlorodihydrofluorescein diacetate (DCFH-DA, Cayman chemicals), which fluoresces following reaction with ROS such as H2O2, OH·, O2 (1Δg) and ROO− [81,82]. The probe was activated by removing acetate groups using an alkaline solution [83]. Specifically, 1 mM DCFH-DA in ethanol was added to 0.01 N sodium hydroxide (NaOH, Sigma) for 30 min. The resultant DCFH solution was then neutralized with 25 mM sodium phosphate buffer (pH 7.2, Alfa Aesar) and stored on ice until use. Fresh DCFH was prepared for each experiment and used the same day. To measure plasma-produced ROS, 150 µl PBS or serum-free medium containing 20 µM DCFH was added to a glass bottom dish and plasma-treated (0–9.24 J cm−2). The 100 µl treated liquid was then transferred to a 96-well plate, and fluorescence was measured at ex/em: 488/525 nm using an Infinite 200 Tecan microplate reader. H2O2 was the positive control.
Intracellular ROS was assessed using the Image-iT LIVE Green ROS Detection Kit (Invitrogen). The assay is based on 5-(and 6)-carboxy-2′, 7′-dichlorodihydrofluorescein diacetate (carboxy-H2DCFDA), a fluorogenic marker for ROS in live cells [81,82]. Carboxy-H2DCFDA is internalized by cells and metabolized by intracellular esterases to carboxy-DCFH, which then reacts with ROS to form fluorescent carboxy-DCF. A total of PAEC (100 000 cells) were seeded in 50 mm glass bottom dishes and cultured for 2 days to confluence. Following plasma treatment (0–8.4 J cm–2), cells were labelled for ROS according to manufacturer's protocol. Briefly, cells were incubated with 25 µM carboxy-H2DCFDA in HEPES buffer (20 mM Hepes, 137 mM NaCl, 5 mM KCl, 1 mM KH2PO4, 2 mM CaCl2, 1 mM MgCl2 and 10 mM glucose, pH 7.4) for 30 min at 37°C. The tert-butylhydroperoxide (tBHP, Invitrogen), which induces intracellular ROS production, was the positive control. Samples were imaged using an Olympus IX81 inverted confocal microscope at ex/em: 488/520 nm.
2.4. Proliferation assay
Endothelial cell proliferation in response to plasma treatment was measured by cell counts. Ten thousand cells were seeded on 18 mm glass coverslips and incubated for 24 h at 37°C. Cells were treated with plasma (0–8.4 J cm–2, day 0) or 10 ng ml–1 FGF-2 (positive control) and returned to the incubator. Medium was changed on days 2 and 4. On days 1, 3 and 5 after plasma treatment, PAEC were trypsinized and counted using a Coulter counter. Cell proliferation was defined as fold change in attached cell number on day 5 compared with day 1.
2.5. Two-dimensional migration assay
Endothelial cell migration in response to plasma treatment was measured using a two-dimensional migration assay [84,85]. Thirty thousand cells were seeded inside 4 mm cloning rings on gridded cover slips. A 5 mm glass bead was used to maintain tight contact between the ring and the coverslip. After 3 h, the bead and ring were removed, and unattached cells were washed away. This process produced a confluent circle of attached PAEC. Cell samples were then treated with 4.2 J cm–2 plasma, since this dose showed maximal cell response in our previous work [74]. Untreated samples were the negative control, and FGF-2 (10 ng ml–1) was the positive control. Images were captured at 0, 24, 48 and 72 h after treatment in each direction (0, 90, 180 and 270°) using a Nikon Eclipse TS100 phase contrast microscope with a Nikon DS-Fi1 CCD camera. Migration distance, defined as the difference between cell migration fronts at 0 and 72 h, was analysed using ImageJ. After 72 h, cells were trypsinized and counted using a Coulter counter to determine cell number.
2.6. Modified Boyden chamber three-dimensional migration assay
Three-dimensional migration was quantified using the QCM cell invasion assay kit (Millipore). In this assay, cells migrate through a collagen-coated Transwell membrane with 8 µm pores and attach to the membrane bottom surface. Migrated cells are then fluorescently quantified by staining DNA following cell lysis. PAEC (1 × 105 cells) in 100 µl serum-free medium were plasma-treated (4.2 J cm–2) and then added to the chamber upper compartment. Cells were plasma-treated prior to being added to the chamber because the DBD plasma electrode was too large for direct cell treatment in the chamber. One hundred and fifty microlitres DMEM with 10 per cent FBS was added to the lower compartment. FGF-2 (10 ng ml–1) was used as positive control and cells without plasma treatment as negative control. Samples were incubated at 37°C for 24 h. Cells that migrated through the membrane were quantified according to manufacturer's protocol. Briefly, residual cells from the upper compartment were removed using a cotton swab. Cells on the membrane bottom surface were removed by incubating inserts in cell detachment solution for 30 min at 37°C. Detached cells were incubated in lysis buffer/CyQuant dye solution for 15 min to stain DNA. One hundred and fifty microlitres cell lysis solution was then transferred to a 96-well plate and fluorescence was measured at ex/em: 480/520 nm using a microplate reader. Fluorescence was converted to cell number using a standard curve.
2.7. Tube formation assay
An in vitro tube formation assay was performed as described previously [86]. Briefly, 4 mg ml–1 rat tail type I collagen (BD Biosciences) was mixed with plasma-treated PAEC (4.2 J cm–2, 3 × 105 cells) in serum-free medium. Cell–collagen mixture (200 µl) was immediately added to a 24-well plate and incubated for 1 h at 37°C. Supplemented medium was then added, and medium was replaced every day without disturbing tubes. Phase contrast microscopy images were taken at 0, 24, 48 and 72 h. Tube formation was quantified using ImageJ [87]. Tube formation was assessed by measuring tube length from a branch point or between branch points. Eight tube lengths were measured in each image, with three images per treatment condition.
2.8. Statistical analysis
Statistical, linear and nonlinear regression analyses were performed with Prism software (Graphpad). All experiments were performed in triplicate and repeated at least three times. Data are expressed as mean ± s.d. Comparisons between two groups were analysed using Student's t-test, and comparisons between more than two groups were analysed by ANOVA.
3. Results
3.1. Reactive oxygen species increased in a dose-dependent manner in liquid and cells following plasma treatment
Previous plasma studies theoretically calculated that a plasma dose of 1 J cm–2 produced 1.88–3.13 × 1016 ROS in gas phase [74]. We now measured plasma-produced ROS in the liquid phase. In both plasma-treated PBS and serum-free medium, ROS concentration increased with plasma dose. ROS levels increased rapidly at low treatment doses and more slowly at higher treatment doses, probably owing to ROS reactions with air and liquid components. ROS concentration was consistently 25–35% higher in plasma-treated serum-free medium compared with PBS (figure 2a). Data were fit to an exponential function by nonlinear regression.
Figure 2.

ROS were produced by DBD plasma in liquid and cells. (a) Cumulative extracellular ROS in PBS (circles) and serum-free (inverted triangles) medium detected using DCFH (ex/em: 488/525 nm). p < 0.001 compared with control for both PBS and serum-free medium, and p < 0.0001 between PBS and serum-free medium. (b) Intracellular ROS were measured using carboxy-H2DCFDA (ex/em: 488/520 nm). PAEC were incubated with carboxy-H2DCFDA for 30 min at 37°C and then treated with plasma or positive control tert-butyl hydroperoxide (100 µM, tBHP). (c) Intracellular ROS levels for 3 h following DBD plasma treatment. (d) Sample ROS images taken by confocal microscopy and quantified using ImageJ. †p < 0.001 compared with untreated control. (Online version in colour.)
Plasma-produced ROS, as well as products from plasma ROS interaction with serum-free medium, either diffuse through the cell membrane into the cytosol or react with the cell membrane to produce intracellular ROS. We therefore quantified intracellular ROS using carboxy-H2DCFDA. Plasma treatment induced a dose-dependent increase in intracellular oxidative stress immediately after treatment (figure 2b). A plasma dose of 4.2 J cm–2 increased intracellular ROS 14 per cent compared with untreated cells, and 8.4 J cm–2 plasma produced an 8 per cent further increase. Cells incubated with positive control tert-butyl hydroperoxide (100 µM) showed 1.5 times more ROS than untreated cells. Intracellular ROS over time following plasma treatment was then quantified for 4.2 J cm–2 plasma. Plasma-induced intracellular ROS peaked by 1 h after treatment and then returned to near baseline levels by 3 h after treatment (figure 2c,d).
3.2. Plasma enhanced endothelial cell proliferation, migration and tube formation through reactive oxygen species-induced fibroblast growth factor-2 release
We previously demonstrated that plasma increased endothelial cell proliferation through FGF-2 release [74]. We now show that plasma-produced ROS are critical to enhanced cell proliferation. Cell proliferation increased in a dose-dependent manner up to 4.2 J cm–2 plasma and then decreased at higher plasma doses (figure 3). Cells treated with 4.2 J cm–2 plasma on day 1 had 10 times as many cells on day 5, whereas control cells had only six times as many cells. Cells incubated with a 10 ng ml–1 FGF-2 bolus on day 1 had 12 times as many cells on day 5. An FGF-2 neutralizing antibody, the intracellular ROS scavenger N-acetyl cysteine, and the extracellular ROS scavenger sodium pyruvate abrogated the plasma-induced cell proliferation enhancement. Data for FGF-2 Ab, NAC and SP were fit to a linear function by linear regression.
Figure 3.
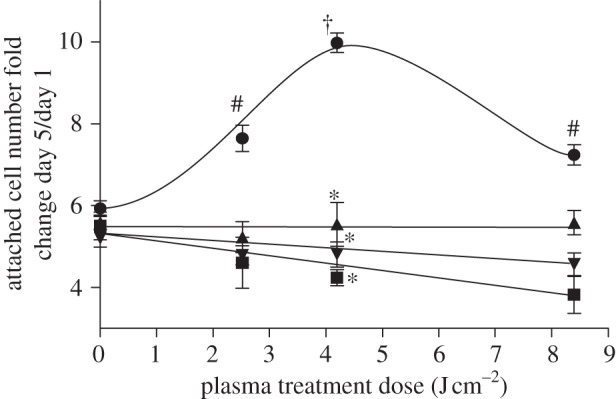
DBD plasma treatment enhanced endothelial cell proliferation through ROS-induced FGF-2 release. PAEC were treated with plasma doses of 0–8.4 J cm–2 and counted 1, 3 and 5 days after plasma treatment. Cells were treated with an FGF-2 neutralizing antibody to block FGF-2 effects, and 10 mM N-acetyl cysteine (NAC) or 50 mM sodium pyruvate (SP) to block intracellular and extracellular ROS, respectively. Data are represented as fold change in cell number on day 5 compared with day 1. †p < 0.001 compared with control. *p < 0.0001 compared with plasma. #p < 0.05 compared with control. Circles, plasma; squares, +FGF Ab; triangles, +NAC; inverted triangles, +SP.
Since maximum cell proliferation was observed at 4.2 J cm–2 plasma, two-dimensional cell migration was measured at this dose. Endothelial two-dimensional migration increased with plasma as early as 24 h after treatment compared with untreated control (390 versus 190 µm), and the difference was maintained up to 72 h after plasma treatment (902 versus 624 µm). Plasma-induced cell migration distance at 72 h was less than positive control FGF-2 (1709 µM). To elucidate whether enhanced cell migration was attributed to ROS-induced FGF-2 release, cells were plasma-treated in the presence of an FGF-2 neutralizing antibody, N-acetyl cysteine or sodium pyruvate. FGF-2 blockade decreased PAEC migration distance by 23 per cent (figure 4a). Similarly, both N-acetyl cysteine and sodium pyruvate inhibited plasma-induced cell migration distance by 14 and 22 per cent, respectively (figure 4b). No significant difference in cell number was observed between control and plasma-treated samples at 72 h, suggesting that these differences are related to cell migration rather than proliferation.
Figure 4.
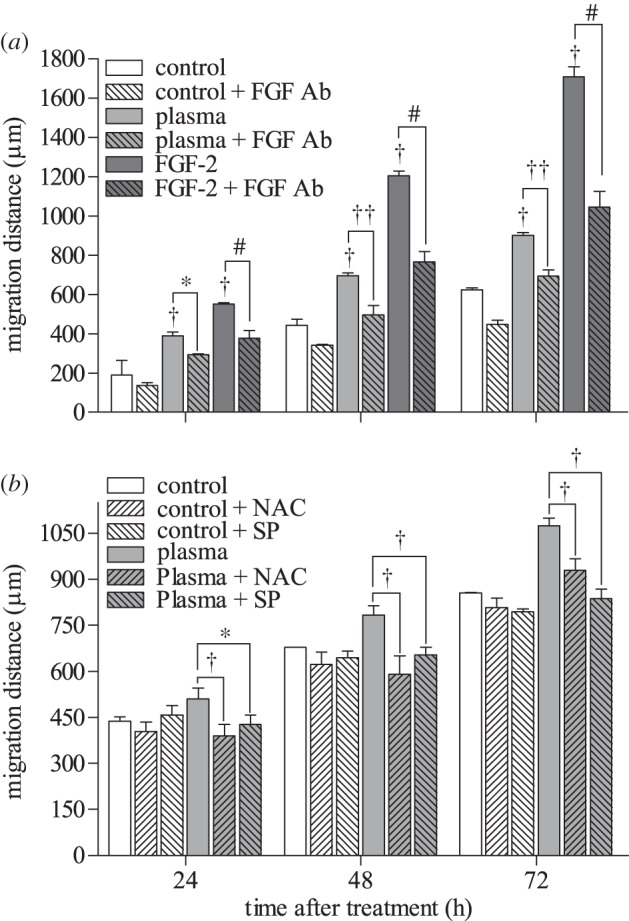
Low-dose DBD plasma treatment enhanced two-dimensional endothelial cell migration, which was abrogated by FGF-2 and ROS blockade. (a) PAEC migration after 4.2 J cm–2 plasma treatment was assessed using a cage assay, with and without an FGF-2 neutralizing antibody. Migration distance was assessed by phase contrast microscopy 0, 24, 48 and 72 h after treatment. † p < 0.001 compared with control. *p < 0.01, ††p < 0.001. #p < 0.001 compared with FGF-2 (10 ng ml–1). (b) NAC and SP were used to block intracellular and extracellular ROS. *p < 0.01, †p < 0.001.
Three-dimensional cell migration, which provides additional physiological relevance compared with two-dimensional migration, also increased with plasma treatment. Twenty-four hours after plasma treatment (4.2 J cm–2), the PAEC number that migrated through the chamber increased significantly. Ten nanograms per millilitre FGF-2, used as positive control, increased migrated PAEC number by 13 per cent. An FGF-2 neutralizing antibody reduced the number of plasma-induced migrated cells by 7 per cent (figure 5a), while sodium pyruvate caused a 22 per cent reduction in migrated cell number (figure 5b).
Figure 5.
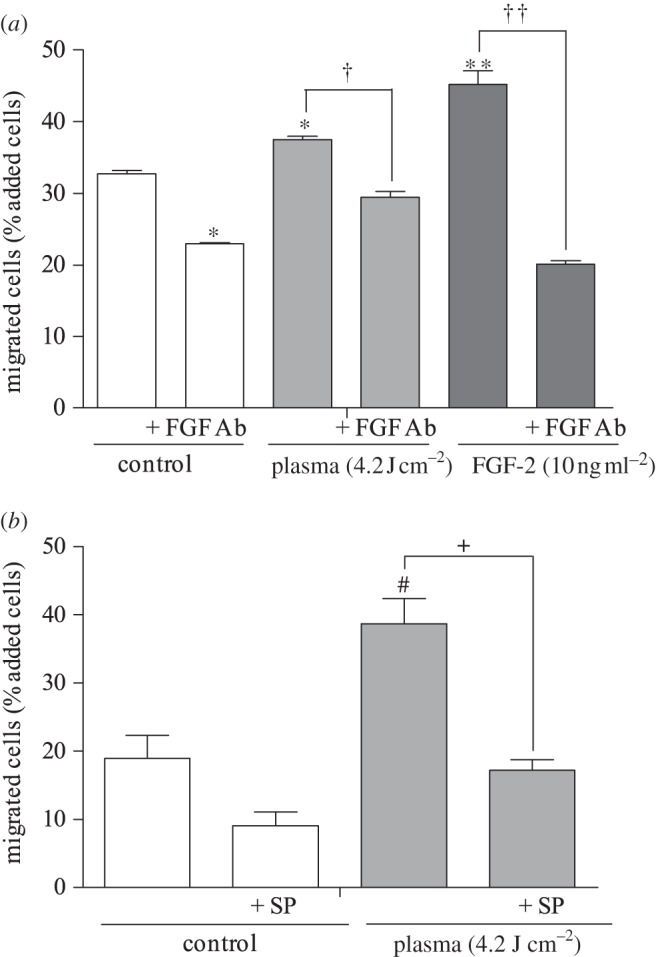
Three-dimensional endothelial cell migration was enhanced by low-dose plasma treatment via ROS-induced FGF-2 release. (a) Plasma-treated PAEC were added to the upper compartment of a Boyden chamber, with or without a neutralizing FGF-2 antibody, and migrated cells on the well bottom were measured. *p < 0.01 compared with control. †p < 0.01 compared with plasma. **p < 0.001 compared with control. ††p < 0.001. (b) To block the ROS effects, cells were plasma-treated in the presence of extracellular ROS scavenger SP. #p < 0.05 compared with control, +p < 0.05 compared with plasma.
Finally, endothelial cell tube formation following plasma treatment was tested. Cells treated with 4.2 J cm−2 plasma formed an extensive tube network 24 h after treatment, while untreated samples formed only tube segments (figure 6a). Average tube length in plasma-treated samples was 121 ± 22 µm, approximately 2.6 times longer than average tube length in control samples (figure 6b). Samples treated with positive control FGF-2 had an average tube length of 134 ± 16 µm. The FGF-2 neutralizing antibody reduced tube length by 70 per cent (37 ± 8 µm). Similarly, a 67 per cent decrease in tube length occurred after plasma treatment in the presence of ROS scavenger sodium pyruvate.
Figure 6.

Endothelial cell tube formation was enhanced by plasma treatment. PAEC were plasma-treated (4.2 J cm–2) and polymerized in a collagen gel. (a) Phase contrast images of tube formation at 24 h. Samples were plasma-treated in the presence of an FGF-2 neutralizing antibody or the extracellular ROS scavenger SP. FGF-2 (10 ng ml–1) was the positive control. (b) Tube length was quantified using ImageJ. †p < 0.001 compared with control, +p < 0.0001 compared with plasma.
4. Discussion
Vascularization is critical to a variety of processes, including wound healing and tissue engineering. Low ROS doses promote vascularization through angiogenic growth factor mechanisms [59,88]. Non-thermal plasma produces a variety of ROS in gas phase, and our group previously demonstrated that non-thermal plasma enhanced endothelial cell proliferation through FGF-2 release [74]. We now show that non-thermal plasma treatment produces ROS in liquid and cells in a dose-dependent manner. These ROS enhance endothelial cell proliferation, migration and tube formation through FGF-2 release and this increase was abrogated by a neutralizing FGF-2 antibody as well as intracellular and extracellular ROS scavengers. These data suggest that plasma may be a useful tool for promoting vascularization.
Plasma produces ROS in gas phase, and these ROS then diffuse into liquid where they react with organic matter. We measured higher ROS levels in plasma-treated serum-free medium when compared with PBS. We believe this occurred because plasma ROS react with medium components such as amino acids, which are not present in PBS. Medium components are readily oxidized by ROS [89–92] to form organic hydroperoxides [91], or ROS can also carbonylate medium amino acids such as lysine, arginine, proline and threonine [92–94]. Since many ROS are short lived, plasma ROS effects on cells may be extended through these ROS interactions with organic medium components.
Plasma-produced ROS (or their reaction products) in liquid then diffuse through the plasma membrane or react with the plasma membrane to produce intracellular ROS through lipid peroxidation. ROS diffusion through the cell membrane depends on molecule size, charge and reactivity. For example, whereas organic hydroperoxides are too large to diffuse through the cell membrane, small molecules such as H2O2 can diffuse through specific membrane aquaporins [95,96]. However, other small molecules, such as 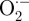 and OH· radicals, cannot diffuse through the cell membrane owing to negative charge and high reactivity, respectively. Once ROS enter cells, they can damage intracellular components [97] or promote or inhibit intracellular signalling pathways [98,99]. Cells may also activate anti-oxidant mechanisms such as glutathione, catalase, ascorbate and superoxide dismutase, which remove the ROS influx and maintain cell redox state [100]. We observed that the intracellular ROS increase immediately after plasma treatment decreased to control values within 3 h. This could be partially due to ROS removal by cellular anti-oxidants.
and OH· radicals, cannot diffuse through the cell membrane owing to negative charge and high reactivity, respectively. Once ROS enter cells, they can damage intracellular components [97] or promote or inhibit intracellular signalling pathways [98,99]. Cells may also activate anti-oxidant mechanisms such as glutathione, catalase, ascorbate and superoxide dismutase, which remove the ROS influx and maintain cell redox state [100]. We observed that the intracellular ROS increase immediately after plasma treatment decreased to control values within 3 h. This could be partially due to ROS removal by cellular anti-oxidants.
Plasma ROS, whether extracellular or intracellular, can cause sub-lethal cell membrane damage and subsequent FGF-2 release [74]. Extracellularly, ROS such as hydroperoxides or carbonylated amino acids damage the plasma membrane through lipid peroxidation [101]. Lipid peroxidation reaction products such as malondialdehyde cause membrane protein and lipid cross-linking, thus disrupting the cell membrane [102]. Intracellularly, ROS may cause cell membrane damage through membrane lipid and protein oxidation. Disruption size may range from 1 nm to greater than 1 µm, thus allowing large molecule movement in and out of the cell [103]. Whereas at low ROS doses, endothelial cell membrane damage can be repaired, at high ROS doses irreparable membrane damage and apoptosis occur [104,105]. However, ROS may actually promote membrane damage repair. Wong et al. [93] showed that ROS cause carbonylation of Annexin-A1, an apoptotic protein present on the cytosolic side of the plasma membrane, thus inactivating it and leading to cell survival.
Sub-lethal cell membrane damage by plasma leads to endothelial cell FGF-2 release [74]. Since FGF-2 lacks a classic secretion sequence, it is only known to be released when plasma membrane is damaged [106]. Cell membrane disruption and FGF-2 release are induced in vivo by mechanical forces [107,108], and in vitro by trypsinization [107]. In our previous work, FGF-2 release peaked 3 h after plasma treatment and decreased to half the maximum value within another 3 h [74]. Released FGF-2 promotes damaged cell survival, proliferation, and FGF-2 mRNA and protein expression [37,73,76]. Since ROS are known to enhance FGF-2 affinity to its receptor, the presence of plasma-produced ROS could accelerate released FGF-2 binding [71].
In this study, FGF-2 released following plasma treatment not only induced cell proliferation, but it also induced cell migration and tube formation. Compared to cells treated with FGF-2, plasma-treated cells proliferated 17 per cent less. However, plasma-treated cells released much less FGF-2 (approx. 0.4% of the positive control FGF-2 dose of 10 ng ml–1). In typical migration assays, a growth factor gradient is used to induce directional migration. In both our two- and three-dimensional migration assays, cell FGF-2 release occurred in all directions, therefore, our migration results may not be as high as if a growth factor gradient were induced. We additionally observed a decrease in cell migration when either a neutralizing FGF-2 antibody or a ROS scavenger was added to both plasma-treated and control cells. Both FGF-2 and ROS are constitutively present in medium and cells, therefore, blockade of these factors is expected to affect even untreated samples.
Non-thermal plasma has many advantages over other ROS vascularization methods. Plasma is safe, cheap and portable compared with other mechanisms of inducing ROS or releasing FGF-2, such as electrical stimulation [109], pulsed electromagnetic fields [34] and ionizing radiation [110]. Unlike these technologies, non-thermal plasma does not cause measurable damage to surrounding tissue. Since plasma devices are portable, they can be used in hospitals, disaster areas and in the military. Plasma devices can also be scaled to meet specific treatment requirements and modified to produce specific reactive species [45]. As shown in our previous work, if direct plasma treatment is not possible, conditioned medium from plasma-treated cells containing growth factors can also be used to promote angiogenesis [74].
While plasma can apply ROS to wounds or tissue constructs and thereby promote vascularization, it is unlikely that plasma alone will be produce sufficient angiogenesis. Angiogenesis is a complex physiological process involving several angiogenic factors that must be released at specific times and in specific concentration gradients. Hence, it might not be possible to achieve angiogenesis of clinical significance through one treatment technique but rather multiple treatment techniques may be necessary. Growth factor therapy for promoting angiogenesis has been disappointing in clinical studies, even though it showed promise in animal studies [28,111]. This could be attributed to insufficient growth factor concentrations for the time needed to generate a response due to difficulties in delivery methods and dosing [111]. Non-thermal plasma enables localized ROS application in precise doses, which may be optimized to release sufficient growth factor concentration. Thus, plasma may be an adjuvant to conventional vascularization techniques. Adding plasma-treated cells could also accelerate vascularization in wounds and tissue-engineering scaffolds.
Our goal was to understand plasma-induced FGF-2 release in endothelial cell proliferation, migration and tube formation. However, these studies are not without limitations. The ROS dye DCFH reacts with a wide variety of ROS, thus we are not certain which plasma-produced ROS induce the angiogenic response. Complexity is further enhanced since plasma-produced ROS interact with each other as well as air and liquid components to produce additional reactive species. Other angiogenic factors may also be upregulated by plasma treatment, and these could also play a role in plasma-induced vascularization. The two-dimensional cell monolayer model employed in our research for plasma treatment is significantly different from in vivo conditions. We also used serum-free medium in our studies, since serum is difficult to characterize and may have anti-oxidant properties [112]. Since serum would be present in a wound environment, specific serum and fluid thickness effects on plasma treatment should be determined.
We now show that non-thermal DBD plasma can stimulate angiogenesis. Plasma dose can be varied by changing treatment time, and plasma composition can be modified by changing electrical parameters. Plasma treatment could be used to promote tissue-engineering scaffold vascularization as well as accelerate wound healing via enhanced angiogenesis. For tissue-engineering scaffolds, cells could be treated with plasma prior to seeding them in the scaffold. For wound healing, plasma ROS could be used initially at higher doses to sterilize the wound and later at lower doses to promote healing. This novel tool could make ROS a practical, physiological method to create new blood vessels either in vitro or in vivo.
Acknowledgements
We thank Dr Gregory Fridman for the plasma device.
References
- 1.Folkman J. 2006. Angiogenesis. Annu. Rev. Med. 57, 1–18 10.1146/annurev.med.57.121304.131306 (doi:10.1146/annurev.med.57.121304.131306) [DOI] [PubMed] [Google Scholar]
- 2.Martin A., Komada M. R., Sane D. C. 2003. Abnormal angiogenesis in diabetes mellitus. Med. Res. Rev. 23, 117–145 10.1002/med.10024 (doi:10.1002/med.10024) [DOI] [PubMed] [Google Scholar]
- 3.Mulligan-Kehoe M. J., Drinane M. C., Mollmark J., Casciola-Rosen L., Hummers L. K., Hall A., Rosen A., Wigley F. M., Simons M. 2007. Antiangiogenic plasma activity in patients with systemic sclerosis. Arthritis Rheum. 56, 3448–3458 10.1002/art.22861 (doi:10.1002/art.22861) [DOI] [PubMed] [Google Scholar]
- 4.Reed M. J., Corsa A., Pendergrass W., Penn P., Sage E. H., Abrass I. B. 1998. Neovascularization in aged mice: delayed angiogenesis is coincident with decreasing levels of transforming growth factor beta1 and type 1 collagen. Am. J. Pathol. 152, 113–123 [PMC free article] [PubMed] [Google Scholar]
- 5.Schäfer M., Werner S. 2008. Oxidative stress in normal and impaired wound repair. Pharmacol. Res. 58, 165–171 10.1016/j.phrs.2008.06.004 (doi:10.1016/j.phrs.2008.06.004) [DOI] [PubMed] [Google Scholar]
- 6.Bland K. I., Palin W. E., Fraunhofer J. A. V., Morris R. R., Adcock R. A., Tobin G. R. 1984. Experimental and clinical observations of the effects of cytotoxic chemotherapeutic drugs on wound healing. Annu. Surg. 199, 782–790 10.1097/00000658-198406000-00017 (doi:10.1097/00000658-198406000-00017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tibbs M. K. 1997. Wound healing following radiation therapy: a review. Radiother. Oncol. 42, 99–106 10.1016/s0167-8140(96)01880-4 (doi:10.1016/s0167-8140(96)01880-4) [DOI] [PubMed] [Google Scholar]
- 8.Shimizu T., Sekine H., Yang J., Isoi Y., Yamato M., Kikuchi A., Okano T. 2006. Polysurgery of cell sheet grafts overcomes diffusion limits to produce thick, vascularized myocardial tissues. FASEB J. 20, 708–710 10.1096/fj.05-4715fje (doi:10.1096/fj.05-4715fje) [DOI] [PubMed] [Google Scholar]
- 9.Lovett M., Lee K., Edwards A., Kaplan D. L. 2009. Vascularization strategies for tissue engineering. Tissue Eng. Part B Rev. 15, 353–370 10.1089/ten.teb.2009.0085 (doi:10.1089/ten.teb.2009.0085) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rouwkema J., Rivron N. C., vanBlitterswijk C. A. 2008. Vascularizaton in tissue engineering. Trends Biotechnol. 26, 434–441 10.1016/j.tibtech.2008.04.009 (doi:10.1016/j.tibtech.2008.04.009) [DOI] [PubMed] [Google Scholar]
- 11.Mikos A. G., et al. 2006. Engineering complex tissues. Tissue Eng. 12, 3307–3339 10.1089/ten.2006.12.3307 (doi:10.1089/ten.2006.12.3307) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferrara N., Gerber H.-P., LeCouter J. 2003. The biology of VEGF and its receptors. Nat. Med. 9, 669–676 10.1038/nm0603-669 (doi:10.1038/nm0603-669) [DOI] [PubMed] [Google Scholar]
- 13.Neufeld G., Cohen T., Gengrinovitch S., Poltorak Z. 1999. Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 13, 9–22 [PubMed] [Google Scholar]
- 14.Bikfalvi A., Klein S., Pintucci G., Rifkin D. B. 1997. Biological roles of fibroblast growth factor-2. Endocr. Rev. 18, 26–45 10.1210/er.18.1.26 (doi:10.1210/er.18.1.26) [DOI] [PubMed] [Google Scholar]
- 15.Presta M., Dell'Era P., Mitola S., Moroni E., Ronca R., Rusnati M. 2005. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 16, 159–178 10.1016/j.cytogfr.2005.01.004 (doi:10.1016/j.cytogfr.2005.01.004) [DOI] [PubMed] [Google Scholar]
- 16.Passaniti A., Taylor R. M., Pili R., Guo Y., Long P. V., Haney J. A., Pauly R. R., Grant D. S., Martin G. R. 1992. A simple, quantitative method for assessing angiogenesis and antiangiogenic agents using reconstituted basement membrane, heparin, and fibroblast growth factor. Lab. Invest. 67, 519–528 [PubMed] [Google Scholar]
- 17.Tong S., Yuan F. 2008. Dose response of angiogenesis to basic fibroblast growth factor in rat corneal pocket assay: I. Experimental characterizations. Microvasc. Res. 75, 10–15 10.1016/j.mvr.2007.06.002 (doi:10.1016/j.mvr.2007.06.002) [DOI] [PubMed] [Google Scholar]
- 18.Jain R. K., Schlenger K., Hockel M., Yuan F. 1997. Quantitative angiogenesis assays: progress and problems. Nat. Med. 3, 1203–1208 10.1038/nm1197-1203 (doi:10.1038/nm1197-1203) [DOI] [PubMed] [Google Scholar]
- 19.Cano M., Karagiannis E. D., Soliman M., Bakir B., Zhuang W., Popel A. S., Gehlbach P. L. 2009. A peptide derived from type 1 thrombospondin repeat-containing protein WISP-1 inhibits corneal and choroidal neovascularization. Invest. Ophthalmol. Vis. Sci. 50, 3840–3845 10.1167/iovs.08-2607 (doi:10.1167/iovs.08-2607) [DOI] [PubMed] [Google Scholar]
- 20.Adya R., Tan B. K., Punn A., Chen J., Randeva H. S. 2008. Visfatin induces human endothelial VEGF and MMP-2/9 production via MAPK and PI3K/Akt signalling pathways: novel insights into visfatin-induced angiogenesis. Cardiovasc. Res. 78, 356–365 10.1093/cvr/cvm111 (doi:10.1093/cvr/cvm111) [DOI] [PubMed] [Google Scholar]
- 21.Ma X., Ottino P., Bazan H. E. P., Bazan N. G. 2004. Platelet-activating factor (PAF) induces corneal neovascularization and upregulates VEGF expression in endothelial cells. Invest. Ophthalmol. Vis. Sci. 45, 2915–2921 10.1167/iovs.04-0128 (doi:10.1167/iovs.04-0128) [DOI] [PubMed] [Google Scholar]
- 22.Camussi G., Montrucchio G., Lupia E., De Martino A., Perona L., Arese M., Vercellone A., Toniolo A., Bussolino F. 1995. Platelet-activating factor directly stimulates in vitro migration of endothelial cells and promotes in vivo angiogenesis by a heparin-dependent mechanism. J. Immunol. 154, 6492–6501 [PubMed] [Google Scholar]
- 23.Rau C., Yang J. C.-S., Jeng S.-F., Chen Y.-C., Lin C.-J., Wu C.-J., Lu T.-H., Hsieh C.-H. 2011. Far-infrared radiation promotes angiogenesis in human microvascular endothelial cells via extracellular signal-regulated kinase activation. Photochem. Photobiol. 87, 441–446 10.1111/j.1751-1097.2010.00853.x (doi:10.1111/j.1751-1097.2010.00853.x) [DOI] [PubMed] [Google Scholar]
- 24.Kanda S., Mochizuki Y., Kanetake H. 2003. Stromal cell-derived factor-1α induces tube-like structure formation of endothelial cells through phosphoinositide 3-kinase. J. Biol. Chem. 278, 257–262 10.1074/jbc.M204771200 (doi:10.1074/jbc.M204771200) [DOI] [PubMed] [Google Scholar]
- 25.Edelman E. R., Mathiowitz E., Langer R., Klagsbrun M. 1991. Controlled and modulated release of basic fibroblast growth factor. Biomaterials 12, 619–626 10.1016/0142-9612(91)90107-L (doi:10.1016/0142-9612(91)90107-L) [DOI] [PubMed] [Google Scholar]
- 26.Borselli C., Oliviero O., Battista S., Ambrosio L., Netti P. A. 2007. Induction of directional sprouting angiogenesis by matrix gradients. J. Biomed. Mater. Res. A 80A, 297–305 10.1002/jbm.a.30896 (doi:10.1002/jbm.a.30896) [DOI] [PubMed] [Google Scholar]
- 27.Dor Y., Djonov V., Abramovitch R., Itin A., Fishman G. I., Carmeliet P., Goelman G., Keshet E. 2002. Conditional switching of VEGF provides new insights into adult neovascularization and pro-angiogenic therapy. EMBO J. 21, 1939–1947 10.1093/emboj/21.8.1939 (doi:10.1093/emboj/21.8.1939) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boodhwani M., Sodha N. R., Laham R. J., Sellke F. W. 2006. The future of therapeutic myocardial angiogenesis. SHOCK 26, 332–341 10.1097/01.shk.0000225318.08681.a7 (doi:10.1097/01.shk.0000225318.08681.a7) [DOI] [PubMed] [Google Scholar]
- 29.Robson M. C., Mustoe T. A., Hunt T. K. 1998. The future of recombinant growth factors in wound healing. Am. J. Surg. 176(Suppl. 2, 1), S80–S82 10.1016/s0002-9610(98)00186-x (doi:10.1016/s0002-9610(98)00186-x) [DOI] [PubMed] [Google Scholar]
- 30.Simons M., et al. 2000. Clinical trials in coronary angiogenesis: issues, problems, consensus: an expert panel summary. Circulation 102, e73–e86 [DOI] [PubMed] [Google Scholar]
- 31.Cross M. J., Claesson-Welsh L. 2001. FGF and VEGF function in angiogenesis: signaling pathways, biological responses and therapeutic inhibition. Trends Pharmacol. Sci. 22, 201–207 10.1016/S0165-6147(00)01676-X (doi:10.1016/S0165-6147(00)01676-X) [DOI] [PubMed] [Google Scholar]
- 32.Nugent M. A., Iozzo R. V. 2000. Fibroblast growth factor-2. Int. J. Biochem. Cell Biol. 32, 115–120 10.1016/S1357-2725(99)00123-5 (doi:10.1016/S1357-2725(99)00123-5) [DOI] [PubMed] [Google Scholar]
- 33.Houchen C. W., George R. J., Sturmoski M. A., Cohn S. M. 1999. FGF-2 enhances intestinal stem cell survival and its expression is induced after radiation injury. Am. J. Physiol. Gastrointest. Liver Physiol. 276, G249–G258 [DOI] [PubMed] [Google Scholar]
- 34.Tepper O. M., et al. 2004. Electromagnetic fields increase in vitro and in vivo angiogenesis through endothelial release of FGF-2. FASEB J. 18, 1231–1233 10.1096/fj.03-0847fje (doi:10.1096/fj.03-0847fje) [DOI] [PubMed] [Google Scholar]
- 35.Fuks Z., Persaud R. S., Alfieri A., McLoughlin M., Ehleiter D., Schwartz J. L., Cordon-Cardo A. P. S. C., Haimovitz-Friedman A. 1994. Basic fibroblast growth factor protects endothelial cells against radiation-induced programmed cell death in vitro and in vivo. Cancer Res. 54, 2582–2590 [PubMed] [Google Scholar]
- 36.Haimovitz-Friedman A., Balaban N., McLoughlin M., Ehleiter D., Michaeli J., Vlodavsky I., Fuks Z. 1994. Protein kinase C mediated basic fibroblast growth factor protection of endothelial cells against radiation-induced apoptosis. Cancer Res. 54, 2591–2597 [PubMed] [Google Scholar]
- 37.Morss A. S., Edelman E. R. 2007. Glucose modulates basement membrane fibroblast growth factor-2 via alterations in endothelial cell permeability. J. Biol. Chem. 282, 14 635–14 644 10.1074/jbc.M608565200 (doi:10.1074/jbc.M608565200) [DOI] [PubMed] [Google Scholar]
- 38.Cheng G. C., Briggs W. H., Gerson D. S., Libby P., Grodzinsky A. J., Gray M. L., Lee R. T. 1997. Mechanical strain tightly controls fibroblast growth factor-2 release from cultured human vascular smooth muscle cells. Circ Res. 80, 28–36 [DOI] [PubMed] [Google Scholar]
- 39.Ku P. T., D'Amore P. A. 1995. Regulation of basic fibroblast growth factor (bFGF) Gene and protein expression following its release from sublethally injured endothelial cells. J. Cell Biochem. 58, 328–343 10.1002/jcb.240580307 (doi:10.1002/jcb.240580307) [DOI] [PubMed] [Google Scholar]
- 40.Finklestein S. P., Apostolides P. J., Caday C. G., Prosser J., Philips M. F., Klagsbrun M. 1988. Increased basic fibroblast growth factor (bFGF) immunoreactivity at the site of focal brain wounds. Brain Res. 460, 253–259 10.1016/0006-8993(88)90370-8 (doi:10.1016/0006-8993(88)90370-8) [DOI] [PubMed] [Google Scholar]
- 41.Fischer T. A., et al. 1997. Regulation of bFGF expression and Ang II secretion in cardiac myocytes and microvascular endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 272, H958–H968 [DOI] [PubMed] [Google Scholar]
- 42.Weich H. A., Iberg N., Klagsbrun M., Folkman J. 1991. Transcriptional regulation of basic fibroblast growth factor gene expression in capillary endothelial cells. J. Cell Biochem. 47, 158–164 10.1002/jcb.240470209 (doi:10.1002/jcb.240470209) [DOI] [PubMed] [Google Scholar]
- 43.Jimenez S. K., Sheikh F., Jin Y., Detillieux K. A., Dhaliwal J., Kardami E., Cattini P. A. 2004. Transcriptional regulation of FGF-2 gene expression in cardiac myocytes. Cardiovasc. Res. 62, 548–557 10.1016/j.cardiores.2004.01.032 (doi:10.1016/j.cardiores.2004.01.032) [DOI] [PubMed] [Google Scholar]
- 44.Seghezzi G., et al. 1998. Fibroblast growth factor-2 (FGF-2) induces vascular endothelial growth factor (VEGF) expression in the endothelial cells of forming capillaries: an autocrine mechanism contributing to angiogenesis. J. Cell Biol. 141, 1659–1673 10.1083/jcb.141.7.1659 (doi:10.1083/jcb.141.7.1659) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kong M. G., Kroesen G., Morfill G., Nosenko T., Shimizu T., Dijk J. v., Zimmermann J. L. 2009. Plasma medicine: an introductory review. New J. Phys. 11, 1 15012. 10.1088/1367-2630/11/11/115012 (doi:10.1088/1367-2630/11/11/115012) [DOI] [Google Scholar]
- 46.Chirokov A., Gutsol A., Fridman A. 2005. Atmospheric pressure plasma of dielectric barrier discharges. Pure Appl. Chem. 77, 487–495 10.1351/pac200577020487 (doi:10.1351/pac200577020487) [DOI] [Google Scholar]
- 47.Kalghatgi S., et al. 2007. Mechanism of blood coagulation by nonthermal atmospheric pressure dielectric barrier discharge plasma. IEEE Trans. Plasma Sci. 35, 1559–1566 10.1109/TPS.2007.905953 (doi:10.1109/TPS.2007.905953) [DOI] [Google Scholar]
- 48.Fridman G., Peddinghaus M., Ayan H., Fridman A., Balasubramanian M., Gutsol A., Brooks A., Friedman G. 2006. Blood coagulation and living tissue sterilization by floating-electrode dielectric barrier discharge in air. Plasma Chem. Plasma Process. 26, 425–442 10.1007/s11090-006-9024-4 (doi:10.1007/s11090-006-9024-4) [DOI] [Google Scholar]
- 49.Kuo S. P., et al. 2009. Contribution of a portable air plasma torch to rapid blood coagulation as a method of preventing bleeding. New J. Phys. 11, 115016. 10.1088/1367-2630/11/11/115016 (doi:10.1088/1367-2630/11/11/115016) [DOI] [Google Scholar]
- 50.Raiser J., Zenker M. 2006. Argon plasma coagulation for open surgical and endoscopic applications: state of the art. J. Phys. D: Appl. Phys. 39, 3520–3523 10.1088/0022-3727/39/16/S10 (doi:10.1088/0022-3727/39/16/S10) [DOI] [Google Scholar]
- 51.Shekhter A. B., Serezhenkov V. A., Rudenko T. G., Pekshev A. V., Vanin A. F. 2005. Beneficial effect of gaseous nitric oxide on the healing of skin wounds. Nitric Oxide Biol. Chem. 12, 210–219 10.1016/j.niox.2005.03.004 (doi:10.1016/j.niox.2005.03.004) [DOI] [PubMed] [Google Scholar]
- 52.Soloshenko I. A., Tsiolko V. V., Khomich V. A., Shchedrin A. I., Ryabtsev A. V., Bazhenov V. Y., Mikhno I. L. 2000. Sterilization of medical products in low-pressure glow discharges. Plasma Phys. Rep. 26, 792–800 10.1134/1.1309476 (doi:10.1134/1.1309476) [DOI] [Google Scholar]
- 53.Sladek R. E. J., Stoffels E., Walraven R., Tielbeek P. J. A., Koolhoven R. A. 2004. Plasma treatment of dental cavities: a feasibility study. IEEE Trans. Plasma Sci. 32, 1540–1543 10.1109/TPS.2004.832636 (doi:10.1109/TPS.2004.832636) [DOI] [Google Scholar]
- 54.Fridman G., Friedman G., Gutsol A., Shekter A. B., Vasilets V., Fridman A. 2008. Applied plasma medicine. Plasma Process. Polym. 5, 503–533 10.1002/ppap.200700154 (doi:10.1002/ppap.200700154) [DOI] [Google Scholar]
- 55.Sensenig R., et al. 2010. Non-thermal plasma induces apoptosis in melanoma cells via production of intracellular reactive oxygen species. Ann. Biomed. Eng. 39, 674–687 10.1007/s10439-010-0197-x (doi:10.1007/s10439-010-0197-x) [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 56.Yildrim E. D., Ayan H., Vasilets V. N., Fridman A., Guceri S., Friedman G., Sun W. 2008. Effect of dielectric barrier discharge plasma on the attachment and proliferation of osteoblasts cultured over poly(ε-caprolactone) scaffolds. Plasma Process. Polym. 5, 58–66 10.1002/ppap.200700041 (doi:10.1002/ppap.200700041) [DOI] [Google Scholar]
- 57.Eliasson B., Egli W., Kogelschatz U. 1994. Modelling of dielectric barrier discharge chemistry. Pure Appl. Chem. 66, 1275–1286 10.1351/pac199466061275 (doi:10.1351/pac199466061275) [DOI] [Google Scholar]
- 58.Kuchenbecker M., Bibinov N., Kaemlimg A., Wandke D., Awakowicz P., Viol W. 2009. Characterization of DBD plasma source for biomedical applications. J. Phys. D: Appl. Phys. 42, 045212. 10.1088/0022-3727/42/4/045212 (doi:10.1088/0022-3727/42/4/045212) [DOI] [Google Scholar]
- 59.Ushio-Fukai M., Alexander R. W. 2004. Reactive oxygen species as mediators of angiogenesis signaling: role of NAD(P)H oxidase. Mol. Cell Biochem. 264, 85–97 10.1023/B:MCBI.0000044378.09409.b5 (doi:10.1023/B:MCBI.0000044378.09409.b5) [DOI] [PubMed] [Google Scholar]
- 60.Fraticelli A., Serrano C., Bochner B., Capogrossi M., Zweier J. 1996. Hydrogen peroxide and superoxide modulate leukocyte adhesion molecule expression and leukocyte endothelial adhesion. Biochim. Biophys. Acta 1310, 251–259 10.1016/0167-4889(95)00169-7 (doi:10.1016/0167-4889(95)00169-7) [DOI] [PubMed] [Google Scholar]
- 61.Roy S., Khanna S., Nallu K., Hunt T. K., Sen C. K. 2006. Dermal wound healing is subject to redox control. Mol. Ther. 13, 211–220 10.1016/j.ymthe.2005.07.684 (doi:10.1016/j.ymthe.2005.07.684) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sen C. K., Khanna S., Babior B. M., Hunt T. K., Ellison E. C., Roy S. 2002. Oxidant-induced vascular endothelial growth factor expression in human keratinocytes and cutaneous wound healing. J. Biol. Chem. 277, 33 284–33 290 10.1074/jbc.M203391200 (doi:10.1074/jbc.M203391200) [DOI] [PubMed] [Google Scholar]
- 63.Wlaschek M., Scharffetter-Kochanek K. 2005. Oxidative stress in chronic venous leg ulcers. Wound Repair Regen. 13, 452–461 10.1111/j.1067-1927.2005.00065.x (doi:10.1111/j.1067-1927.2005.00065.x) [DOI] [PubMed] [Google Scholar]
- 64.Black S. M., DeVol J. M., Wedgwood S. 2008. Regulation of fibroblast growth factor-2 expression in pulmonary arterial smooth muscle cells involves increased reactive oxygen species generation. Am J. Physiol. Cell Physiol. 294, C345–C354 10.1152/ajpcell.00216.2007 (doi:10.1152/ajpcell.00216.2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pechan P. A., Chowdhury K., Seifert W. 1992. Free radicals induce gene expression of NGF and bFGF in rat astrocyte culture. Neuroreport 3, 469–472 10.1097/00001756-199206000-00003 (doi:10.1097/00001756-199206000-00003) [DOI] [PubMed] [Google Scholar]
- 66.Eyries M., Collins T., Khachigian L. M. 2004. Modulation of growth factor gene expression in vascular cells by oxidative stress. Endothelium 11, 133–139 10.1080/10623320490482691 (doi:10.1080/10623320490482691) [DOI] [PubMed] [Google Scholar]
- 67.Delafontaine P., Ku L. 1997. Reactive oxygen species stimulate insulin-like growth factor-1 synthesis in vascular smooth muscle cells. Cardiovasc. Res. 33, 216–222 10.1016/S0008-6363(96)00179-4 (doi:10.1016/S0008-6363(96)00179-4) [DOI] [PubMed] [Google Scholar]
- 68.Vivekananda J., Lin A., Coalson J. J., King R. J. 1994. Acute inflammatory injury in the lung precipitated by oxidant stress induces fibroblasts to synthesize and release transforming growth factor-α. J. Biol. Chem. 269, 25 057–25 061 [PubMed] [Google Scholar]
- 69.Colavitti R., Pani G., Bedogni B., Anzevino R., Borrello S., Waltenberger J., Galeotti T. 2002. Reactive oxygen species as downstream mediators of angiogenic signaling by vascular endothelial growth factor receptor-2/KDR. J. Biol. Chem. 277, 3101–3108 10.1074/jbc.M107711200 (doi:10.1074/jbc.M107711200) [DOI] [PubMed] [Google Scholar]
- 70.Goldkorn T., Balaban N., Matsukuma K., Chea V., Gould R., Last J., Chan C., Chavez C. 1998. EGF-receptor phosphorylation and signaling are targeted by H2O2 redox stress. Am. J. Respir. Cell Mol. Biol. 19, 786–798 [DOI] [PubMed] [Google Scholar]
- 71.Herbert J., Bono F., Savi P. 1996. The mitogenic effect of H2O2 for vascular smooth muscle cells is mediated by an increase of the affinity of basic fibroblast growth factor for its receptor. FEBS Lett. 395, 43–47 10.1016/0014-5793(96)00998-2 (doi:10.1016/0014-5793(96)00998-2) [DOI] [PubMed] [Google Scholar]
- 72.Martin K. R., Barrett J. C. 2002. Reactive oxygen species as double-edged swords in cellular processes: low-dose cell signaling versus high-dose toxicity. Hum. Exp. Toxicol. 21, 71–75 10.1191/0960327102ht213oa (doi:10.1191/0960327102ht213oa) [DOI] [PubMed] [Google Scholar]
- 73.Witte L., Fuks Z., Haimovitz-Friedman A., Vlodavsky I., Goodman D. S., Eldor A. 1989. Effects of irradiation on the release of growth factors from cultured bovine, porcine and human endothelial cells. Cancer Res. 49, 5066–5072 [PubMed] [Google Scholar]
- 74.Kalghatgi S., Friedman G., Fridman A., Clyne A. M. 2010. Endothelial cell proliferation is enhanced by low dose non-thermal plasma through fibroblast growth factor-2 release. Ann. Biomed. Eng. 38, 748–757 10.1007/s10439-009-9868-x (doi:10.1007/s10439-009-9868-x) [DOI] [PubMed] [Google Scholar]
- 75.Wong M. K., Gotlieb A. I. 1984. In vitro reendothelialization of a single-cell wound. Role of microfilament bundles in rapid lamellipodia-mediated wound closure. Lab. Invest. 51, 75–81 [PubMed] [Google Scholar]
- 76.Clyne A. M., Zhu H., Edelman E. R. 2008. Elevated fibroblast growth factor-2 increases tumor necrosis factor-alpha induced endothelial cell death in high glucose. J. Cell Physiol. 217, 86–92 10.1002/jcp.21476 (doi:10.1002/jcp.21476) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fridman G., Brooks A., Balasubramanian M., Fridman A., Gutsol A., Vasilets V., Ayan H., Friedman G. 2007. Comparison of direct and indirect effects of non-thermal atmospheric pressure plasma on bacteria. Plasma Process. Polym. 4, 370–375 10.1109/PPPS.2007.4345628 (doi:10.1109/PPPS.2007.4345628) [DOI] [Google Scholar]
- 78.Ayan H., Fridman G., Staack D., Gutsol A. F., Vasilets V. N., Fridman A. A., Friedman G. 2009. Heating effect of dielectric barrier discharges for direct medical treatment. Plasma Science IEEE Trans. 37, 113–120 10.1109/TPS.2008.2006899 (doi:10.1109/TPS.2008.2006899) [DOI] [Google Scholar]
- 79.Bibinov N., Rajasekaran P., Mertmann P., Wandke D., Viol W., Awakowicz P. 2011. Basics and biomedical applications of dielectric barrier discharge (DBD). In Biomedical engineering, trends in materials science (ed. Laskovski A. N.). Rijeka, Croatia: InTech [Google Scholar]
- 80.Morfill G. E., Kong M. G., Zimmermann J. L. 2009. Focus on plasma medicine. New J. Phys. 11, 1 15011. 10.1088/1367-2630/11/11/115011 (doi:10.1088/1367-2630/11/11/115011) [DOI] [Google Scholar]
- 81.Ferrer A. S., Santema J., Hilhorst R., Visser A. 1990. Fluorescence detection of enzymatically formed hydrogen peroxide in aqueous solution and in reversed micelles. Anal. Biochem. 187, 129–132 10.1016/0003-2697(90)90429-D (doi:10.1016/0003-2697(90)90429-D) [DOI] [PubMed] [Google Scholar]
- 82.LeBel C. P., Ischiropoulos H., Bondy S. C. 1992. Evaluation of the probe 2′,7′-dichlorofluorescin as an indicator of reactive oxygen species formation and oxidative stress. Chem. Res. Toxicol. 5, 227–231 10.1021/tx00026a012 (doi:10.1021/tx00026a012) [DOI] [PubMed] [Google Scholar]
- 83.Cathcart R., Schwiers E., Ames B. N. 1983. Detection of picomoles of hydroperoxides using a fluorescence dichlorofluorescein assay. Anal. Biochem. 134, 111–116 10.1016/0003-2697(83)90270-1 (doi:10.1016/0003-2697(83)90270-1) [DOI] [PubMed] [Google Scholar]
- 84.Dixit P., Hern-Anderson D., Ranieri J., Schmidt C. E. 2001. Vascular graft endothelialization: comparative analysis of canine and human endothelial cell migration on natural biomaterials. J. Biomed. Mater. Res. 56, 545–555 (doi:10.1002/1097-4636(20010915)56:4<545::AID-JBM1126>3.0.CO;2-V) [DOI] [PubMed] [Google Scholar]
- 85.Dollé J., Rezvan A., Allen F. D., Lazarovici P., Lelkes P. I. 2005. Nerve growth factor-induced migration of endothelial cells. J. Pharmacol. Exp. Ther. 315, 1220–1227 10.1124/jpet.105.093252 (doi:10.1124/jpet.105.093252) [DOI] [PubMed] [Google Scholar]
- 86.Koh W., Stratman A. N., Sacharidou A., Davis G. E. 2008. Chapter 5 in vitro three dimensional collagen matrix models of endothelial lumen formation during vaculogenesis and angiogenesis. Methods Enzymol. 433, 83–101 10.1016/S0076-6879(08)02005-3 (doi:10.1016/S0076-6879(08)02005-3) [DOI] [PubMed] [Google Scholar]
- 87.Cai W., Wang M., Moore P. K., Jin H., Yao T., Zhu Y. 2007. The novel proangiogenic effect of hydrogen sulfide is dependent on Akt phosphorylation. Cardiovasc. Res. 76, 29–40 10.1016/j.cardiores.2007.05.026 (doi:10.1016/j.cardiores.2007.05.026) [DOI] [PubMed] [Google Scholar]
- 88.Yasuda M., Ohzeki Y., Shimizu S., Naito S., Ohtsuru A., Yamamoto T., Kuroiwa Y. 1999. Stimulation of in vitro angiogenesis by hydrogen peroxide and the relation with ETS-1 in endothelial cells. Life Sci. 64, 249–258 10.1016/S0024-3205(98)00560-8 (doi:10.1016/S0024-3205(98)00560-8) [DOI] [PubMed] [Google Scholar]
- 89.Stadtman E. R., Berlett B. S. 1991. Fenton chemistry. amino acid oxidation. J. Biol. Chem. 266, 17 201–17 211 [PubMed] [Google Scholar]
- 90.Violand B. N., Siegel N. R. 2000. Protein and peptide chemical and physical stability. In Peptide and protein drug analysis (ed. Reid R. E.), pp. 257–284 Boca Raton, FL: CRC Press [Google Scholar]
- 91.Gebicki S., Gebicki J. M. 1993. Formation of peroxides in amino acids and proteins exposed to oxygen free radicals. Biochem. J. 289, 743–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Davies K. J. A., Delsignore M. E., Lin S. W. 1987. Protein damage and degradation by oxygen radicals II. Modification of amino acids. J. Biol. Chem. 262, 9902–9907 [PubMed] [Google Scholar]
- 93.Wong C. M., Cheema A. K., Zhang L., Suzuki Y. J. 2008. Protein carbonylation as a novel mechanism in redox signaling. Circ. Res. 102, 310–318 10.1161/CIRCRESAHA.107.159814 (doi:10.1161/CIRCRESAHA.107.159814) [DOI] [PubMed] [Google Scholar]
- 94.Suzuki Y. J., Carini M., Butterfield D. A. 2010. Protein carbonylation. Antioxid. Redox Signal. 12, 323–325 10.1089/ars.2009.2887 (doi:10.1089/ars.2009.2887) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bienert G., Moller A., Kristiansen K., Schulz A., Moller I., Schjoerring J., Jahn T. 2007. Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J. Biol. Chem. 282, 1183–1192 10.1074/jbc.M603761200 (doi:10.1074/jbc.M603761200) [DOI] [PubMed] [Google Scholar]
- 96.Bienert G. P., Schjoerring J. K., Jahn T. P. 2006. Membrane transport of hydrogen peroxide. Biochim. Biophys. Acta 1758, 994–1003 10.1074/jbc.M603761200 (doi:10.1074/jbc.M603761200) [DOI] [PubMed] [Google Scholar]
- 97.Slater T. F. 1984. Free-radical mechanisms in tissue injury. Biochem. J. 222, 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Finkel T. 1999. Signal transduction by reactive oxygen species in non-phagocytic cells. J. Leukoc. Biol. 65, 337–340 [DOI] [PubMed] [Google Scholar]
- 99.Finkel T. 2000. Redox-dependent signal transduction. FEBS Lett. 476, 52–54 10.1016/S0014-5793(00)01669-0 (doi:10.1016/S0014-5793(00)01669-0) [DOI] [PubMed] [Google Scholar]
- 100.Salganik R. I. 2001. The benefits and hazards of antioxidants: controlling apoptosis and other protective mechanisms in cancer patients and the human population. J. Am. Coll. Nutr. 20, S464–S472 [DOI] [PubMed] [Google Scholar]
- 101.Snider R. L., Moore R. B. 1986. The role of lipid peroxidation in tert-butyl hydroperoxide inhibition of human erythrocyte Ca2++Mg2+-ATPase. Bios 57, 63–74 [Google Scholar]
- 102.Jain S. K. 1984. The accumulation of malonyldialdehyde, a product of fatty acid peroxidation, can disturb aminophospholipid organization in the membrane bilayer of human erythrocytes. J. Biol. Chem. 259, 3391–3394 [PubMed] [Google Scholar]
- 103.McNeil P. L., Steinhardt R. A. 1997. Loss, restoration and maintenace of plasma membrane integrity. J. Cell Biol. 137, 1–4 10.1083/jcb.137.1.1 (doi:10.1083/jcb.137.1.1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cho K. S., Lee E. H., Choi J. S., Joo C. K. 1999. Reactive oxygen species-induced apoptosis and necrosis in bovine corneal endothelial cells. Invest. Ophthalmol. Vis. Sci. 40, 911–919 [PubMed] [Google Scholar]
- 105.Simon H. U., Haj-Yehia A., Levi-Schaffer F. 2000. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 5, 415–418 10.1023/A:1009616228304 (doi:10.1023/A:1009616228304) [DOI] [PubMed] [Google Scholar]
- 106.Muthukrishnan L., Warder E., McNeil P. L. 1991. Basic fibroblast growth factor is efficiently released from a cytosolic storage site through plasma membrane disruptions of endothelial cells. J. Cell Physiol. 148, 1–16 10.1002/jcp.1041480102 (doi:10.1002/jcp.1041480102) [DOI] [PubMed] [Google Scholar]
- 107.McNeil P. L., Muthukrishnan L., Warder E., D'Amore P. A. 1989. Growth factors are released by mechanically wounded endothelial cells. J. Cell Biol. 109, 811–822 10.1083/jcb.109.2.811 (doi:10.1083/jcb.109.2.811) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yu Q. C., McNeil P. L. 1992. Transient disruptions of aortic endothelial cell plasma membranes. Am. J. Pathol. 141, 1349–1360 [PMC free article] [PubMed] [Google Scholar]
- 109.Bourguignon G., Bourguignon L. 1987. Electric stimulation of protein and DNA synthesis in human fibroblasts. FASEB J. 1, 398–402 [DOI] [PubMed] [Google Scholar]
- 110.Kataoka Y., Murley J. S., Baker K. L., Grdina D. J. 2007. Relationship between phosphorylated histone H2AX formation and cell survival in human microvascular endothelial cells (HMEC) as a function of ionizing radiation exposure in the presence or absence of thiol-containing drugs. Radiat. Res. 168, 106–114 10.1667/RR0975.1 (doi:10.1667/RR0975.1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Annex B., Simons M. 2005. Growth factor-induced therapeutic angiogenesis in the heart: protein therapy. Cardiovasc. Res. 65, 649–655 10.1016/j.cardiores.2004.09.004 (doi:10.1016/j.cardiores.2004.09.004) [DOI] [PubMed] [Google Scholar]
- 112.Roche M., Rondeau P., Singh N. R., Tarnus E., Bourdon E. 2008. The antioxidant properties of serum albumin. FEBS Lett. 582, 1783–1787 10.1016/j.febslet.2008.04.057 (doi:10.1016/j.febslet.2008.04.057) [DOI] [PubMed] [Google Scholar]