

Abstract
Current paradigms suggest that two macrophage subsets, termed M1 and M2, are involved in inflammation and host defense. While the distinct functions of M1 and M2 macrophages have been intensively studied — the former are considered proinflammatory and the latter antiinflammatory — the determinants of their speciation are incompletely understood. Here we report our studies that identify Krüppel-like factor 4 (KLF4) as a critical regulator of macrophage polarization. Macrophage KLF4 expression was robustly induced in M2 macrophages and strongly reduced in M1 macrophages, observations that were recapitulated in human inflammatory paradigms in vivo. Mechanistically, KLF4 was found to cooperate with Stat6 to induce an M2 genetic program and inhibit M1 targets via sequestration of coactivators required for NF-κB activation. KLF4-deficient macrophages demonstrated increased proinflammatory gene expression, enhanced bactericidal activity, and altered metabolism. Furthermore, mice bearing myeloid-specific deletion of KLF4 exhibited delayed wound healing and were predisposed to developing diet-induced obesity, glucose intolerance, and insulin resistance. Collectively, these data identify KLF4 as what we believe to be a novel regulator of macrophage polarization.
Introduction
Clinical and experimental studies support an important role for the macrophage in a broad spectrum of acute (e.g., pathogen infection, sepsis) and chronic inflammatory conditions (e.g., insulin resistance, atherosclerosis, chronic wounds, and tumorigenesis) (1–6). Macrophages exhibit remarkable plasticity that allows them to modulate their phenotype and efficiently respond to environmental signals (7). For purposes of simplicity, researchers have established a model system that classifies macrophages on a continuum in which M1 macrophages represent one extreme — the proinflammatory state — and M2 macrophages represent the contradistinct antiinflammatory state. Thus M1 and M2 macrophages can serve distinct functions in the regulation of the inflammatory response (7, 8). While the classification of the functions of M1 and M2 macrophages has provided an important tool for understanding the regulation of the inflammatory process, at present the molecular mechanisms that govern M1/M2 polarization remain incompletely understood.
Macrophages stimulated with LPS or INF-γ assume an M1 proinflammatory phenotype characterized by a high expression level of iNOS, a high capacity to present antigen, and production of proinflammatory cytokines such as TNF-α, IL-1β, and monocyte chemoattractant protein–1 (MCP-1) (9). Mechanistically, stimuli such as LPS can activate TLR, resulting in the activation of key transcriptional mediators including NF-κB that, in turn, regulate the production of proinflammatory factors. M1 macrophages are critical effector cells that kill microorganisms and thus benefit the host. However, this proinflammatory activity must be carefully titrated, as sustained activation can predispose to chronic inflammatory states such as obesity and insulin resistance. Indeed, evidence derived from murine experimental models suggests that infiltration of adipose tissue by M1 macrophages contributes to the development of obesity and insulin resistance (1, 4). Corroborative evidence has also been found in studies of human obesity. For example, clinical data indicate that macrophages residing in adipose tissue of obese human subjects can produce robust amounts of inflammatory cytokines and thus contribute to the development of insulin resistance (10). In contrast, M2 macrophages are involved in the resolution of inflammation. This cell type is more heterogeneous and is further classified into at least 3 subcategories — namely M2a, M2b, and M2c — that express different subsets of M2 marker genes and have specialized functions (8). Macrophages are differentiated to M2a by IL-4 and IL-13; M2b by immunoglobulin complexes in combination with TLR agonists; and M2c by IL-10, TGF-β, or glucocorticoids (8, 11). Accumulating evidence suggests that M2 macrophages can protect against insulin resistance, eliminate parasites, and promote tissue remodeling and repair. Interestingly, weight loss, known to improve insulin sensitivity and to induce remodeling of adipose tissue, is associated with an increase in macrophages labeled with M2 surface markers (e.g., CD206) (12). Mechanistically, IL-4 stimulation leads to phosphorylation of Stat6, which then translocates to the nucleus and activates transcription of target genes (13). More recently, studies from several laboratories support a critical role for nuclear receptors and specific coactivators (e.g., PPARγ, PPARδ, and PPARγ coactivator 1α [PGC-1α]) and the transcriptional factor HIF-2α in regulating the M2a phenotype (4, 14–17). However, the precise molecular mechanisms governing M2a polarization remain incompletely understood. Krüppel-like factors (KLFs) are a subfamily of the zinc finger class of DNA-binding transcriptional regulators. Members of this gene family have been shown to play important roles in a diverse array of cellular processes including hematopoiesis (18–20). Our group and others have reported that KLF4 is expressed in a stage-specific pattern during myelopoiesis and functions to promote monocyte differentiation (21, 22). While several studies have evaluated the role of KLF4 in macrophage cell lines (23–28), the physiologic role of myeloid KLF4 in vivo has not been elucidated. Given the important roles of the KLF family members in cellular differentiation, we hypothesized that KLF4 might be involved in the transcriptional control of macrophage polarization. Here we present data identifying KLF4 as an essential regulator of macrophage M1/M2 polarization and attendant functions. These results may have implications for the physiology and pathophysiology associated with a broad spectrum of inflammatory states.
Results
KLF4 expression is differentially regulated by M1/M2 stimuli.
To determine whether members of the KLF gene family are critical in macrophage polarization, we first performed quantitative RT-PCR (qPCR) analysis for KLF1-17 in thioglycollate-elicited mouse peritoneal macrophages (PMs) (purified by differential adhesion, Supplemental Figure 1; supplemental material available online with this article; doi: 10.1172/JCI45444DS1) and M-CSF–differentiated mouse bone marrow–derived macrophages (BMDMs) after stimulation with well-established M1 (LPS) or M2 (IL-4) polarizing agents. In both PMs and BMDMs, treatment with these two stimuli differentially regulated the expression of several KLF family members (Figure 1, A and B). The expression of Klf4 was particularly noteworthy, as it was greatly increased by IL-4 and strongly attenuated following LPS treatment. Time course experiments revealed that a significant increase in Klf4 expression was maintained for at least 16 hours after treatment with M2 stimuli such as IL-4 or IL-13 but not IL-10 (Supplemental Figure 2). In contrast, treatment with M1 stimuli (LPS or INF-γ) caused a mild, transient increase in Klf4 expression between 1 and 4 hours after stimulation, followed by a significant reduction in expression at 16 hours after stimulation (Supplemental Figure 2). These differential effects on Klf4 mRNA expression were confirmed at the protein level by Western blot analyses (Figure 1, C and D). Similar effects on KLF4 mRNA expression were also obtained in primary human macrophages (Figure 1E). Next, we sought to determine how M2 stimuli induce KLF4 expression. A key mechanism by which the effects of IL-4 and IL-13 are mediated is the activation of the Stat6 pathway (13, 29). As shown in Figure 1F, the IL-4–mediated induction of Klf4 was abolished in Stat6-null PMs and BMDMs, demonstrating that Klf4 induction by M2 agonists is Stat6 dependent.
Figure 1. Klf4 expression is augmented by M2 and inhibited by M1 stimuli in macrophages.

(A and B) KLF mRNA levels in (A) mouse PMs and (B) BMDMs after 16 hours of stimulation with LPS (50 ng/ml) or mouse IL-4 (5 ng/ml). Gene expression levels were assessed by qPCR and normalized to those in untreated cells (dashed line). Klf1, Klf15, and Klf17 were not detectable in PMs or BMDMs. Klf5 and Klf14 were not detectable in BMDMs. n = 5. (C and D) KLF4 protein levels after LPS and IL-4 treatment. n = 3. (E) KLF4 mRNA level in M-CSF–differentiated primary human macrophages (from 3 donors) stimulated with LPS (50 ng/ml) or human IL-4 (10 ng/ml) for 16 hours. (F) IL-4–mediated induction of the Klf4 gene was diminished in Stat6-null PMs and BMDMs. n = 3. C57BL/6J mice were used as control (WT). *P < 0.05, Student’s t test.
KLF4 promotes macrophage M2 polarization.
To study the function of KLF4 in the process of macrophage polarization, we performed both gain- and loss-of-function studies. Adenoviral overexpression of KLF4 in RAW264.7 cells was employed as a gain-of-function model. For the loss-of-function model, we developed myeloid-specific KLF4-deficient mice (LysMCre/CreKlf4fl/fl, designated Mye-KO). Mye-KO mice demonstrated greater than 90% deletion of KLF4 in PMs and BMDMs compared with the LysMCre/Cre controls (designated Mye-WT), but maintained normal complete blood count (CBC) parameters and distribution of Ly6ChiLy6Clo monocyte populations (Supplemental Figure 3). IL-4 induces expression of prototypical target genes that characterize the M2 phenotype, including arginase-1 (Arg1), the mannose receptor (Mrc1), resistin-like α (Retnla, Fizz1), chitinase 3–like 3 (Chi3l3, Ym1), and other molecules that mediate several critical functions, including deposition of ECM, macrophage adhesion, and clearance of cell debris (30–32). In addition, PPARγ has been shown to be induced by M2 stimuli in macrophages and thereby promote the M2 phenotype (4, 17, 30). As shown in Figure 2A, the IL-4–mediated induction of characteristic M2 marker genes was significantly attenuated by approximately 42.2%–69.4% in KLF4-deficient macrophages, indicating that this factor is required for optimal M2 activation. Concordant effects were seen at the protein level (Figure 2, C and D). Consistent with these observations, overexpression of KLF4 in RAW264.7 cells enhanced basal and/or IL-4–mediated induction of Arg1, Mrc1, programmed cell death 1 ligand 2 (Pdcd1lg2), and Pparg by 2.0- to 4.7-fold (Figure 2B). KLF4 overexpression also augmented Arg-1 protein levels (Supplemental Figure 4A), and co-transfection of KLF4 induced Pparg promoter–driven luciferase activity (Supplemental Figure 4B). Arg1 encodes an l-arginine–degrading enzyme that limits NO production and promotes polyamine synthesis (13, 33). The Arg1 promoter has been well characterized (13), and therefore we focused our mechanistic studies on understanding the molecular basis for its regulation by KLF4. Sequence analysis revealed two consensus KLF-binding sites (CACCC) within a previously identified IL-4–responsive enhancer region of the Arg1 promoter (13, 30) (Figure 2E and Supplemental Figure 4C). This region contains a Stat6-binding site that is essential for IL-4–mediated induction of the Arg1 gene (13). Thus, gene reporter assays were undertaken using luciferase constructs under control of a 4-kb mouse Arg1 promoter with either the normal (WT) or mutated enhancer region (ΔKLF, ΔStat6, as described in Figure 2E). As expected, IL-4 strongly induced the Arg1 promoter (Figure 2E); this effect was strongly attenuated with mutation of either the KLF4- or Stat-binding site. The IL-4–mediated induction of promoter activity was enhanced further by transfection of either KLF4 or Stat6 and maximally induced following co-transfection of both plasmids (Figure 2E). While mutation of the individual KLF sites conferred a mild reduction in KLF4-mediated transactivation, a more significant loss of activity was observed when both KLF sites were mutated (ΔKLF) (Figure 2E and Supplemental Figure 4C). Further, activation of Arg-1 by Stat6 was maintained with mutation of the KLF sites, but the synergistic activation of Arg-1 by KLF4 and Stat6 required both of the KLF4 sites and Stat6 to be intact (Figure 2E). Finally, to determine whether KLF4 is recruited to the Arg1 enhancer region, we performed ChIP assays. As shown in Figure 2F, recruitment of KLF4 to the Arg1 enhancer region was enhanced following IL-4 treatment, and this increase was abrogated in Stat6-null cells. In contrast, the binding of Stat6 to the Arg1 promoter was independent of KLF4 as revealed by ChIP assay with KLF4-deficient PMs and KLF4-overexpressing RAW264.7 cells (Supplemental Figure 4D). Additionally, deficiency of KLF4 did not affect IL-4–mediated Stat6 phosphorylation, a key upstream event during Stat6 activation (Supplemental Figure 4E). Thus, we conclude that KLF4 is induced by IL-4 in a Stat6-dependent manner and that these two factors cooperate to induce M2 genes such as Arg1. This cooperative interaction between KLF4 and Stat6 was also found on the Pparg promoter (Supplemental Figure 4B).
Figure 2. KLF4 is essential for IL-4–mediated macrophage M2 polarization.

(A) Impairment of M2 marker gene expression in KLF4-deficient macrophages. PMs isolated from Mye-WT and Mye-KO mice were treated with IL-4 for 16 hours. n = 3 in each group. (B) KLF4 overexpression enhances M2 gene expression. RAW264.7 cells were infected with either Ad-GFP (Ad-EV) or Ad-KLF4 for 24 hours prior to treatment with IL-4 for 16 hours. n = 3 in each group. (C) Representative Western blot showing IL-4–mediated protein induction of Arg-1, Retnla, Chi3l3, and PPARγ in Mye-WT and Mye-KO macrophages. (D) Quantification of Western blot data by densitometry. For Arg-1, protein levels were normalized to Mye-WT control group. For Retnla, Chi3l3 and PPARγ, only IL-4–induced protein levels were calculated and normalized to the IL-4–treated Mye-WT group, due to extremely low levels of expression at baseline. Data were calculated from 3–5 independent blots. (E) Synergistic activation of the mouse Arg1 promoter by KLF4 and Stat6 as assayed by transient transfection. WT, ~4 kb WT mouse Arg1 promoter; ΔKLF, Arg1 promoter with both KLF-binding sites mutated; ΔStat6: Arg1 promoter with Stat6-binding site mutated. Transient transfection experiments were performed in RAW264.7 cells. n = 3. (F) KLF4 binding to Arg1 promoter detected by ChIP assay in WT and Stat6-null BMDMs with or without IL-4 treatment (4 hours). *P < 0.05, **P < 0.01, Student’s t test with Bonferroni correction.
KLF4 inhibits macrophage M1 polarization.
M1 macrophages produce characteristic molecules such as prostaglandins (through induction of prostaglandin-endoperoxide synthase 2 [Cox-2]), nitric oxide (through induction of iNOS), and proinflammatory cytokines (e.g., TNF-α and IL-1β), that are critical for the immune response (8). Because KLF4 expression was reduced by M1 stimuli, we posited that this reduction in expression may be important for the M1 phenotype. As shown in Figure 3A, in response to LPS stimulation, KLF4-deficient macrophages demonstrated a marked enhancement (~1.63- to 3.24-fold) in the expression of several M1 genes such as Cox-2, Tnfa, MCP-1 (Ccl2), RANTES (Ccl5), and iNOS (Nos2). Conversely, overexpression of KLF4 in RAW264.7 cells attenuated the LPS-mediated induction of M1 genes (~33.6%–53.4%; Figure 3B). These changes in mRNA levels were recapitulated by parallel changes in protein levels, as determined by Western blot and ELISA assays (Figure 3C and Supplemental Figure 5). Further, KLF4-deficient macrophages exhibit enhanced NO production following LPS stimulation as assessed by the Griess assay (Figure 3D). Of note, we did not observe any enhancement of LPS-induced M1 gene expression or NO production in Stat6-null macrophages (Supplemental Figure 6), suggesting that the enhanced expression of some M1 targets seen in KLF4-deficient macrophages is not a consequence of global impairment in M2 activation. To elucidate the molecular mechanism by which KLF4 inhibits M1 gene expression, we focused our attention on Cox-2, a classic proinflammatory target. Transient transfection assays in RAW264.7 cells showed that KLF4 inhibited basal and LPS-induced Cox-2 promoter activity (Figure 3E). However, sequence analysis of the Cox-2 promoter did not reveal a canonical KLF-binding site. Thus, we reasoned that KLF4 may function indirectly by altering pathways that mediate LPS induction of the Cox-2 promoter. Indeed, the increased levels of acetylated histone H3 associated with the Cox-2 promoter in LPS-treated KLF4-deficient macrophages would correspond to greater transcriptional activation of the Cox-2 gene (Figure 3F). Because activation of the NF-κB pathway is critical for LPS-mediated Cox-2 induction (34), we assessed whether KLF4 affects this pathway. KLF4 overexpression or deficiency did not significantly affect IκB phosphorylation or degradation (data not shown) and resulted in a modest increase in p65 recruitment to the Cox-2 promoter in LPS-treated cells (Figure 3, G and H). However, KLF4 deficiency enhanced and overexpression attenuated the recruitment of the coactivators p300 and PCAF to the Cox-2 promoter following LPS stimulation (Figure 3, G and H). The recruitment of p300 and PCAF is specific for the NF-κB site on the Cox-2 promoter (35), as no enrichment was observed to a different region of the Cox-2 promoter (Supplemental Figure 7). These data suggest that KLF4 deficiency allows for increased recruitment of cofactors known to augment NF-κB transcriptional activity. Finally, because KLF4 induces the M2 and inhibits the M1 phenotype, we sought to determine whether KLF4 may be required for suppression of inflammatory gene expression in M2 macrophages. For these studies, PMs from Mye-WT and Mye-KO mice were treated with IL-4 for 16 hours, and LPS was added for an additional 6 hours. As expected, pretreatment with IL-4 significantly inhibited LPS-mediated M1 gene induction (e.g., iNOS, Tnfa, MCP-1, and RANTES) by approximately 42.8%–51.3% in WT macrophages. However, this inhibitory effect was strongly attenuated in KLF4-deficient macrophages (Supplemental Figure 8). These data suggest that KLF4 may also be important in suppressing inflammatory gene expression in M2 macrophages.
Figure 3. KLF4 deficiency enhances macrophage M1 polarization.

(A) Enhanced expression of M1 marker genes in KLF4-deficient macrophages stimulated by LPS for 16 hours. n = 3 in each group. (B) Attenuated expression of M1 marker genes in RAW264.7 cells with overexpression of KLF4. n = 3 in each group. (C) Protein levels of Cox-2 and iNOS in PMs (left) and RAW264.7 cells (right). (D) KLF4-deficient macrophages generate more NO after LPS stimulation. n = 3 in each group. (E) Inhibition of Cox-2 promoter by KLF4. Transient transfection experiments were performed in RAW264.7 cells. n = 3. (F) Enhanced acetylation of histone H3 (AcH3) in LPS-treated KLF4-deficient macrophages demonstrated by ChIP. (G and H) KLF4 regulates LPS-induced recruitment of p300/PCAF as shown by deficiency (G) and overexpression (H) experiments. *P < 0.05, **P < 0.01, Student’s t test with Bonferroni correction.
KLF4-deficient macrophages exhibit enhanced bactericidal activity.
The bactericidal activity of macrophages is generally viewed as within the purview of M1-polarized macrophages (9). Because KLF4-deficient macrophages exhibit enhanced M1 polarization, we hypothesized that these cells may exhibit enhanced bactericidal activity. Thus, we performed phagocytosis and bacterial killing assays in control and KLF4-deficient primary macrophages. Indeed, KLF4-deficient macrophages exhibited significantly greater bactericidal activity toward both Gram-negative (E. coli) (Figure 4, A and B) and Gram-positive (Staphylococcus aureus; Supplemental Figure 9, A and B) bacteria. This effect was not due to alterations in phagocytosis, as assessed by fluorescence microscopy and FACS analysis (Supplemental Figure 10). To determine the basis for the enhanced bactericidal activity, we assessed for expression of effector molecules known to be involved in pathogen killing. Exposure of KLF4-deficient macrophages to E. coli (Figure 4C) or S. aureus (Supplemental Figure 9C) increased expression of the ROS-generating enzyme NADPH oxidase 1 (Nox1) by approximately 2.0-fold. In addition, exposure to E. coli also increased expression of several additional bactericidal targets such as cathelicidin-related antimicrobial peptide (Cramp; ~2.3-fold), iNOS (~5.7-fold), Ifng (~2.1-fold), and Tnfa (~1.4-fold) (Figure 4, D–G). Consistent with the effect on iNOS, nitric oxide production was approximately 3.4-fold higher in KLF4-deficient macrophages infected by E. coli (Figure 4H).
Figure 4. KLF4-deficient macrophages exhibit enhanced bactericidal activity.

(A and B) KLF4-deficient macrophages exhibit enhanced bactericidal activity against E. coli ex vivo. n = 3 in each group. (C–G) Expression of genes involved in bactericidal activity is augmented in KLF4-deficient macrophages after incubation with E. coli. n = 3 in each group. (H) NO production in macrophage was determined by nitrite (NO2–) levels in conditioned medium. n = 3 in each group. *P < 0.05, Student’s t test with Bonferroni correction.
Myeloid KLF4 deficiency results in delayed wound healing.
Macrophages are rapidly recruited to sites of tissue injury and become key regulators of inflammation, tissue repair, and cellular debris clearance (8, 36). The initial inflammatory response is regulated by M1 macrophages, while the healing/repair response is thought to lie in the purview of M2 macrophages (37). Importantly, excess or dysregulated inflammation in the skin can lead to delayed wound closure and further tissue destruction. Because KLF4-deficient macrophages exhibit enhanced M1 polarization, we hypothesized that these cells may increase proinflammatory cytokine expression in the surrounding tissue and lead to delayed wound closure due to tissue destruction. Thus, we assessed the time for wound closure in Mye-WT and Mye-KO mice following cutaneous wounding. Data from wound healing curves showed that Mye-KO mice have delayed wound healing (Figure 5A). At 1 week after wounding, Mye-WT mice demonstrated a 49.3% total reduction in average wound size, in contrast to a 34.9% reduction in Mye-KO mice. Ultimately, complete closure of wounds was delayed by 2 days in Mye-KO mice compared with Mye-WT mice (Figure 5A). Histologic examination revealed that the skin adjacent to wounds in both genotypes had equivalent monocyte/macrophage infiltration in the dermal and subcutaneous layers (Figure 5B and Supplemental Figure 11A). Furthermore, ex vivo studies confirmed that migration of KLF4-deficient macrophages in response to the prototypical monocyte/macrophage chemokine MCP-1 was similar to that of control cells (Supplemental Figure 11B). To determine whether delayed wound healing is mediated by enhanced M1-polarized macrophages in Mye-KO mice, we analyzed the skin surrounding the wound for proinflammatory factors. Data from qPCR analysis of RNA harvested from skin at 48 hours after wounding show that iNOS and Tnfa expression was significantly increased (>17-fold) in Mye-KO mice compared with Mye-WT mice (Figure 5C). These data demonstrate that Mye-KO mice have increased proinflammatory factors in the skin associated with a concomitant delay in wound healing.
Figure 5. Myeloid KLF4 deficiency results in delayed wound healing.
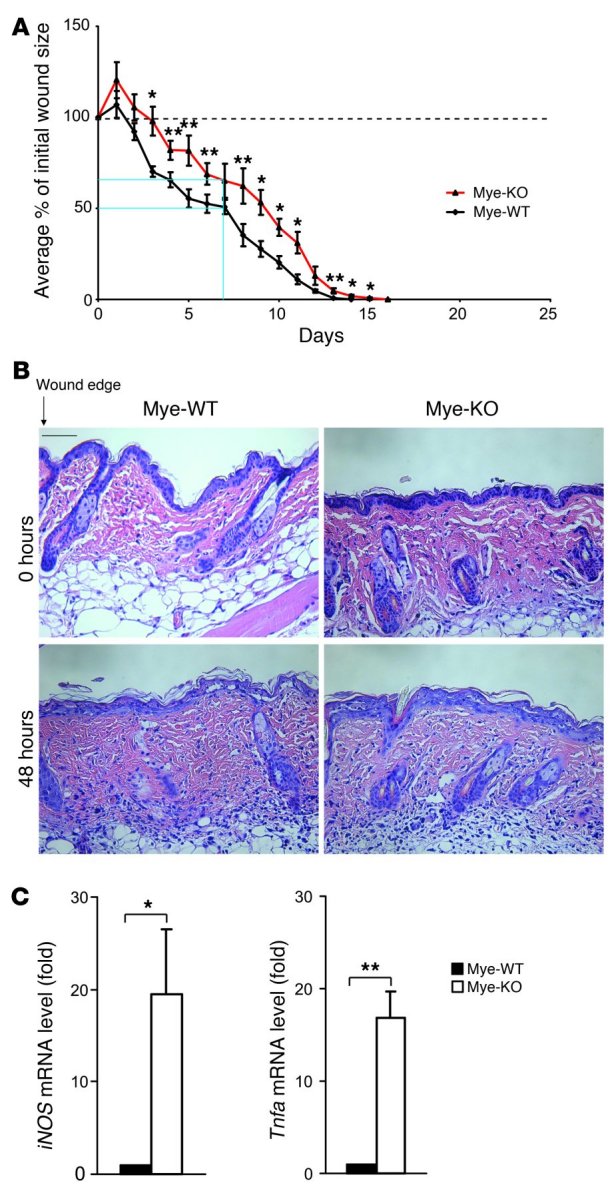
(A) Wound healing graphs of the mean percentage of initial wound area demonstrate a delay in wound healing in Mye-KO mice. At 1 week after wounding, Mye-WT mice had a 49.3% total reduction in average wound size, in contrast to a 34.9% reduction in Mye-KO mice (blue lines). Furthermore, complete wound closure in Mye-KO mice occurred on day 16, compared with day 14 for WT mice. Dashed line at 100% indicates “baseline” percentage of wound area. n = 9 in each group. (B) H&E staining of skin surround wound edge at 48 hours demonstrates similar levels of myeloid cells in the perilesional wound tissue. Scale bar: 150 μm. (C) Expression of iNOS and Tnfa from skin surrounding the wound edge as assessed by qPCR at 48 hours after wounding. n = 3 per genotype. *P < 0.05, **P < 0.01, Student’s t test.
KLF4 expression is regulated in human obesity.
Macrophages are abundantly present in obese adipose tissue, and these adipose tissue macrophages (ATMs) contribute to many of the circulating inflammatory molecules postulated to be involved in insulin resistance and type 2 diabetes progression (1, 14). Increased presence of M1 macrophages has been described in adipose tissue from obese human subjects (12). Thus, we assessed KLF4 gene expression in subcutaneous adipose tissue (needle biopsies) from nondiabetic obese (n = 48), diabetic obese (n = 37), and lean patients (n = 20) (Supplemental Table 1). Data from qPCR analysis showed that KLF4 expression was significantly reduced by 50% in obese patients compared with lean subjects (P < 0.05), regardless of their diabetic status (Figure 6A). Age adjustments did not change this relationship. The expression of KLF2, a closely related family member (38, 39), was unchanged (Figure 6A). Consistent with previous studies (40), IL6 expression was increased, while adiponectin (ADPN) levels were reduced in obese adipose tissue (Figure 6, B and C). Further, a significant positive correlation (r2 = 0.27, P < 0.0001, Pearson correlation test) was observed between mRNA levels of KLF4 and ADPN in human adipose tissue (Figure 6D). Finally, since visceral fat is more metabolically active and closely linked to insulin resistance, we compared KLF4 expression in this compartment versus subcutaneous fat. KLF4 (but not KLF2) expression in visceral fat was significantly lower (~50%) than that in subcutaneous fat samples from obese subjects (Supplemental Figure 12A).
Figure 6. Relationship of KLF4 to obesity in human subjects.
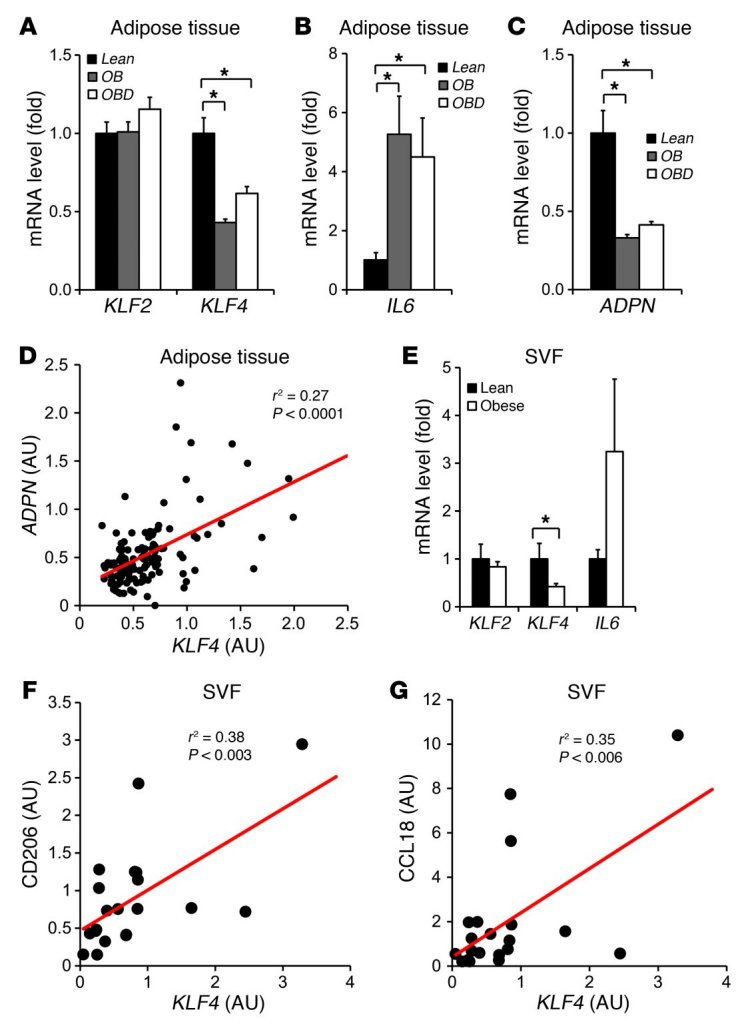
(A–C) Gene expression of KLF2, KLF4, IL6, and ADPN in subcutaneous adipose tissue (needle biopsies) from 48 nondiabetic obese (OB), 37 obese diabetic (OBD), and 20 control (Lean) subjects. (D) Significant correlation between gene expression of KLF4 and ADPN in human adipose tissue. r2 and P values by Pearson correlation test. (E) KLF2, KLF4, and IL6 gene expression in SVF from 9 non-obese and 12 obese patients. (F and G) Significant correlations between gene expression of KLF4 and CD206 and CCL18 in SVF from 9 non-obese and 12 obese patients. r2 and P values by Pearson correlation test. *P < 0.05, Kruskal-Wallis test (vs. lean group). Gene expression in SVF samples was normalized to CD68.
Macrophages are typically present in the stromal vascular fraction (SVF), and thus we also assessed KLF4 expression in the SVF of lean and obese subjects. Cellular fractionation of human SVF revealed that KLF4 expression in macrophages was higher than in other cellular constituents (Supplemental Figure 12B). Consistent with the adipose tissue data (Figure 6A), a 50% reduction of KLF4 expression was observed in SVF from obese subjects (Figure 6E). Finally, in SVF samples, the expression of KLF4 was positively correlated with that of prototypical human M2 macrophage makers such as CD206 (mannose receptor) (r2 = 0.38, P < 0.003, Pearson correlation test) and CCL18 (r2 = 0.35, P < 0.006, Pearson correlation test) (Figure 6, F and G). Collectively, these data indicate that KLF4 expression is regulated in human obesity.
Myeloid KLF4 deficiency promotes insulin resistance.
We next asked whether myeloid-specific deficiency of KLF4 could affect macrophage metabolism and systemic metabolic homeostasis. M2 stimuli, such as IL-4, induce a metabolic switch to fatty acid oxidation, while M1 stimuli promote glycolysis in macrophages (15, 30). As shown in Figure 7A, in the absence of KLF4, the IL-4–mediated uptake and β-oxidation of oleic acid by macrophages was significantly attenuated. However, in response to an M1 stimulus such as LPS, KLF4-deficient macrophages exhibited higher glucose uptake both in untreated and LPS-treated conditions and released more L(+)-lactate, indicating an enhanced rate of glycolysis (Figure 7B). To determine the role of myeloid KLF4 in the development of obesity and insulin resistance, we subjected Mye-WT and Mye-KO mice to diet-induced obesity (DIO) via a high-fat diet (HFD). After 10 weeks of HFD (started at age of 8 weeks), the body weight of Mye-KO mice (37.7 ± 1.1 g) exceeded that of Mye-WT controls (33.7 ± 1.2 g) by approximately 12% (Figure 7C). Mye-KO mice accumulated more adipose tissue than the Mye-WT controls (60% more subcutaneous fat and 110% more visceral fat) as revealed by MRI (Figure 7D and Supplemental Figure 13A). This higher fat mass was associated with a moderate increase in circulating triglyceride levels (Mye-KO: 91.7 ± 8.6 mg/dl vs. Mye-WT: 75.2 ± 5.2 mg/dl, P = 0.067) (Supplemental Figure 13B). HDL and LDL levels were similar in the two groups (Supplemental Figure 13, C and D). These data indicated that Mye-KO mice developed more pronounced DIO. This phenotype was not due to differences in food consumption (Supplemental Figure 14A), indicating that total body energy expenditure may be reduced in the Mye-KO mice. Indeed, indirect calorimetric assays confirmed that Mye-KO mice exhibited reduced oxygen consumption, heat production, and respiratory quotient (Supplemental Figure 14B). To determine whether myeloid KLF4 deficiency resulted in insulin resistance, we performed glucose and insulin tolerance tests in HFD-fed Mye-WT and Mye-KO mice. Serum insulin levels in HFD-fed Mye-KO mice were significantly increased, indicating that these mice may have a propensity toward insulin resistance (Supplemental Figure 15A). Glucose tolerance tests revealed that Mye-KO mice had increased glucose levels 30 minutes after challenge (Figure 7E). As expected with decreased insulin sensitivity, Mye-KO mice were more resistant to the glucose-lowering effects of exogenous insulin (Figure 7F). Lean mice on a normal chow diet showed a similar trend, in that Mye-KO mice were also less glucose tolerant than Mye-WT controls, though the difference did not reach statistical significance (Supplemental Figure 15B). Finally, in keeping with an insulin-resistant state, phosphorylation of Akt was reduced in liver and skeletal muscle of Mye-KO mice after HFD (Supplemental Figure 15C).
Figure 7. Myeloid-specific deficiency of KLF4 exaggerates HFD-induced obesity and insulin resistance state in mice.

(A) Impairment of IL-4–mediated lipid uptake (left panel) and β-oxidation (right panel) in KLF4-deficient macrophages. [3H] counts were normalized to protein content and expressed as fold increase relative to untreated controls. n = 8 in each group. (B) KLF4-deficient macrophages exhibit enhanced glucose uptake before and after LPS stimulation (left panel) and release more L(+)-lactate (right panel), a product of glycolysis. [3H] counts from 2-deoxy-[3H]-d-glucose in the cell lysate and L(+)-lactate concentration in the conditioned medium were normalized to protein content and expressed as fold increase over untreated controls. n = 8 in each group. (C and D) Mye-KO mice on HFD gained more weight (C) and stored more fat (D) than Mye-WT controls. n = 6 in each group. Subc., subcutaneous. (E and F) After 10–12 weeks of HFD, Mye-KO mice show impaired glucose metabolism as revealed by glucose intolerance (E) and resistance to exogenous insulin (F). n = 6 per genotype. *P < 0.05, **P < 0.01, Student’s t test with Bonferroni correction.
To better define the HFD-induced alterations in adipose tissue and muscle, we quantified transcript levels of genes important in fatty acid metabolism and inflammation. As shown in Figure 8A, mRNA levels of a large number of genes encoding key enzymes in nutrient uptake (Slc4a, sodium-coupled bicarbonate transporter 4A), fatty acid binding (Fabp4), fatty acid synthesis (Fasn, fatty acid synthase; Acaca, acetyl-coenzyme A carboxylase α), and β-oxidation (Cpt1a, carnitine palmitoyltransferase 1a), as well as transcriptional factors known to regulate fatty acid metabolism (Pparg; Srebf1, sterol regulatory element–binding protein 1), were reduced by 33.3%–68.0% in white adipose tissue from HFD-fed Mye-KO mice. To determine whether macrophage infiltration and the polarization status of infiltrated macrophages contributed to adiposity, we analyzed macrophage-specific gene expression in the white adipose tissue of HFD-fed Mye-WT and Mye-KO mice. Notably, expression of the macrophage-specific gene F4/80 increased more than 2.3-fold in adipose tissue of HFD-fed Mye-KO mice, indicating enhanced infiltration of macrophages into adipose tissue of these mice (Figure 8A). The inflammatory genes Il6 and Vcam1 were increased 1.7- and 1.9-fold, respectively, while Rentla, a marker of M2 macrophages, was reduced by 73.4% in adipose tissue (Figure 8A). In skeletal muscle, genes encoding fatty acid β-oxidation enzymes (Acox1, peroxisomal acyl-coenzyme A oxidase 1; Acadm, acyl-coenzyme A dehydrogenase for medium-chain fatty acids; Cpt1b, carnitine palmitoyltransferase 1b) and the insulin-sensitive glucose transporter gene Glut4 were reduced by 29.4% to 51.3% in HFD-fed Mye-KO mice (Figure 8B and Supplemental Figure 16A). F4/80 expression was increased by 66.3% in skeletal muscle, indicating enhanced macrophage infiltration. Moreover, the inflammatory genes (MCP-1, Il6) were increased, and the M2 macrophage marker gene Rentla was reduced significantly in skeletal muscle (Figure 8B and Supplemental Figure 16A). These gene expression data demonstrate that, after HFD, Mye-KO mice have enhanced macrophage infiltration in adipose tissue and skeletal muscle and that the infiltrating macrophages exhibit an inflammatory M1 phenotype. The enhanced infiltration of macrophages in adipose tissue and skeletal muscle of obese Mye-KO mice was further confirmed by immunohistochemistry for Mac-3 (Figure 8, C and D). By comparison, livers from Mye-KO mice did not show significant changes in metabolic genes, but did show a trend toward increased expression of inflammatory genes (e.g., MCP-1, Vcam1) (Supplemental Figure 16B). Finally, we assessed for M1/M2 targets in the SVF of Mye-WT and Mye-KO mice following HFD feeding. As shown in Figure 8E, many M1 genes (e.g., Vcam1, Il1b, iNOS, RANTES, Tnfa, Cox-2) were expressed at significantly higher levels in Mye-KO SVF, while M2 genes (e.g., Arg1, Rentla, Pparg) were significantly reduced. Collectively, these data suggest that KLF4 deficiency may affect the polarization status of ATMs.
Figure 8. Mye-KO mice exhibit altered inflammatory and metabolic gene expression in tissues following HFD.

(A and B) Myeloid deficiency of KLF4 in HFD-mice affects transcription of many genes involved in nutrition uptake, β-oxidation, oxidative phosphorylation and inflammation in white adipose tissue (A) and skeletal muscle (B). n = 6 in each group. (C) Increased macrophage infiltration of white fat tissues and muscle tissues as determined by Mac-3 antibody staining. Original magnification, ×400. (D) Quantification of Mac-3 staining. n = 6 in each group. (E) Myeloid deficiency of KLF4 in HFD-fed mice affects expression of macrophage M1 and M2 genes in SVF. n = 6 in each group. *P < 0.05, Student’s t test. (F) Schematic of proposed mechanism. See text for details.
Discussion
Transcriptional control of macrophage differentiation is currently the subject of intense investigation. To date, control of macrophage polarization has largely been attributed to the function of a small group of factors including NF-κB, AP-1, HIFs, STATs, and PPARs. Our observations introduce the KLF family as contributors to this process and, more specifically, identify KLF4 as critical in regulating M1/M2 polarization (Figure 8F). Since the first identification of mammalian KLF family members in 1993 (41), a total of 17 KLF factors have been identified. Several recent reports have implicated KLFs in myeloid cell biology. With respect to KLF4, Feinberg et al. showed that KLF4 promotes monocyte differentiation (21). These observations were extended by elegant studies from the Civin laboratory that determined, by transplantation of hematopoietic Klf4-KO fetal liver cells into lethally irradiated WT mice, that KLF4 has an essential role in the differentiation of mouse monocytes (22). Specifically, these studies identified an essential role for KLF4 in Ly6Chi and Ly6Clo monocyte development. In light of these observations, and in order to minimize developmental effects, we employed the LysM-Cre mouse line, in which the Cre gene is under control of the endogenous LysM promoter. Owing to the temporal and spatial nature of LysM promoter activation in myeloid cells, the Cre gene is expressed at low levels during myeloid development but is highly induced upon transformation of mature monocytes into macrophages (42, 43). Consistent with our expectation, KLF4 deficiency, as achieved in the LysMCre/CreKlf4fl/fl mouse, did not significantly affect the peripheral blood cell differential or monocyte subsets (Supplemental Figure 3). Thus, our approach enabled a detailed evaluation of KLF4 in macrophage biology and led to our identification of KLF4 as an important regulator of M1/M2 polarization. Collectively, the results of the current study coupled with previous work implicate KLF4 in multiple stages of monocyte/macrophage biology, from cellular differentiation to activation.
A key observation provided by our studies is that KLF4 can differentially affect a large repertoire of genes that characterize the M1 and M2 phenotype. With respect to M2 polarization, our studies indicate that KLF4 is both induced by and cooperates with Stat6 to induce quintessential M2 targets such as Arg-1 following IL-4 stimulation. Our studies of the Arg1 promoter reveal that maximal transcriptional activity of Stat6 requires intact KLF4 sites (and vice versa). This type of inductive and cooperative relationship has also been observed between Stat6 and PPARγ in macrophages during M2 activation (30). The fact that PPARγ levels were reduced in Klf4-null cells and tissues raises the possibility that KLF4 and Stat6 may also cooperate to augment PPARγ expression — a possibility that is supported by the inductive effect of KLF4 and Stat6 on the Pparg promoter (Supplemental Figure 4B). The importance of this relationship between PPARγ and KLF4 is also supported by the fact that myeloid deletion of PPARγ leads to a phenotype that is similar to that of KLF4 Mye-KO mice observed in this study (30, 44). While additional studies are clearly warranted, current observations raise the possibility that a cascade of inductive and cooperative interactions in the Stat6/KLF4/PPARγ axis may allow for optimal and sustained M2 activation. Finally, while much of our work here has focused on M2 polarization in the context of IL-4 stimulation, a recent study suggested that alternative IL-4–independent mechanisms may also regulate the M2 phenotype (45). Whether KLF4 participates in this pathway is an important issue that will require further investigation.
The induction of KLF4 provides a molecular mechanism to not only induce the M2 phenotype but also inhibit the M1 pathway. The importance of KLF4’s inhibition of proinflammatory targets is highlighted by gain- and loss-of-function studies in vitro (Figures 3 and 4) and by corroborative evidence in disease models (Figures 5, 7, and 8). Furthermore, the observation that KLF4 deficiency attenuates the ability of IL-4 to inhibit M1 targets (Supplemental Figure 8) suggests that KLF4 may also be important in suppression of proinflammatory genes in M2 macrophages. Our mechanistic studies demonstrate that this antiinflammatory effect occurs at least in part through the ability of KLF4 to inhibit NF-κB transcriptional activity. Importantly, altering KLF4 levels did not significantly affect activation of IKK/IκBα. However, optimal NF-κB transcriptional activity requires key coactivators such as p300/CBP and associated factors such as PCAF. Because KLF4 interacts with p300/CBP (46, 47), we reasoned that it may compete with NF-κB for these rate-limiting amounts of this coactivator, a notion supported by our ChIP studies (Figure 3, G and H). Finally, in addition to competing for coactivators, the KLF4-mediated induction of PPARγ likely contributes to the potent inhibition of M1 polarization by KLF4. Efforts to understand whether these two factors cooperate to inhibit classical macrophage activation is an important issue that is currently being investigated. Our studies also provide insights regarding discrepant observations in the literature regarding the kinetics of KLF4 expression and its targets in response to proinflammatory stimuli. It is likely that (a) idiosyncrasies of various cell lines employed (e.g., J774a, THP-1, RAW, and BV-2) and (b) differences in the concentration (ranging from 10 ng/ml to 1,000 ng/ml) and source of LPS employed (e.g., E. coli, Porphyromonas gingivalis, and Salmonella enterica) may account for many of the disparate results seen among various studies (23–28). For example, several studies in cell lines and/or primary cells have reported an increase in KLF4 expression. Most of these studies assessed expression at early time points following stimulation. In the current study, the most dramatic effect on KLF4 expression in primary murine macrophage was a reduction that occurred at late time points. While this appears to be contradictory, we note that a modest but reproducible increase (statistical significance achieved in PMs [10 ng/ml at 4 hours] and BMDMs [50 ng/ml at 4 hours]; Supplemental Figure 2) was also seen in primary cells. The magnitude is clearly modest but is consistent with observations in cell lines. Importantly, our studies suggest that the reduction in KLF4 expression, observed in both murine and human macrophages as well as human tissue samples (SVF fraction; Figure 6E), is critical, as evidenced by the in vitro and in vivo gene expression and functional studies. A second issue relates to the regulation of iNOS expression by KLF4. The increase in this M1 marker in the setting of KLF4 deficiency was surprising given the previous finding by our group and others that KLF4 induces iNOS expression (23). An important difference between these two studies is that the previous work was performed in the mouse monocyte/macrophage tumor cell line J774a. Indeed, repeat studies have confirmed that overexpression of KLF4 in this cell line robustly induces iNOS. Interestingly, in contrast to the induction of iNOS, KLF4 overexpression reduced the expression of MCP-1 in J774a cells (Kapadia and Jain, unpublished observation). Thus, while gain-of-function studies (in cell lines) and loss-of-function studies (in primary cells) revealed the anticipated anti-parallel effect on some M1 markers such as MCP-1, the basis for the induction of iNOS in both contexts remains incompletely understood. It is noteworthy that differential effects of KLF4 on target genes and cellular functions have been reported in other systems. In the cancer literature, a number of studies support both a pro- and anti-tumorigenic role for KLF4. A potential explanation based on the p21 status of cancer cells was put forth by Rowland and colleagues. This study and others suggest that the cellular context likely affects KLF4 action (48, 49). While additional studies are required to elucidate the molecular basis for the differential effect of KLF4 on iNOS expression in J774a cells versus primary cells, the concordant effects on an entire cassette of M1 genes in primary macrophages observed in the current study suggest that KLF4 inhibits the M1 phenotype. Furthermore, the fact that iNOS was robustly induced in the skin wounds of myeloid-specific KLF4-deficient mice (Figure 5C) strongly supports this in vitro finding. Finally, our observations reinforce the notion that the use of primary cells and corroboration of findings in vivo will be critical in future studies aimed at understanding the biology of myeloid KLF4.
Converging studies reveal that phenotypic polarization of macrophages by the inflammatory microenvironment is critical for both induction and resolution of the inflammatory response. For example, in lean adipose tissue, production of IL-13 can induce the M2 phenotype in ATMs (14). However, obesity can induce a proinflammatory state that promotes the M1 phenotype and initiates a feed-forward circuit that amplifies the inflammatory response and contributes to insulin resistance (1). Similarly, Th1 cytokines predominate early in the course of an infection and thereby enhance macrophage bactericidal effects, while Th2 cytokines confer an M2 phenotype to help in the resolution and repair following an inflammatory insult. In this regard, our observations that KLF4 expression is differentially regulated by Th1 and Th2 cytokines is of particular interest. Furthermore, these studies predict that in the absence of KLF4, the balance would be tipped in favor of an M1 phenotype. Our functional studies are aligned with this hypothesis, as we observed that KLF4-deficient macrophages exhibit robust bactericidal activity. However, proinflammatory activation can be deleterious in the setting of chronic inflammatory conditions, resulting in delayed wound healing and, if sustained, insulin resistance. Indeed, the observation that KLF4 Mye-KO mice exhibited delayed wound healing and susceptibility to DIO/insulin resistance is consistent with this idea.
An important aspect of this work is the association of KLF4 with human obesity. Consistent with the view that ATMs in obese subjects assume an M1 phenotype, a significant reduction in KLF4 expression was seen in adipose tissue from obese human subjects. Furthermore, the observation that KLF4 expression is reduced in visceral versus subcutaneous fat in obese subjects is particularly intriguing given the epidemiologic association between visceral fat and insulin resistance. Our studies also extended our assessment of KLF4 in obesity to the SVF fraction where the macrophage population resides. We note that although murine and human M1 macrophages express similar M1 markers, there are distinct differences in M2 markers (33). For example, some studies indicate that human monocytes and monocyte-derived macrophages do not induce Arg-1, YM1, or FIZZ1 in response to IL-4/IL-13 (31, 33, 50). However, like murine macrophages, alternatively activated human macrophages do express CD206 (C-type receptor 1; MRC1) and CCL18. This receptor belongs to the group of pattern recognition receptors involved in host cell immune responses through phagocytosis and endocytosis. Previous studies demonstrate that the ratio of CD40+ to CD206+ macrophages was 2-fold lower after weight loss in the subcutaneous adipose tissue of obese subjects due to a concomitant decrease in CD40+ and increase in CD206+ macrophages (12). Furthermore, we also assessed CCL18, which was identified in the late 1990s by several groups (5–9, 51) and given different names owing to the manner of discovery, including alternative macrophage activation–associated CC chemokine–1 (AMAC-1). Although a rodent counterpart has not been identified, CCL18 expression by macrophages has been implicated in lymphocyte and immature dendritic cell recruitment. CCL18 has also been identified as being differentially expressed in adipose tissue of patients with nonalcoholic steatohepatitis with fibrosis (52). Interestingly, we found a significant correlation between both human M2 markers and KLF4 in SVF samples. In addition, and consistent with the observation in adipose tissue, KLF4 expression was reduced in the SVF of obese subjects (Figure 6E). Collectively, these observations suggest that the reduction in KLF4 expression may have important implications for human obesity.
The ability for KLF4 to regulate both M1 and M2 polarization has potentially broad implications for numerous inflammatory disease states beyond insulin resistance and wound healing. The fact that KLF4-deficient macrophages exhibited enhanced bactericidal activity (Figure 4) supports a role for this factor in disease processes such as sepsis. Importantly, since KLF4 is also expressed in granulocytes, and LysM-Cre deletes floxed genes in both cell types (53, 54), such studies will need to account for effects of KLF4 deficiency in both macrophages and granulocytes in order to fully elucidate the integrated host response to bacterial infection. In addition, since macrophages are critical in combating chronic infections (e.g., Mycobacterium tuberculosis) and parasites (e.g., Listeria monocytogenes), investigation of these pathologic states is also likely to be fruitful. Furthermore, given the importance of macrophages in atherosclerosis and tumorigenesis, assessment of these processes in the myeloid KLF4-deficient model is clearly warranted. Finally, as KLF4 levels can be manipulated by diverse agonists such as statins, resveratrol, proteosome inhibitors (e.g., bortezomib), and dietary compounds (e.g., broccoli), KLF4 is a potentially promising therapeutic target.
Methods
Human subjects.
The study enrolled 114 morbidly obese subjects involved in a gastric surgery program and 20 lean controls recruited at the Department of Nutrition, Hôtel-Dieu Hospital, Paris, France. Obese subjects met the criteria for obesity surgery and were categorized into diabetic and nondiabetic groups (Supplemental Table 1). Fat specimens were obtained by needle aspiration in the periumbilical area under local anesthesia (12). The Ethics Committee, Hôtel-Dieu Hospital, approved all clinical investigations, and informed written consent was obtained from all subjects.
Animals.
To generate macrophage-specific Klf4-knockout mice, we crossed Klf4fl/fl mice with LysMCre/Cre mice (53, 54). LysMCre/Cre mice (designated Mye-WT) were used as control animals, and LysMCre/CreKlf4fl/fl mice (designated Mye-KO) were used as experimental animals. Klf4fl/fl mice were generated as described previously (55, 56). LysMCre/Cre mice, C57BL/6J wide-type mice, and Stat6-null mice were obtained from The Jackson Laboratory. All experimental procedures were approved by the IACUC of Case Western Reserve University.
Cell culture and transfection assays.
Mouse peritonitis was induced by i.p. injection of 3% thioglycolate broth in 8- to 20-week-old mice as described previously (57). Peritoneal cells were harvested at 72 hours after i.p. injection, and macrophages were enriched by quick adhesion. BMDMs were differentiated with M-CSF as described previously (58). Cells were cultured in DMEM supplemented with 10% fetal bovine serum and antibiotics. Macrophage culture purity was assessed by CD115-based flow cytometry. All transient transfection and adenoviral overexpression procedures were performed in the RAW264.7 cell line (ATCC). For overexpression, RAW264.7 cells were infected with the empty control virus (Ad-EV) or the adenovirus carrying the human KLF4 gene (Ad-KLF4). Transient transfection assays were done with FuGENE 6 transfection reagent (Roche Applied Science) in RAW264.7 cells. The Arg1 promoter luciferase construct was a gift from Peter Murray (St. Jude Children’s Research Hospital, Memphis, Tennessee, USA). KLF- and Stat6-binding sites (13, 30) were mutated using a site-directed mutagenesis kit (Agilent Technologies). The Stat6 expression plasmid was a gift from Thomas A. Hamilton (Cleveland Clinic). KLF4 and p65 expression plasmids and Cox-2 luciferase plasmid were described previously (21, 38).
Phagocytosis and bacteria killing.
To assess the phagocytic capability of macrophages, opsonized Alexa Fluor 488–conjugated E. coli (strain K-12) bio-particles (Invitrogen) were incubated with macrophages and intracellular uptake was visualized by fluorescence microscopy and quantified by flow cytometry. To determine the bactericidal function of macrophages, 2 × 106 adherent PMs were incubated with 5 × 107 E. coli (strain ATCC-29522) (59) or 1 × 106 S. aureus (strain Newman) (57) for 2 hours, and then cells were thoroughly washed with PBS and incubated for 24 hours in DMEM medium containing antibiotics before harvesting of intracellular bacteria. Cell lysate from macrophages containing intracellular bacteria was serially diluted with PBS and spread onto agar plates to determine bacterial viability (expressed as CFU).
Wound preparation, treatment, and measurement.
Each mouse was subjected to three 6-mm excisional wounds, 1 midline and 2 on each side of the midline, followed by a single dose of 72 mJ/cm2 UVB. This dose of UVB has been previously shown to recruit monocytes and macrophages into the skin (60, 61). Wounds were measured daily (length × width), and total wound area was calculated, until they healed (loss of serum crust and reepithelialization).
Mouse HFD model and assessment of metabolic status.
Male mice were fed either standard rodent chow or Clinton/Cybulsky High Fat Rodent Diet (HFD: D12108C, Research Diet) (62) starting at 8 weeks of age for 8–12 weeks. These mice were subjected to a glucose tolerance test or an insulin tolerance test at 10 weeks after initiation of HFD (44) (Case Mouse Metabolic Phenotyping Center, Case Western Reserve University). After 12 weeks of HFD, animals were assessed for body composition on a 7T small animal MRI scanner in the Case Center for Imaging Research (Case Western Reserve University) and sacrificed for harvesting of serum, fat tissue (perigonadal, visceral, and subcutaneous), skeletal muscle (quadriceps and soleus), and liver. To investigate the potential sites of insulin resistance, HFD-fed Mye-WT and Mye-KO mice were injected with insulin (5 mU/g) through the inferior vena cava, and liver and skeletal muscle were quickly harvested for biochemical analysis. Samples were stored at –80°C or fixed in 10% formalin for subsequent analysis. Food intake and energy expenditure were determined after 10 weeks of HFD at the Mouse Metabolic Phenotyping Center, University of Cincinnati, Cincinnati, OH.
RNA isolation and qPCR.
Total RNA from cultured cells, human tissue samples or mouse tissue samples was isolated using either TRIzol reagent (Invitrogen) or RNeasy kit (QIAGEN). First-strand cDNA was synthesized and subjected to qPCR with either SYBR green or Roche universal probe reagents (Universal ProbeLibrary, Roche Applied Science) on a StepOnePlus Real-Time PCR System (Applied Biosystems). Gene expression was normalized to GAPDH, 18SRNA, or β-actin using the ΔΔCt method.
Western blot analysis and immunohistochemistry.
Western blots were performed following standard protocols. Chicken anti–arginase-1 antibody was a gift of Sidney M. Morris Jr. (University of Pittsburgh, Pittsburgh, Pennsylvania, USA). Other antibodies were iNOS (SC-8310), Cox-2 (SC-7951), PPARγ (SC-7273), and HRP-conjugated anti-goat IgG (SC-2020) from Santa Cruz Biotechnology Inc.; Stat6 (catalog 9362) and phospho–Tyr641-Stat6 (catalog 9361) from Cell Signaling Technology; goat anti-KLF4 (AF3158) from R&D Systems; mouse anti–β-actin (A1978) from Sigma-Aldrich; HRP-conjugated anti-rabbit IgG (NA934V) and anti-mouse-IgG (NA931V) from GE Healthcare; and HRP-conjugated anti-chicken IgY (SA1-300) from Affinity BioReagents. Mac-3 antibody (ab22506) was from Abcam. Immunohistochemical studies on tissue samples were performed as described previously (44), and images were collected with a ×40 objective.
ChIP.
ChIP assays were performed as previously described (38, 47). In brief, 2 × 107 cells were stimulated with LPS (50 ng/ml) for 30 minutes or IL-4 (5 ng/ml) for 1–4 hours prior to crosslinking for 10 minutes with 1% formaldehyde. Antibodies recognizing p65 (SC-372), p300 (SC-585), PCAF (SC-8999), and Stat6 (SC-981) were from Santa Cruz Biotechnology Inc. Acetyl-histone 3 antibody (06-599B) and Magna ChIP Protein A Magnetic Beads (catalog 16-661) are products of Millipore. Normal rabbit IgG (PP64B, Upstate) was used as negative control. Mouse KLF4 ChIP was performed with an ExactaChIP Mouse KLF4 Chromatin Immunoprecipitation Kit (ECP3158, R&D Systems). Arg1 promoter PCR was performed with the specific primers flanking the Stat6 enhancer region (sense: TCACGCGTGGTAGCCGACGAGAG; antisense: CGCACGCGTAAAGTGGCACAACTCACGTA) and Cox-2 promoter PCR was performed with specific primers flanking the NF-κB–binding site (sense: ATGTGGACCCTGACAGAGGA; antisense: TCTCCGGTTTCCTCCCAGTC) (13, 35). Nontargeting primers for Arg1 promoter (sense: CGCCTCGAGGCTGCATGTGCTCGG; antisense: CGCACGCGTAGAACTGCTTTGGGTTGTCA), and Cox-2 promoter (sense: ATTCAAGCAGCAGAAGAGGGCAG; antisense: CTGGGATGCCAGAGCACACTG) were used to perform nonspecific control PCR.
In vitro metabolic assays.
Macrophage polarization was induced by treating cells with either LPS (50 ng/ml) or IL-4 (5 ng/ml) for 16 hours prior to performing metabolic assays. For glucose uptake, 2-deoxy-[3H]-d-glucose (0.4 μCi/ml) was added to the cells for 10 minutes at 37°C, and the reaction was terminated by washing cells twice in ice-cold PBS, followed by cell lysis for radioactivity determination (30). To determine fatty acid uptake, cells were incubated at 25°C for 5 minutes with 1-[3H]-oleic acid (0.2 μCi/ml), followed by washing with cold PBS and lysis with RIPA buffer. Fatty acid oxidation was determined by radioassay for [3H]-H2O in the aqueous phase of cell lysate chloroform extracts (63). Nitrite derived from NO was determined with the Griess Reagent System (Promega) in macrophage-conditioned medium per the manufacturer’s instructions. Glycolysis was determined by an L(+)-lactate release assay (K627-100, BioVision) following the manufacturer’s instructions. All sample values were normalized for protein content and expressed as fold change relative to control samples.
Statistics.
Results are presented as mean ± SEM. To analyze the difference between 2 groups, 2-tailed Student’s t test was used. Bonferroni correction was used when more than 2 groups were present. A P value less than 0.05 was considered significant. Human gene expression data were analyzed with Kruskal-Wallis test and Pearson correlation test.
Supplementary Material
Acknowledgments
This work supported by NIH grants 1 K23 CA109115-01A3 (to J.A. Kim), HL097593, HL086548, HL076754 HL084154, HL075427 (to M.K. Jain), and K08HL083090 (to A. Hamik); the Visconsi Research Scholars’ Fund (to A. Hamik); American Heart Association postdoctoral award 09POST2060203 (to N. Sharma) and NIH grant K99/R00 HL097023 (to G.H. Mahabeleshwar). The clinical part was supported by the “Programme Hospitalier de Recherche Clinique,” Assistance Publique — Hôpitaux de Paris (AOR 02076) and the Commission of the European Communities (Collaborative Project ADAPT, contract number HEALTH-F2-2008-201100) (to K. Clément).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2011;121(7):2736–2749. doi:10.1172/JCI45444.
References
- 1.Heilbronn LK, Campbell LV. Adipose tissue macrophages, low grade inflammation and insulin resistance in human obesity. Curr Pharm Des. 2008;14(12):1225–1230. doi: 10.2174/138161208784246153. [DOI] [PubMed] [Google Scholar]
- 2.Johnson JL, Newby AC. Macrophage heterogeneity in atherosclerotic plaques. Curr Opin Lipidol. 2009;20(5):370–378. doi: 10.1097/MOL.0b013e3283309848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23(11):549–555. doi: 10.1016/S1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- 4.Charo IF. Macrophage polarization and insulin resistance: PPARgamma in control. Cell Metab. 2007;6(2):96–98. doi: 10.1016/j.cmet.2007.07.006. [DOI] [PubMed] [Google Scholar]
- 5.Evans TJ. The role of macrophages in septic shock. Immunobiology. 1996;195(4–5):655–659. doi: 10.1016/S0171-2985(96)80029-5. [DOI] [PubMed] [Google Scholar]
- 6.Lucas T, Abraham D, Aharinejad S. Modulation of tumor associated macrophages in solid tumors. Front Biosci. 2008;13:5580–5588. doi: 10.2741/3101. [DOI] [PubMed] [Google Scholar]
- 7.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5(12):953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 8.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8(12):958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Benoit M, Desnues B, Mege JL. Macrophage polarization in bacterial infections. J Immunol. 2008;181(6):3733–3739. doi: 10.4049/jimmunol.181.6.3733. [DOI] [PubMed] [Google Scholar]
- 10. Zeyda M, et al. Human adipose tissue macrophages are of an anti-inflammatory phenotype but capable of excessive pro-inflammatory mediator production. Int J Obes (Lond). 2007. 31 9 1420 1428 . 10.1038/sj.ijo.0803632 [DOI] [PubMed] [Google Scholar]
- 11.Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25(12):677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 12.Aron-Wisnewsky J, et al. Human adipose tissue macrophages: m1 and m2 cell surface markers in subcutaneous and omental depots and after weight loss. . J Clin Endocrinol Metab. 2009;94(11):4619–4623. doi: 10.1210/jc.2009-0925. [DOI] [PubMed] [Google Scholar]
- 13.Pauleau AL, Rutschman R, Lang R, Pernis A, Watowich SS, Murray PJ. Enhancer-mediated control of macrophage-specific arginase I expression. J Immunol. 2004;172(12):7565–7573. doi: 10.4049/jimmunol.172.12.7565. [DOI] [PubMed] [Google Scholar]
- 14.Kang K, et al. Adipocyte-derived Th2 cytokines and myeloid PPARdelta regulate macrophage polarization and insulin sensitivity. Cell Metab. 2008;7(6):485–495. doi: 10.1016/j.cmet.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vats D, et al. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. 2006;4(1):13–24. doi: 10.1016/j.cmet.2006.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takeda N, et al. Differential activation and antagonistic function of HIF-{alpha} isoforms in macrophages are essential for NO homeostasis. Genes Dev. 2010;24(5):491–501. doi: 10.1101/gad.1881410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bouhlel MA, et al. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007;6(2):137–143. doi: 10.1016/j.cmet.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 18.Takahashi K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 19.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 20.Aziz A, Soucie E, Sarrazin S, Sieweke MH. MafB/c-Maf deficiency enables self-renewal of differentiated functional macrophages. Science. 2009;326(5954):867–871. doi: 10.1126/science.1176056. [DOI] [PubMed] [Google Scholar]
- 21.Feinberg MW, et al. The Kruppel-like factor KLF4 is a critical regulator of monocyte differentiation. EMBO J. 2007;26(18):4138–4148. doi: 10.1038/sj.emboj.7601824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alder JK, et al. Kruppel-like factor 4 is essential for inflammatory monocyte differentiation in vivo. . J Immunol. 2008;180(8):5645–5652. doi: 10.4049/jimmunol.180.8.5645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feinberg MW, Cao Z, Wara AK, Lebedeva MA, Senbanerjee S, Jain MK. Kruppel-like factor 4 is a mediator of proinflammatory signaling in macrophages. J Biol Chem. 2005;280(46):38247–38258. doi: 10.1074/jbc.M509378200. [DOI] [PubMed] [Google Scholar]
- 24.Liu J, et al. KLF4 regulates the expression of interleukin-10 in RAW264.7 macrophages. Biochem Biophys Res Commun. 2007;362(3):575–581. doi: 10.1016/j.bbrc.2007.07.157. [DOI] [PubMed] [Google Scholar]
- 25.Liu J, Liu Y, Zhang H, Chen G, Wang K, Xiao X. KLF4 promotes the expression, translocation, and release of HMGB1 in RAW264.7 macrophages in response to LPS. Shock. 2008;30(3):260–266. doi: 10.1097/shk.0b013e318162bef7. [DOI] [PubMed] [Google Scholar]
- 26.Liu Y, et al. Induction of KLF4 in response to heat stress. Cell Stress Chaperones. 2006;11(4):379–389. doi: 10.1379/CSC-210.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaushik DK, Gupta M, Das S, Basu A. Kruppel-like factor 4, a novel transcription factor regulates microglial activation and subsequent neuroinflammation. J Neuroinflammation. 2010;7:68. doi: 10.1186/1742-2094-7-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou Q, Amar S. Identification of signaling pathways in macrophage exposed to Porphyromonas gingivalis or to its purified cell wall components. J Immunol. 2007;179(11):7777–7790. doi: 10.4049/jimmunol.179.11.7777. [DOI] [PubMed] [Google Scholar]
- 29.Hou J, Schindler U, Henzel WJ, Ho TC, Brasseur M, McKnight SL. An interleukin-4-induced transcription factor: IL-4 Stat. Science. 1994;265(5179):1701–1706. doi: 10.1126/science.8085155. [DOI] [PubMed] [Google Scholar]
- 30.Odegaard JI, et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature. 2007;447(7148):1116–1120. doi: 10.1038/nature05894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raes G, De Baetselier P, Noel W, Beschin A, Brombacher F, Hassanzadeh GG. Differential expression of FIZZ1 and Ym1 in alternatively versus classically activated macrophages. J Leukoc Biol. 2002;71(4):597–602. [PubMed] [Google Scholar]
- 32.Raes G, Noel W, Beschin A, Brys L, De Baetselier P, Hassanzadeh GH. FIZZ1 and Ym as tools to discriminate between differentially activated macrophages. Dev Immunol. 2002;9(3):151–159. doi: 10.1080/1044667031000137629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raes G, et al. Arginase-1 and Ym1 are markers for murine, but not human, alternatively activated myeloid cells. J Immunol. 2005;174(11):6561–6562. doi: 10.4049/jimmunol.174.11.6561. [DOI] [PubMed] [Google Scholar]
- 34.Jobin C, Morteau O, Han DS, Balfour SR. Specific NF-kappaB blockade selectively inhibits tumour necrosis factor-alpha-induced COX-2 but not constitutive COX-1 gene expression in HT-29 cells. Immunology. 1998;95(4):537–543. doi: 10.1046/j.1365-2567.1998.00646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ki SH, Choi MJ, Lee CH, Kim SG. Galpha12 specifically regulates COX-2 induction by sphingosine 1-phosphate. Role for JNK-dependent ubiquitination and degradation of IkappaBalpha. J Biol Chem. 2007;282(3):1938–1947. doi: 10.1074/jbc.M606080200. [DOI] [PubMed] [Google Scholar]
- 36.Nathan C. Metchnikoff’s Legacy in 2008. Nat Immunol. 2008;9(7):695–698. doi: 10.1038/ni0708-695. [DOI] [PubMed] [Google Scholar]
- 37.Deonarine K, et al. Gene expression profiling of cutaneous wound healing. J Transl Med. 2007;5:11. doi: 10.1186/1479-5876-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Das H, et al. Kruppel-like factor 2 (KLF2) regulates proinflammatory activation of monocytes. Proc Natl Acad Sci U S A. 2006;103(17):6653–6658. doi: 10.1073/pnas.0508235103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Senbanerjee S, et al. KLF2 Is a novel transcriptional regulator of endothelial proinflammatory activation. J Exp Med. 2004;199(10):1305–1315. doi: 10.1084/jem.20031132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matsuzawa Y. Adiponectin: a key player in obesity related disorders. Curr Pharm Des. 2010;16(17):1896–1901. doi: 10.2174/138161210791208893. [DOI] [PubMed] [Google Scholar]
- 41.Miller IJ, Bieker JJ. A novel, erythroid cell-specific murine transcription factor that binds to the CACCC element and is related to the Krüppel family of nuclear proteins. Mol Cell Biol. 1993;13(5):2776–2786. doi: 10.1128/mcb.13.5.2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ye M, et al. Hematopoietic stem cells expressing the myeloid lysozyme gene retain long-term, multilineage repopulation potential. Immunity. 2003;19(5):689–699. doi: 10.1016/s1074-7613(03)00299-1. [DOI] [PubMed] [Google Scholar]
- 43.Faust N, Varas F, Kelly LM, Heck S, Graf T. Insertion of enhanced green fluorescent protein into the lysozyme gene creates mice with green fluorescent granulocytes and macrophages. Blood. 2000;96(2):719–726. [PubMed] [Google Scholar]
- 44.Hevener AL, et al. Macrophage PPAR gamma is required for normal skeletal muscle and hepatic insulin sensitivity and full antidiabetic effects of thiazolidinediones. J Clin Invest. 2007;117(6):1658–1669. doi: 10.1172/JCI31561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Satoh T, et al. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat Immunol. 2010;11(10):936–944. doi: 10.1038/ni.1920. [DOI] [PubMed] [Google Scholar]
- 46.Evans PM, Chen X, Zhang W, Liu C. KLF4 interacts with beta-catenin/TCF4 and blocks p300/CBP recruitment by beta-catenin. Mol Cell Biol. 2010;30(2):372–381. doi: 10.1128/MCB.00063-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Evans PM, Zhang W, Chen X, Yang J, Bhakat KK, Liu C. Kruppel-like factor 4 is acetylated by p300 and regulates gene transcription via modulation of histone acetylation. J Biol Chem. 2007;282(47):33994–34002. doi: 10.1074/jbc.M701847200. [DOI] [PubMed] [Google Scholar]
- 48.Rowland BD, Bernards R, Peeper DS. The KLF4 tumour suppressor is a transcriptional repressor of p53 that acts as a context-dependent oncogene. Nat Cell Biol. 2005;7(11):1074–1082. doi: 10.1038/ncb1314. [DOI] [PubMed] [Google Scholar]
- 49.Rowland BD, Peeper DS. KLF4, p21 and context-dependent opposing forces in cancer. Nat Rev Cancer. 2006;6(1):11–23. doi: 10.1038/nrc1780. [DOI] [PubMed] [Google Scholar]
- 50.Munder M, et al. Arginase I is constitutively expressed in human granulocytes and participates in fungicidal activity. Blood. 2005;105(6):2549–2556. doi: 10.1182/blood-2004-07-2521. [DOI] [PubMed] [Google Scholar]
- 51.Schutyser E, Richmond A, Van DJ. Involvement of CC chemokine ligand 18 (CCL18) in normal and pathological processes. J Leukoc Biol. 2005;78(1):14–26. doi: 10.1189/jlb.1204712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Estep JM, et al. Expression of cytokine signaling genes in morbidly obese patients with non-alcoholic steatohepatitis and hepatic fibrosis. Obes Surg. 2009;19(5):617–624. doi: 10.1007/s11695-009-9814-x. [DOI] [PubMed] [Google Scholar]
- 53.Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8(4):265–277. doi: 10.1023/A:1008942828960. [DOI] [PubMed] [Google Scholar]
- 54.Cramer T, et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell. 2003;112(5):645–657. doi: 10.1016/S0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Katz JP, et al. The zinc-finger transcription factor Klf4 is required for terminal differentiation of goblet cells in the colon. Development. 2002;129(11):2619–2628. doi: 10.1242/dev.129.11.2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Katz JP, et al. Loss of Klf4 in mice causes altered proliferation and differentiation and precancerous changes in the adult stomach. Gastroenterology. 2005;128(4):935–945. doi: 10.1053/j.gastro.2005.02.022. [DOI] [PubMed] [Google Scholar]
- 57.Shi C, et al. Down-regulation of the forkhead transcription factor Foxp1 is required for monocyte differentiation and macrophage function. Blood. 2008;112(12):4699–4711. doi: 10.1182/blood-2008-01-137018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lowell CA, Soriano P, Varmus HE. Functional overlap in the src gene family: inactivation of hck and fgr impairs natural immunity. Genes Dev. 1994;8(4):387–398. doi: 10.1101/gad.8.4.387. [DOI] [PubMed] [Google Scholar]
- 59.Pinheiro da Silva F, et al. CD16 promotes Escherichia coli sepsis through an FcR gamma inhibitory pathway that prevents phagocytosis and facilitates inflammation. Nat Med. 2007;13(11):1368–1374. doi: 10.1038/nm1665. [DOI] [PubMed] [Google Scholar]
- 60.Baadsgaard O, Wulf HC, Wantzin GL, Cooper KD. UVB and UVC, but not UVA, potently induce the appearance of T6– DR+ antigen-presenting cells in human epidermis. J Invest Dermatol. 1987;89(1):113–118. doi: 10.1111/1523-1747.ep12580461. [DOI] [PubMed] [Google Scholar]
- 61.Meunier L, Bata-Csorgo Z, Cooper KD. In human dermis, ultraviolet radiation induces expansion of a CD36+ CD11b+ CD1- macrophage subset by infiltration and proliferation; CD1+ Langerhans-like dendritic antigen-presenting cells are concomitantly depleted. J Invest Dermatol. 1995;105(6):782–788. doi: 10.1111/1523-1747.ep12326032. [DOI] [PubMed] [Google Scholar]
- 62.Atkins GB, et al. Hemizygous deficiency of Kruppel-like factor 2 augments experimental atherosclerosis. Circ Res. 2008;103(7):690–693. doi: 10.1161/CIRCRESAHA.108.184663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dagher Z, Ruderman N, Tornheim K, Ido Y. Acute regulation of fatty acid oxidation and amp-activated protein kinase in human umbilical vein endothelial cells. Circ Res. 2001;88(12):1276–1282. doi: 10.1161/hh1201.092998. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.