

Abstract
Reported are the synthesis of two intermediates for derivatization at position 6 of 7,7-dimethyl-7,8-dihydropterin: 6-carboxylic acid ethyl ester-7,7-dimethyl-7,8-dihydropterin, which is a novel compound, and 6-aldehyde-7,7-dimethyl-7,8-dihydropterin, which is synthesized by a new method with a yield of 90%.
Keywords: Pterin, Carboxylation, Bromination, Ester, Aldehyde
The folate biosynthetic pathway is essential for bacterial growth. Targeting key enzymes in the pathway, sulfonamides and diaminopyrimidines were developed as antibacterial drugs.1–4 As bacteria have developed resistance to those drugs, new agents are needed for the treatment of bacterial infection. 6-Hydroxymethyl-7,8-dihydropterin pyrophosphokinase (HPPK) is another key enzyme in the folate pathway. It catalyzes the transfer of pyrophosphate from ATP to 6-hydroxymethyl-7,8-dihydropterin (HP) to form AMP and 6-hydroxymethyl-7,8-dihydropterin pyrophosphate (HPPP).5–8
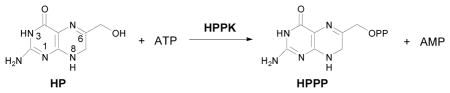
Targeting HPPK, we previously synthesized a series of bisubstrate analogs by linking 6-hydroxymethylpterin (1c, Scheme 1) to adenosine through 2, 3, or 4 phosphate groups, and submicromolar affinity to the enzyme was obtained for the tetraphosphate inhibitor.9 To improve the affinity and linker properties of such bisubstrate analogs, we have designed new compounds by replacing the pterin moiety (1a, Scheme 1) with 7,7-dimethyl-7,8-dihydropterin (2a, Scheme 1). Mimicking HP, 2a has one more hydrogen bond donor than 1a at position 8, which is important for binding to HPPK by forming a hydrogen bond with the carbonyl oxygen of a leucine residue of the protein.10,11
Scheme 1.

Pterin compounds: (a) R is absent, (b) R=CH3, (c) R=CH2OH, (d) R=OH, (e) R=CH2NHR′.
Stuart, Suckling, Wood, and colleagues12–15 developed methods for the synthesis of both 6,7,7-trimethyl-7,8-dihydropterin (2b, Scheme 1) and 6-hydroxymethyl-7,7-dimethyl-7,8-dihydropterin (2c, Scheme 1), but 2c was found to be degraded to 6-hydroxy-7,7-dimethyl-7,8-dihydropterin (2d, Scheme 1) upon prolonged incubation with HPPK.11 In addition, the synthesis of 2c was troublesome and the yield was low. A number of modifications were made, but the yield was still low.16 In contrast, 2b was easier to synthesize and stable, and the yield was high.12 In this study, we focused on the derivatization of 2b at position 6.
The first reaction of 2b derivatization was bromination. Previously, Stuart13,14 prepared 6-bromomethyl-7,7-dimethyl-7,8-dihydropterin (3a, Scheme 2) and 6-dibromomethyl-7,7-dimethyl-7,8-dihydropterin (3b) with bromine in glacial acetic acid (Scheme 2, i and ii, respectively). Compound 3b could be used to derive 6-aldehyde-7,7-dimethyl-7,8-dihydropterin (4b) at high temperature,17–21 but we found that the yield of the reaction was poor (Scheme 2iii). In general, bromination of methyl aromatic compounds could give tribromomethyl aromatic compounds that could be hydrolyzed to carboxylic acid aromatic compounds with silver nitrate in aqueous ethanol or sulfuric acid,22 but we found that although 2b gave 6-tribromomethyl-7,7-dimethyl-7,8-dihydropterin (3c, Scheme 2iv), compound 3c did not yield the suggested 6-carboxy-7,7-dimethyl-7,8-dihydropterin (4c, Scheme 2v). Instead, multiple products were found in the reaction mixture.
Scheme 2.

Derivatization of compound 2b: (i, ii) as reported,13,14 (iii–viii) this work, (ix, x) as reported.23,12
We attempted to make compound 2e by reacting 3a with amines (Scheme 2vi). The desired products could be detected by LC-MS, but the products decomposed to a yellow fluorescent compound that was identified as 4b (Scheme 2vii). To test whether we could derive 2e from 4b, we needed a better method for the synthesis of 4b. Using direct oxidation of methyl group by SeO2 as O’Neal and Jacobi reported previously,24 we derived 4b from 2b and the yield of 4b was 90% (Scheme 3).
Scheme 3.

New method of deriving 6-aldehyde-7,7-dimethyl-7,8-dihydropterin (4b) from 6,7,7-trimethyl-7,8-dihydropterin (2b).
Our method for the synthesis of 4b: A solution of 2b (207 mg, 1.0 mmol) in DMF (10 mL) and pyridine (105 uL, 1.30 mmol) was treated with SeO2 (145 mg, 1.30 mmol) and stirred at room temperature for 5 h. The reaction was then heated to 80 °C for 15 min. The solvent was evaporated under high vacuum and the residue purified by flash chromatography (silica gel, methanol:dichloromerhane = 2:8) to give 4b (199 mg, 0.9 mmol, 90%) as a yellowish powder. HRMS (ESI) calculated for C9H11N5O2 ([M+H]+) 222.0981, found 222.0991; 1H NMR (400 MHz, CD3OD) δ 1.53 (6 H, s), 9.33 (1 H, s); UV (0.1%AcOH), 400 nm.
Indeed, with 4b, compound 2e could be made by reductive amination reacting with an amine (Scheme 2viii). Again, the desired products, which could be detected by LC-MS, decomposed to the starting material (Scheme 2vii). Taken together, compound 2e could be made with either 3a or 4b, but the products would still hydrolyze to 4b (Scheme 2). Previously, Suckling and co-workers23,12 reported that 4b was obtained by gently oxidizing 2c or a pterin ether analog (5a) (Scheme 2, ix and x, respectively).
Compound 2e was not stable. We thought that the methylene group conjugating C=N could be a problem, and that an amide could improve the stability. Therefore, our second derivative of 2b contains a carboxyl group linked to the pterin moiety. For the synthesis of carboxylic acid substituted aromatic compounds from methyl or aldehyde substituted aromatic compounds, various methods with the use of oxidation reagents can be found in the literature.22 However, we found that these methods were not applicable for the synthesis of 2b derivatives. When 2b was mixed with the oxidation reagents, the characterized fluorescence of the compound disappeared.
For the synthesis of 3a, a variety of bromination reagents were tested, including bromine, triphenylphosphine dibromide, N-bromosuccinimide, and phosphorus tribromide. We also tested a variety of solvents such as chloroform, carbon tetrachloride, ethanol, N,N-dimethylformamide, and tetrahydrofuran. Usually, this kind of reaction gives a mixture of mono, bi, and tribromomethyl compounds. However, when ethanol was used as the solvent and the reaction mixture was separated by silica gel chromatography, light-yellow plate-shaped crystals were found in the collecting tubes. These crystals had a blue fluorescence, different from 4b (yellow fluorescent) and 3a (green fluorescent). The crystals were collected and identified as a new compound, 6-carboxylic ethyl ester-7,7-dimethyl-7,8-dihydropterin (6a, Scheme 4). We report here that 6a was produced by heating 2b with bromine in an ethanol solution (Scheme 4).
Scheme 4.

Preparation of 6-carboxylic ethyl ester-7,7-dimethyl-7,8-dihydropterin (6a).
To generate 6a, 2b (0.5 g, 2.4 mmol) was dissolved in 80 ml ethanol in a heavy wall pressure vessel, bromine (0.43 ml 8.4 mmol) was added dropwise to the solution, and the pressure vessel was sealed with a Teflon bushing. The solution was heated overnight at 120 °C. The ethanol was evaporated and the residue was purified by a Teledyne column chromatography system. The desired compound 6a (35%) was obtained as a yellow solid. N-bromosuccinimide (NBS) is one of the best bromination reagents. Using NBS instead of bromine in this procedure, however, the yield of 6a was similar. HRMS (ESI) calculated for C11H15N5O3 ([M+H]+) 266.1248, found 266.1270; 1H NMR (400 MHz, CD3OD) δ 1.52 (6 H, s), 1.32 (3 H, t), 4.23 (2 H, q); UV (0.1% AcOH), 380nm.
Goswami and coworkers25 reported the side chain bromination of methyl heteroaromatic and aromatic compounds by solid phase NBS reaction under microwave to produce dibromo and carbaldehyde heterocyclic compounds. We tested the reaction with microwave but in solution. Compound 2b (0.5 g, 2.4 mmol) and NBS (1.28 g, 7.2 mmol) were dissolved in 20 ml ethanol and the reaction mixture was heated in a Biotage microwave initiator for 5–25 min. The ethanol was evaporated and the residue was purified by the Teledyne column chromatography system. The desired compound 6a (52%) was obtained as a yellow solid. We also added azobisisobutyronitrile (AIBN) in the reaction, but the yield (44%) was not improved. Bromine was also tested in the microwave reaction system. Due to a pressure-related issue, we had to decrease the reaction temperature and extend the reaction time; however, the yield (14%) was poor.
Overall, the microwave-assisted reaction had higher yield than conventionally heated reaction, but the microwave gave one byproduct which was identified to be 2d: HRMS (ESI) calculated for C8H11N5O2 ([M+H]+) 210.0986, found 210.0988; 1H NMR (400 MHz, CD3OD) δ 1.39 (s). The removal of the byproduct, however, was difficult in the reaction workup. In this reaction, NBS and bromine played the same role. Without the microwave, we prefer the use of bromine because the reaction was cleaner.
In summary, we synthesized a novel intermediate, 6-carboxylic acid ethyl ester-7,7-dimethyl-7,8-dihydropterin (6a, Scheme 4), and found a new way to derive 6-aldehyde-7,7-dimethyl-7,8-dihydropterin (4b, Scheme 3). With these two compounds, it is easy to extend the side chain at position 6 of 7,7-dimethyl-7,8-dihydropterin (2a, Scheme 1). Such derivatives of these two compounds can be used as antifolate agents (antibacterials, antimalarials, and anticancer drugs),3 as nitric oxide synthase activators for the treatment of cardiovascular diseases,26,27 and as pteridine reductase inhibitors targeting African sleeping sickness and the Leishmaniases.28 The method can also be applied to other systems from methyl heteroaromatic and aromatic compounds to carboxylic ester heteroaromatic and aromatic compounds.
Acknowledgments
We thank Dr. Larry Keefer for critical reading of the manuscript. This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research and the Trans NIH/FDA Intramural Biodenfense Program Y3-RC-8007-01 from the NIAID. Mass spectrometry experiments were conducted with an Agilent 1100 series LC/Mass Selective Detector maintained by the Biophysics Resource in the Structural Biophysics Laboratory, an Agilent 1200 LC/MSD-SL system in the Chemical Biology Laboratory, and a Thermoquest Surveyor Finnigan LCQ deca maintained by the Comparative Carcinogenesis Laboratory of National Cancer Institute at Frederick.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Franklin TJ, Snow GA. Biochemistry and molecular biology of antimicrobial drug action. 6. Springer Verlag; 2005. [Google Scholar]
- 2.Gale EF, Cundliffe E, Reynolds PE, Richmond MH, Waring MJ. The molecular basis of antibiotic action. John Wiley & Sons; 1981. [Google Scholar]
- 3.Kompis IM, Islam K, Then RL. Chem Rev. 2005;105:593–620. doi: 10.1021/cr0301144. [DOI] [PubMed] [Google Scholar]
- 4.Russell AD, Chopra I. Understanding antibacterial action and resistance. Ellis Horwood; London: 1996. [Google Scholar]
- 5.Bermingham A, Derrick JP. Bioessays. 2002;24:637–648. doi: 10.1002/bies.10114. [DOI] [PubMed] [Google Scholar]
- 6.Derrick JP. Vitamins & Hormones. 2008;79:411–433. doi: 10.1016/S0083-6729(08)00415-9. [DOI] [PubMed] [Google Scholar]
- 7.Blaszczyk J, Shi G, Li Y, Yan H, Ji X. Structure (Camb) 2004;12:467–475. doi: 10.1016/j.str.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 8.Yan H, Ji X. Protein Pept Lett. 2011;18:328–335. doi: 10.2174/092986611794654003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shi G, Blaszczyk J, Ji X, Yan H. J Med Chem. 2001;44:1364–1371. doi: 10.1021/jm0004493. [DOI] [PubMed] [Google Scholar]
- 10.Blaszczyk J, Shi G, Yan H, Ji X. Structure. 2000;8:1049–1058. doi: 10.1016/s0969-2126(00)00502-5. [DOI] [PubMed] [Google Scholar]
- 11.Hennig M, Dale GE, D’Arcy A, Danel F, Fischer S, Gray CP, Jolidon S, Muller F, Page MG, Pattison P, Oefner C. J Mol Biol. 1999;287:211–219. doi: 10.1006/jmbi.1999.2623. [DOI] [PubMed] [Google Scholar]
- 12.Al-Hassan SS, Cameron RJ, Curran AWC, Lyall WJS, Nicholson SH, Robinson DR, Stuart A, Suckling CJ, Stirling I, Wood HCS. J Chem Soc, Perkin Trans. 1985;1:1645–1659. [Google Scholar]
- 13.Stuart A. 3,939,160. US Patent. 1976
- 14.Stuart A. 4,036,961. US Patent. 1977
- 15.Wood HC, Stuart A. 3,635,978. US Patent. 1972
- 16.Li G, Felczak K, Shi G, Yan H. Biochemistry. 2006;45:12573–12581. doi: 10.1021/bi061057m. [DOI] [PubMed] [Google Scholar]
- 17.Augustine JK, Naik YA, Mandal AB, Chowdappa N, Praveen VB. Tetrahedron. 2008;64:688–695. [Google Scholar]
- 18.Bankston D. Synthesis. 2004;2:283–289. [Google Scholar]
- 19.Abrishami F, Teimuri-mofrad R. Can J Chem. 2007;85:352–357. [Google Scholar]
- 20.Larock RC. Comprehensive organic transformations: a guide to functional group preparations. VCH; New York: 1989. [Google Scholar]
- 21.Luzzio FA, Wlodarczyk MT. Tetrahedron Lett. 2009;50:580–583. [Google Scholar]
- 22.Hudlicky M. ACS Monograph. p. 186. [Google Scholar]
- 23.Al-Hassan SS, Cameron R, Nicholson SH, Robinson DH, Suckling CJ, Wood HCS. J Chem Soc, Perkin Trans. 1985;1:2145–2150. [Google Scholar]
- 24.O’Neal WG, Jacobi PA. J Am Chem Soc. 2008;130:1102–1108. doi: 10.1021/ja0780075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goswami S, Dey S, Jana S, Adak AK. Chem Lett. 2004;33:916–917. [Google Scholar]
- 26.Suckling CJ, Gibson CL, Huggan JK, Morthala RR, Clarke B, Kununthur S, Wadsworth RM, Daff S, Papale D. Bioorg Med Chem Lett. 2008;18:1563–1566. doi: 10.1016/j.bmcl.2008.01.079. [DOI] [PubMed] [Google Scholar]
- 27.McInnes CR, Suckling CJ, Gibson CL, Morhtala RR, Daff S, Gazur B. Pterieines. 2009;20:27–35. [Google Scholar]
- 28.Tulloch LB, Martini VP, Iulek J, Huggan JK, Lee JH, Gibson CL, Smith TK, Suckling CJ, Hunter WN. J Med Chem. 53:221–229. doi: 10.1021/jm901059x. [DOI] [PMC free article] [PubMed] [Google Scholar]