

Summary
Therapeutic cancer vaccines are emerging as novel and potent approaches to treat cancer. These vaccines enhance the body’s immune response to cancerous cells, and dendritic cells (DCs), an initiator of adaptive immunity, are a key cell type targeted by these strategies. Current DC-based cancer vaccines are based on ex vivo manipulation of the cells following their isolation from the patient, followed by reintroduction to the patient, but this approach has many limitations in practical cancer treatment. However, recent progress in materials science has allowed the design and fabrication of physically and chemically functionalized materials platforms that can specifically target DCs in the body. These materials, through their in vivo modulation of DCs, have tremendous potentials as new cancer therapies. Nanoparticles, which are several orders of magnitude smaller than DCs, can efficiently deliver antigen and danger signals to these cells through passive or active targeting. Three-dimensional biomaterials, with sizes several orders of magnitude larger than DCs, create microenvironments that allow the effective recruitment and programming of these cells, and can be used as local depots of nanoparticles targeting resident DCs. Both material strategies have shown potential in promoting antigen-specific T cell responses of magnitudes relevant to treating cancer.
Keywords: Cytotoxic T Lymphocytes, Dendritic Cells, Hydrogels, Immunotherapy, Nanoparticles, Porous Scaffolds
Introduction
Immunotherapy with protein drugs (e.g., cytokines and monoclonal antibodies) is becoming a standard approach for cancer management. Therapeutic cancer vaccines, another form of immunotherapy, are rapidly emerging as a promising new approach to treat cancer. Cancer vaccines are designed to invoke strong anti-tumor immune activity, and the induction of antigen-specific cytotoxic (CD8+) T lymphocytes (CTLs) are believed to be a critical aspect of their function [1–3]. Activated CD8+ T cells can kill tumor cells upon recognition of specific labels (antigens) present on tumor cells, and this recognition is dependent on binding of the label to a T cell receptor (TCR) specific to that antigen. Dendritic cells (DCs) are believed to be the most important antigen presenting cells (APCs), and play a key role in initiating CTL responses [4–7]. In 2010 April, the first DC-based therapeutic cancer vaccine, known as Provenge, was approved by the Food and Drug Administration [8]. This breakthrough in cancer therapy demonstrated that one can stimulate a patient’s own immune system to fight cancer. However, this therapy is based on ex vivo manipulation of DCs in order to generate large numbers of these cells, and to activate the cells with cancer antigen, and thus suffers from a high cost and significant regulatory burden. In addition, tumors were not eradicated with this therapy, and the increase in patient survival time has been limited to 4 months to this point. While this breakthrough will undoubtedly have a major impact on cancer treatment, it also highlights the need to make further progress on the DC-based cancer vaccine strategy, and to bypass its dependency on ex vivo manipulation.
Recent progress in material science has led to new biomaterials and the applications of materials in a wide range of biomedical applications, including diagnostics, cancer therapy, and tissue regeneration. In particular, nanoparticles and macroscopic, three-dimensional biomaterials have significant potential in many clinical applications. Because of their nanosize and easy surface modification, targeting of nanoparticles to various tissues, including tumors and lymph nodes can be exploited to deliver imaging or therapeutic modalities [9–13]. 3-D macroscale biomaterials, especially porous scaffolds, have been extensively explored for applications involving the controlled release of growth factors, cell delivery, and tissue regeneration [14–22]. These materials create microenvironments that allow the fate of resident cells to be modulated, typically via control over the physical properties and presentation of cell signaling molecules from the walls of the materials. These 3-D macroscale materials and nanoparticles are being increasingly explored for their relevance to therapeutic vaccination in the context of cancer, particularly via the targeting and programming of specific immune cell populations.
In this review, basic concepts in immunity, focused on the important roles of DCs, and efforts to date to develop cancer vaccines are first introduced. Next, the recently approved DC-based cancer vaccine used to stimulate CTL responses will be discussed, as this provides a benchmark for material-based approaches. The majority of this article will then review current efforts in materials science to develop new cancer vaccines based on the modulation of DCs in the body, with a focus on two types of materials systems, nanoparticles and 3D macroscale biomaterials.
Dendritic cells
Since the discovery of DCs in 1973 by Ralph Steinmann and Zanvil Cohn [23], these cells have been the focus of intensive research aimed at understanding their role in the initiation of adaptive responses in the context of cancer. Dendritic cells, named for their dendritic shapes (inset of Fig. 1), are believed to be the most important antigen-presenting cells (APCs) of the body, are both initiators and modulators of immune responses, and have the ability to induce primary immune responses [4–6]. A brief summary of DC biology, relevant to their role in immune responses upon infection, is provided in Fig. 1. DCs normally reside in peripheral tissues (e.g., skin) in an immature state (the state of DCs before they encounter antigen). Immature DCs are not present in high numbers, but are spread over the body and have an extraordinarily high phagocytic ability. When present in the skin and mucosal barriers, they have a high probability of encountering pathogens invading the body, and DCs serve as sentinels that seek out foreign invaders, such as bacteria, viruses, or dangerous toxins. Immature DCs are very active in eliminating pathogens or antigens by non-specific or receptor-mediated phagocytosis (solid antigens) and macropinocytosis (soluble antigens).
Figure 1.

DC biology relevant to creating a cascading cytotoxic T lymphocyte (CTL) response upon infection. Immature DCs (iDC) in peripheral tissues (e.g., skin) encounter and eliminate pathogens by phagocytosis. DCs digest the antigens into small fragments of peptides, present those on their surface coupled to the major histocompatibility complex (MHC), and become mature DCs (mDC). Mature DCs also express receptors (e.g., CCR7) that allow them to migrate to lymph nodes (LNs) in response to gradients of chemokine (e.g., CCL19/21) secreted from LNs. In LNs, DCs transfer the antigenic information to naïve CD8+ T cells via interactions between their surface receptors, including both MHC/antigen-T cell receptor (TCR) binding and CD80/86-CD28 interactions. CTLs are particularly driven by recognition of fragments presented from MHC class I molecules (one particular type of MHC). The activated CTLs leave the LNs to kill infected cells or pathogens. Inset: fluorescent microscope image of dendritic cells [5].
Once immature DCs uptake foreign substances, called antigens, they digest them into small fragments and present them from their surface, in the process typically becoming a mature DC. The fragments of foreign agents are presented from cell surface receptors called the major histocompatibility complex (MHC), and the MHC-antigen complex can be recognized by T cell receptors. The activation of cytotoxic CD8+ T cells (which can kill pathogen-infected cells or cancerous cells) is driven specifically by this binding and recognition of fragments presented from MHC class I molecules. Usually the exogenous molecules taken up by DCs are fragmented by proteases in the DC endosomes, and antigenic peptides are then loaded onto MHC class II molecules that will bind with and activate CD4+ helper cells. Alternatively, the exogenous antigens are also processed through a pathway called “cross-presentation” in which dendritic cells take up antigens and efficiently present the MHC class I-peptide complex on the cell surface to activate CD8+ T cells, and this is believed to be necessary to develop an effective therapeutic cancer vaccine.
The cells of the immune system have been extensively characterized in terms of the various cell surface receptors they express, allowing one to readily probe the state of the cells. Mature DCs express high levels of co-stimulatory molecules (CD80, CD86) and adhesion molecules (ICAM-1) on their surface, and these mediate their interactions with naïve T cells in secondary lymphatic organs, especially the lymph nodes. Mature DCs also express high levels of CCR7, a lymph node homing receptor, as this allows them to migrate to the lymph nodes in response to a gradient of the chemokines, CCL-19 and CCL-21, secreted from lymph node cells [24]. Naïve T and B cells reside in high numbers in the lymph nodes, allowing the migrating DCs to interact with these cells and transfer the antigenic information that they cargo on their surfaces in the form of MHC-antigenic peptide complexes. CD8+ T cells (also called cytotoxic T lymphocytes (CTLs)), are important effector cells for effective cancer vaccine. In the lymph node, the DCs present cancer antigenic peptides coupled to MHC molecule to the TCR of CD8+ T cells. Binding interactions between costimulatory molecules (CD80, CD86) on DCs and CD28 on the T cells are also required to activate the CD8+ cells, and cause these cells to become cytotoxic and expand their number in presence of cytokines secreted by DCs and other T cells. The activated CTLs leave the lymph nodes via efferent lymphatic vessels, enter the blood stream, and migrate to peripheral sites of inflamed tissue in response to various inflammatory cytokines secreted from infected cells and tissue around the site of infection. Finally CTLs specifically kill the pathogens or infected cells that express the same antigentic peptide molecules presented by the DCs.
An important aspect of DC biology is that antigenic information alone is not sufficient to generate strong CTL responses. A variety of other cues, generically termed danger signals, are required to efficiently activate DCs. These danger signals contain conserved molecular patterns present in common infectious agents, termed pathogen associated molecular patterns (PAMPs), and bind to DC receptors as they process antigen. Toll like receptors (TLRs) are one family of these danger signal receptors in DCs [25,26]. Various TLR agonists including lipopolysaccharide (LPS), polyinosinic:polycytidylic acid (poly I:C), or unmethylated cytosine-phosphate-guanosine oligodeoxynucleotide (CpG-ODN), bind to TLRs to activate DCs [25,26]. The presence of danger signals greatly enhances the resulting expression levels of costimulatory molecules (e.g., CD80, CD86) and cytokines by mature DCs that are needed to activate CD8+ T cells to kill the cancer cells.
DC-Based Cancer Vaccine
Since the historical success of Jenner’s first vaccination strategy against smallpox [27], the development of vaccines for other infectious diseases has been pursued intensively and has often been successful. It has also become apparent that cancer can be prevented or treated by antigen specific immunization, as the same mechanism of CTL response introduced in the last section applies to cancerous cells. However, the immune system faces several hurdles that subvert its ability to detect and kill the cancer cells, in part because the microenvironment in cancer is more complex than other infectious diseases. Cancer cells possess various immune-escape mechanisms, including high antigenic mutation, secretion of immunosuppressive cytokines, and down-regulation of MHC molecule expression [28,29]. In general, cancers that are better at escaping host immunity will be more malignant than those that inefficiently evade immune responses. Even if DCs successfully take up antigen from cancer cells, tolerizing signals secreted from tumor cells can suppress the immune reactions of DCs and CTLs [30]. Cells comprising tumors are often heterogeneous, and have low expression of MHC I molecules, rendering them invisible to CTLs [31]. These factors together can circumvent the ability of the immune system to spontaneously generate a potent attack on the tumor. The immune suppressive environment of tumors suggests active interventions are required to induce potent therapeutic cancer vaccines.
To overcome the intrinsic immunosuppressive capacity of tumors, the first FDA-approved cancer vaccine isolates immune cell precursors from the patient’s body, and thus from the influence of the tumor, and matures and loads the DCs with antigen before returning them to the patient (Fig. 2). First, a patient’s own monocytes [32] are isolated and cultured in the presence of cytokine cocktails (granulocyte-macrophage colony stimulating factor (GM-CSF) and interleukin (IL)-4) to generate autologous DCs. The resulting immature DCs in culture are pulsed with tumor antigens and TLR agonists. The activated DCs presenting tumor antigen on their surface are then injected back to patient’s body, where it is expected they will migrate to LNs to invoke tumor-specific CTL responses.
Figure 2.

DC-based cancer vaccine via ex vivo manipulation of DCs. A patient’s own peripheral blood mononuclear cells (PBMCs) are taken from the blood, followed by isolation of monocytes from PBMCs. Monocytes are cultured in the presence of cytokine cocktails (GM-CSF and IL-4) to generate autologous DCs. The immature DCs are pulsed with tumor antigens and TLR agonists to generate mature DCs presenting tumor antigen on their surface. Finally the mature DCs are injected back to patient’s body, where it is expected they will migrate to LNs to invoke tumor-specific CTL responses.
Although the effectiveness of this lab-based DC vaccine has been demonstrated in both preclinical animal models and in human clinical studies [33], the improvement in patient survival is limited, and it is a labor-intensive and expensive process. The vast majority of the cells injected back into the patient rapidly die, with few migrating to the LNs (estimated at ~ 0.5–2 %), and their activation may be transient [2,3,34]. Together, these issues likely underlie the limited effectiveness of this first generation therapeutic cancer vaccine, and provide practical obstacles to its large-scale application. In contrast, the modulation of DCs already present in the body, without any laboratory manipulation, may allow one to more efficiently and effectively activate the cells, allow for a more inexpensive treatment, require less time to initiate the immune cascade, and generate more potent immune responses.
Material-Based Cancer Vaccines
The in vivo activation of DCs, obviating the need for manipulating cells outside of the body, could potentially overcome many of the limitations of current cell-based cancer vaccines. There have been attempts to use simple formulations of synthetic peptides or proteins as vaccines, similar to what is done with infectious disease vaccination. These are much simpler than cell-based vaccines, but their clinical efficacy has not been satisfactory [35–37]. One of the major obstacles of peptide- or protein-based vaccines is that uptake and/or presentation of the antigens is not performed by professional APCs, lead to insufficient stimulation of T cells [38]. For efficient priming and activation of antigen-specific CTLs, sufficient quantities of antigens should be presented to T cells by functionally activated, professional APCs for an appropriate period of time. Material-based delivery of antigens is a promising approach for cancer vaccines, due to the ability of materials to either passively or actively target cell populations in the body, and to prolong antigen presentation in concert with presentation of costimulatory signals [39].
Material-based antigen delivery offers a number of advantages over the administration of soluble antigens. First, materials can be localized or targeted to specific tissues. Materials can also be surface functionalized with antigen, targeting molecules, or protecting layers. Multiple agents, including antigen, danger signals, and cytokines, can be loaded into a single material delivery system; alternatively, multiple materials, each delivering a single factor, can be readily combined. Antigens and adjuvants are typically protected from degradation by proteases, RNAses, and DNAses in the body following encapsulation in a material, allowing DCs a better chance to acquire the antigen. Material-based antigen delivery also can achieve cross-presentation of the antigen by DC. The simplicity in design and synthesis of materials, coupled with their multifunctionality makes materials attractive tools for cancer vaccine.
The cytotoxity of materials, including various nanoparticles and biodegradable polymers, is a key factor in their evaluation to proceed to clinical use. The materials used in vaccine systems are desired to be biocompatible and have low cytotoxicity, in order to reduce side effects after vaccination. The chemical and physical characteristics, such as size, surface functional group, and composition, should be controlled to reduce the potential cytotoxicity of materials-based cancer vaccine.
Nanoparticles that target either cells in peripheral tissue (Langerhans cells, dermal DCs), or target cells residing in the lymph nodes will be reviewed in the following sections, as will scaffolds that mimic infection by instead recruiting the APCs to themselves, and then presenting antigen and other cues to activate the APCs.
Nanoparticles to target DCs in peripheral tissue
Nanoparticles can be targeted to peripheral DCs by subcutaneous or intradermal injection. Upon injection of nanoparticle via the subcutaneous or intradermal routes, nanoparticles predominantly remain at the injection site, while some fraction enter the draining lymph nodes.[40] The nanoparticles gradually enter into the blood stream over time and finally accumulate in different organs, especially liver, or are excreted via urine and feces. DCs first internalize the nanoparticles and process the antigens, and then migrate to the lymph nodes within 1~2 days (Fig. 3). Antigens and danger signals can be coencapsulated in single nanoparticles and delivered into DCs. DC-specific targeting molecules coupled on the surface of nanoparticles further allow active DC-targeting and more efficient delivery of antigens to DCs.
Figure 3.

Nanoparticles loaded with cancer antigens and danger signals can target DCs in peripheral tissue via passive phagocytosis or active DC targeting with specific antibodies. Following nanoparticle uptake and processing, DCs become mature, present antigens on their surface, and migrate to LNs to activate T cells.
Antigenic protein or peptides can be coupled to the surface of nanoparticles or encapsulated in the particles. Various types of nanoparticles, including liposome-based [41–46], poly(lactic-co-glycolic acid) (PLGA) [47–57], polyglycolic acid (PGA) [58–63], polystyrene (PS) [64], gelatin [65,66] and other polymers [67,68], gold [69], and magnetic [70] nanoparticles have been studied for antigen delivery to DCs. For example, ovalbumin (OVA), a model antigen, was incorporated in pH-sensitive synthetic liposomes (Fig. 4a) [42]. Healthy mice were immunized with ovalbumin-encapsulating liposomes or soluble ovalbumin, and T cells were isolated from the mice’s spleens to test if antigen-specific CTLs were generated. Liposome-encapsulated OVA resulted in an antigen-specific, CD8+ T cell response, while this response was not generated by immunization with equivalent amounts of soluble OVA (Fig. 4b). Similar results were also reported using other nanoparticles encapsulating OVA [41,42,44,46,47,49,50,52,58,59,61,64], supporting the finding that nanoparticle-mediated antigen delivery enhances cross-presentation of exogenous antigens and the induction of a CTL response.
Figure 4.

Immunization with antigen-loaded liposome enhances antigen-specific lysis by CTL in compared to immunization with soluble antigen. (a) Schematic presentation of antigen (OVA)-loaded liposome and soluble OVA used in immunization. (b) OVA-specific in vitro CTL activity using T cells isolated from mouse spleen [42].
In order to efficiently mature DCs, allowing for their migration to the LNs and activation of T cells, antigen presentation from a material can be combined with TLR ligation. For example, the TLR 9 ligand, CpG oligonucleotide, has been encapsulated in various nanoparticles along with antigens to accomplish this goal [43,46,51,53,65,66]. PLGA particles were used to encapsulate both OVA and CpG (Fig. 5a) [53]. In vitro stimulation of DCs with these nanoparticles increased expression of the DC activation markers, CD86, MHC I, and MHC II. The efficiency of CTL priming in mice after vaccination with biodegradable PLGA particles was enhanced when OVA was co-encapsulated with a CpG, as contrasted to co-inoculation of OVA-bearing particles with soluble or separate particles encapsulating CpG (Fig. 5b and 5c), indicating that co-delivery of TLR ligands and antigen (in the same particle) is likely an important paradigm for the generation of potent CTL responses.
Figure 5.

Co-encapsulation of antigen (OVA) and TLR ligand (CpG) leads to efficient CTL response. (a) Schematic presentation of PLGA particles encapsulating OVA and CpG in a single particle, and PLGA particles loaded separately with OVA and CpG, respectively. (b) Representative flow cytometry plots of splenocytes from mice 6 days after subcutaneous vaccination with single PLGA formulation containing coencapsulated OVA and CpG oligonucleotide or physical mixture of OVA- and CpG-loaded PLGA particles [53]. SIINFEKL tetramer is specific peptide sequence for OVA, and the number in the box represents the percentage of T cells specific for this OVA antigen with the two vaccinations approaches. (c) Quantitative representation of in vivo cytolysis of injected tumor cells [53].
Although nanoparticles are generally taken up and endocytosed by DCs, the efficiency is relatively low as there is no specific targeting to DCs. Modification of the surface of nanoparticles with specific DC-targeting groups has been pursued to increase antigen presentation by DCs. A number of DC-specific (or positive) surface markers exist, such as CD11c and DEC-205, and antibodies for these receptors have been used to increase the targeting efficiency of antigen-containing nanoparticles to DCs [44,55,67]. For example, peripheral DCs have been targeted with antigen-containing liposomes that were functionalized with a specific antibody targeting CD11c or DEC-205 (Fig. 6a) [44]. This led to a 4–8-fold increase in binding above control peptide-labeled liposomes, and the majority of DCs in the draining popliteal lymph node exhibit fluorescein fluorescence following footpad injection (Fig. 6b). One can also incorporate various other signals into these targeted liposomes to enhance the maturation of DCs following uptake. For example, mice immunized with DC-targeted liposomes incorporating IFN-γ or LPS had a greater ability to resist the metastases of melanoma cells, as compared to control mice immunized with liposome coupled with non-specific protein (Fig. 6c).
Figure 6.

Targeting DCs with antigen-containing liposome. (a) A schematic presentation of antigen-bearing liposome coupled with DC-specific antibody or non-specific protein. (b) FACS data for lymph node cells of mice injected with fluorescein-labeled liposome coupled with non-specific peptide and anti-DEC-205 [44]. The upper right quadrant of each plot represents the percentage of DCs containing fluorescein-labeled liposome. (c) Representative images of the metastasis of melanoma to lungs in mice when the mice were vaccinated with liposome containing IFN-γ and engrafted with non-specific peptide or anti-DEC-205 (metastasis are black spots) [44].
Nanoparticles to target DCs in lymph node
An alternative approach to targeting peripheral DCs that must subsequently migrate to the LNs is to instead target immature DCs already residing in the LNs. A substantial fraction of resident DCs in the lymph nodes are phenotypically immature and capable of internalizing antigens and particles [71,72]. Further, there is higher density of DCs in lymph nodes than in peripheral tissue, and large numbers of naïve T and B cells available for activation. Various nanoparticles, including quantum dots, magnetic nanoparticles, and radio-labeled nanoparticles have been previously used to image the sentinel lymph node (closest lymph node from diseased tissue, especially cancer) in order to diagnose and treat metastatic cancer [73–77], providing a starting point for designing particles to target LN-resident DCs. The basis for this approach is that fluid and particulates from the interstitial space of tissues flows into the lymphatic circulation, providing an avenue for the accumulation of antigens in LN [78]. Nanoparticles can be readily taken into lymphatic vessels and retained in the draining lymph nodes.
Successful LN targeting of nanoparticles is dependent on their size [79–81]. The effects of nanoparticle size on lymphatic uptake, lymph node retention, and internalization by lymph node DCs were explored following intradermal injections of 20, 45, and 100 nm polypropylene sulfide (PPS) nanoparticles into mice [79,80]. It was found that 20 nm particles were more readily taken up into the lymphatics than 45 and 100nm (Fig. 7a), and that they accumulated in the lymph nodes at earlier times (Fig. 7b). Within the lymph node, nanoparticles were internalized by resident APCs, even without a targeting ligand (Fig. 7c). A large fraction (up to 50%) of lymph node resident DCs internalized the 20 nm particles. Intradermal injection of antigen-conjugated nanoparticles promoted specific humoral and CTL immunity, suggesting that LN targeting with antigen-nanoparticles can be a promising strategy for cancer vaccination (Fig. 7d).
Figure 7.

Targeting of nanoparticles to lymph node. (a) Schematic presentation of 100- and 25-nm fluorescently labeled PPS NPs (left) and corresponding fluorescence lymphangiography after injection of NPs into mouse-tail skin (right) [80]. 25-nm NPs enter the dermal lymphatic capillary network much more efficiently than 100-nm NPs. Scale bar, 1 mm. (b) The 25-nm, but not the 100-nm, nanoparticles are visible in mouse lymph node sections 24 h after injection [80]. Cell nuclei shown in blue (DAPI); scale bar, 200 mm. (c) Quantification of FACS data for uptake of NPs by MHC II + cells and CD11c+ cells (DCs) in lymph node [79]. (d) A restimulation assay data to determine CD8 T-cell memory by IFN-γ production, showing CD8 T-cell memory after treatment with OH-functionalized 25 nm OVA-NPs but not CH3O-functionalized NPs [80].
The size of nanoparticles also influences how they traffic to and where they localize in the LNs [81]. Shortly after injection, 20 nm polystyrene (PS) nanoparticles were found in the subcapsular region of the LN (Fig. 8a, 20 nm 2 h), a pattern consistent with drainage from the afferent lymphatics. At 48 h after injection, these particles were distributed in the subcapsular area, as well as in the paracortex and cortex of the LN in tight proximity with B cell follicles (Fig. 8a, 20 nm 48h). In contrast, 1000 nm nanoparticles were excluded from the subcapsular sinus and found in areas more distal from B cell follicles where DC have been shown to reside (Fig. 8a, 1000 nm 48 h). The delivery route to LNs could be either by lymphatic flow or by migration of DCs that uptake the nanoparticles in peripheral tissue, and this was investigated with mice deficient in DCs. Twenty nm particles still reached the LN 2 h after injection in both the DC-depleted and control animals, while 500-nm nanoparticles were observed only at the injection site of both control and DC-deficient mice (Fig. 8b, upper row), demonstrating that small particles rapidly reach the LN independently of DC. At 48 h after injection, 500-nm particles had reached the LN of control animals, but not in DC-depleted mice (Fig. 8c, lower row), indicating that large particles traffic to the LN in a DC-dependent manner.
Figure 8.

Accumulation of nanoparticles to lymph node according to their size (diameter). (a) Immunofluorescence microscopy on frozen sections of popliteal LN isolated from mice injected with 20- or 1000-nm green fluorescent nanoparticles into footpad [81]. Red fluorescence depicts B220 staining specific for B cell follicles. Scale bar, 100 μm. (b) Trafficking of particles in vivo. Left and right footpads of DC-depleted (left) and control (right) mice were injected with 20- or 500-nm red fluorescent nanoparticles, respectively [81]. Popliteal LN that have acquired nanoparticles are shown with arrowheads and injection sites with arrows.
3D Polymer Scaffolds as Cancer Vaccines
In cancer, the immune system has already been tolerized to the cancer-specific antigens, suggesting a major challenge for cancer vaccines is to reprogram this pre-existing immune response and break tolerance. Materials that can create a highly defined microenvironment to load antigen, to mature DCs, and to minimize the influence of tolerance-inducing signals from tumors may be needed to provide a therapeutic immune response. Three dimensional (3D) scaffolds may be ideal for this purpose. 3D biomaterials have been the focus of much biomaterials research, due to their use in controlled drug and cell delivery [14,16–22], and their ability to control the kinetics and dose of released drugs can be exploited to allow in situ recruitment and manipulation of immune cells (Fig. 9). Immune cells are known to migrate in response to the gradients of several chemokines arising from infection sites and secondary lymphatic organs. These chemokines can be released from biomaterials in order to recruit host immature DCs and other immune cells [82–88]. Although there is burst of release from 3D biomaterials at the beginning, the subsequent sustained release of chemokines over time maintained an appropriate gradient in the outer tissue to recruit DCs. The gradients were preserved over the time frame of the DC activation and programming in vivo. Cancer antigens and TLR ligands can also be incorporated into these 3D biomaterials [82,84,86–88] in either soluble form or as nanoparticles, in order to load recruited DCs with antigen and promote their maturation. This strategy may ultimately provide effective, therapeutic immune responses to cancer.
Figure 9.

Schematic presentation of 3D polymer scaffold cancer vaccine. 3D porous polymer scaffold can load various signals, including chemokines, antigens, and danger signals, to manipulate dendritic cells in situ in the body. Immature DCs migrate to 3D polymer scaffold in response to the gradient of chemokine released, mature, and proliferate in the 3D microenvironment. The mature DCs then leave the scaffolds and migrate to the lymph nodes for activation of naïve T cell.
Solid polymer implants may be used to concentrate DCs in a particular tissue site, load the DCs with antigen, and influence their subsequent homing to LNs. Millimeter sized ethylene vinyl acetate (EVA) polymer rods encapsulating MIP-3β (CCL-19) or tumor antigens in the form of the model antigen OVA, tumor lysate, or antigenic peptide have been implanted subcutaneously to induce anti-tumor CTL response [82]. CCL-19 is a chemokine known for inducing migration of matured DCs expressing CCR7. Upon implant of polymer rods releasing CCL-19 in the presence of haptens (low-molecular weight molecules which contain an antigenic determinant but which are not themselves antigenic) in the implant site, DCs migrated to the site of implantation (Fig. 10a). The subsequent homing of these DCs to LN was also observed over 3 days (Fig. 10b). A CTL response was observed when both polymer rods releasing CCL-19 and antigen proteins were implanted in the presence of haptens (Fig. 10c). Two repeated prophylactic immunization with these polymer rods before tumor inoculation almost completely prevented the development of tumors (Fig. 10d) and four repeated vaccinations after tumor inoculation significantly inhibited tumor growth (Fig. 10e), supporting the potential relevance of this strategy for cancer treatment.
Figure 10.
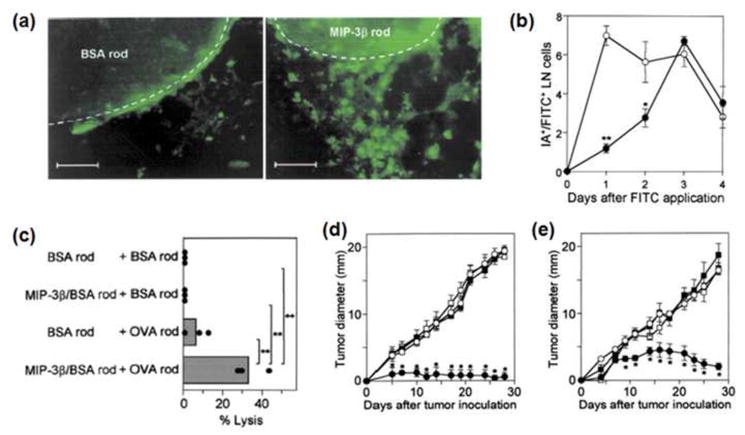
(a) Accumulation of MHC II+ cells (green) around BSA rods (left) and MIP-3β rods (right) [82]. Scale bar, 100 μm. (b) Kinetics of LN-homing DCs from BSA rods (◦) and MIP-3β rods (●)[82]. (c) CTL activities after co-implantation of OVA rods and MIP-3β rods against E.G7-OVA cells as target [82]. Prophylactic (d) and therapeutic (e) immunity through immunization of MIP-3b rods + antigen rods (●), BSA rod + antigen rods (◦), MIP-3b rods + BSA rods (■), or BSA rods + BSA rods (□)[82]. Prophylactic model received two immunization on day −14 and −7 along with hapten (DNFB) application, followed by 3LL tumor inoculation on day 0 [82]. Therapeutic model received 3LL tumor inoculation on day 0 and four immunization on days 2, 9, 16, 23 long with hapten application.
An injectable hydrogel matrix has also been investigated for its ability to recruit host DCs and T cells to the injection site, the periphery of a tumor, for local anticancer immunotherapy [83–85]. Ca2+-loaded alginate beads, providing a depot of Ca2+ ions, were mixed with a soluble alginate solution and mature dendritic cells, and the injected mixture rapidly gelled in vivo as calcium diffused out of the beads into the surrounding solution. The mature DCs in the alginate gels secreted inflammatory chemokines to attract host dendritic cells, and these transplanted DCs and recruited host DCs were capable of migrating to the LN to prime T cells. Activated T cells subsequently migrated back to the injection sites, and local anticancer immunotherapy was achieved by peritumoral injection of alginate gels loaded with mature DCs and an interleukin-15 superagonist (IL-15SA). An additional chemokine IL-15SA was necessary in this approach because the recruitment of CD8+ T cells was significantly decreased in the peritumoral injection site, probably due to immune suppressive microenvironment around the tumor. IL-15SA is known to expand CD8+ T cells, natural killer (NK) cells, and NK-T cells in vivo [89]. This strategy suppressed the growth of large established B16 tumors for up to a week, and similar antitumor effects were achieved by incorporating CpG along with IL-15SA in the gel without the use of mature DCs, suggesting this approach could be modified to develop a cancer vaccine not requiring ex vivo DC manipulation.
It has recently been proposed that porous polymer matrices that provide a temporary residence for DCs can effectively regulate host DC trafficking and activation in situ, while simultaneously preventing upregulation of the tolerizing arm of the immune system, and provide therapeutic protection against cancer [86–88]. Macroporous PLG scaffolds (Fig. 11a) incorporating i) GM-CSF to recruit DCs, ii) nanoparticles containing CpG to mature the DCs, and iii) tumor lysate to provide a mixture of cancer antigens were developed for this purpose. Upon subcutaneous implantation, GM-CSF was released and established a gradient in the surrounding tissue to recruit significant numbers of host DCs (Fig. 11b and c). The presentation of CpG nanoparticles from the polymer to the recruited DCs increased the maturation of DCs in the scaffolds and their LN-homing (Fig. 11d). These scaffolds induced strong specific CTL responses to melanoma in a prophylactic model, with a 90% survival rate (Fig. 11e) as well as in therapeutic models of melanoma and glioblastoma with over a 50% survival rate after vaccinations (Fig. 11f). Strikingly, this systems recruited various DC subsets, including significant numbers of plasmacytoid DCs (pDCs) and CD8+ DCs, which are very important in antigen cross presentation, and the numbers of these DC subsets strongly correlated with the vaccine efficacy [87]. This vaccine also diminished the local concentrations of tolerogenic cytokines (e.g., IL-10, TGF-β), and numbers of T regulatory cells (Fig. 11g), suggesting that a key aspect of its success related to its ability to down-regulate tolerance [87]. These effects were only found when the polymer had the physical form of a macroporous scaffold. When PLG microspheres loaded with GM-CSF, CpG and tumor lysates were used instead of a macroporous PLG scaffold, the vaccine effectiveness was significantly diminished. The decreased effectiveness was likely due to the inability of the microspheres to provide pores in which the recruited cells could reside. This result suggests that creating a microenvironment in which host environmental cues are minimized, and exogenous maturation factors are highly concentrated, may be a key to reprogram immune responses in situations such as cancer where there exist significant, pathology-associated tolerizing cues.
Figure 11.

(a) SEM image of macroporous PLG scaffold. Scale bar, 1000 μm. (b) H&E staining of sectioned PLG scaffolds explanted from subcutaneous pockets in the backs of C57BL/6J mice after 14 days: blank scaffolds (BLANK) and GM-CSF (3000 ng)-loaded scaffolds (GM-CSF) [86]. Scale bar, 500 μm. (c) Number of CD11c+ DCs isolated from PLG scaffolds at day 14 after implantation in response to doses of 0, 1000, 3000, and 7000 ng of GM-CSF [86]. (d) The number of CD11c+CCR7+ host DCs isolated from matrices loaded with PEI-ODN control, 10 μg PEI-CpG-ODN, 400 and 3,000 ng GM-CSF and 400 and 3,000 ng GM-CSF in combination with 10 μg PEI-CpG-ODN at day 7 after implantation [86]. Inset: photographs of inguinal lymph nodes from control mice (right) and after 10 days implantation of matrices incorporating 10 μg CpG-ODN + 3,000 ng GM-CSF. A comparison of the survival time in mice for (e) prophylactic [86] and (f) therapeutic [87] cancer vaccination. (g) Ratio of CD8a+ T cells versus FoxP3+ Treg cells residing within PLG scaffolds loaded with GM-CSF and lysates (GM+Lys) alone or in combination with CpG-ODN (GM+Lys+CpG) at day 12 after implantation [87].
Conclusion
This review documents the potential of nanostructured materials with appropriately engineered physical and chemical properties to generate strong cytotoxic immune responses without ex vivo manipulation of DCs. Diverse materials platforms have been designed to interact with DCs to deliver combinations of inflammatory chemokines, tumor antigens, and danger signals, which enable modulation of DCs in the body. Nanoparticles have been designed to target DCs, and aim to be internalized by DCs in order to release the antigens and danger signals intracellularly. Targeting peripheral DCs can be achieved either passively via non-specific endocytosis, phagocytosis, and micropinosis, or actively via specific targeting to surface receptor of DCs. Nanoparticles less than 50 nm in size can also be targeted to the lymph nodes through passive lymphatic flow in order to regulate DCs already in contact with T and B cells. Alternatively, 3D biomaterials with a size order of magnitude larger than DCs can be used to generate gradients of chemokines to recruit DCs, and to program the recruited cells. DCs targeted by nanoparticles or recruited into 3D cell niches can be matured by co-delivery of antigens and danger signals, allowing cross-presentation of antigen and triggering cytotoxic CD8+ T lymphocytes responses. Recent findings also suggest that the ability of material systems to create a microenvironment in which to program DCs, while excluding host cues promoting tolerance, may be a key to generating robust therapeutic responses. However, while significant advances have been made, our understanding of the molecular and cellular mechanisms underlying the ability of materials to modulate specific immune responses is still rudimentary. As we improve our understanding of these processes, this will allow the design of future generations of material-based vaccines that are even more potent.
Acknowledgments
The authors acknowledge support from the NIH/NIDCR (R01-DE019917), the DoD (BC084682 Idea Award), Harvard Clinical and Translational Science Center (NIH Grant #1 UL1 RR 025758-01), Harvard Institute for Translational Immunology Helmsley T1D Pilot Grants, and the Wyss Institute.
Biographies
 David Mooney is the is the Robert P. Pinkas Family Professor of Bioengineering in the School of Engineering and Applied Sciences at Harvard University, and a Core Faculty Member of the Wyss Institute for Biologically Inspired Engineering. His laboratory is developing materials to direct the trafficking and fate of cell populations in the body to regenerate damaged or diseased tissues, or the targeted destruction of undesirable tissue masses in the body. Dr. Mooney’s education and training is from the University of Wisconsin, Massachusetts Institute of Technology, and Harvard Medical School.
David Mooney is the is the Robert P. Pinkas Family Professor of Bioengineering in the School of Engineering and Applied Sciences at Harvard University, and a Core Faculty Member of the Wyss Institute for Biologically Inspired Engineering. His laboratory is developing materials to direct the trafficking and fate of cell populations in the body to regenerate damaged or diseased tissues, or the targeted destruction of undesirable tissue masses in the body. Dr. Mooney’s education and training is from the University of Wisconsin, Massachusetts Institute of Technology, and Harvard Medical School.
 Jaeyun Kim received his BS (2001), MS (2003), and PhD (2007) in School of Chemical and Biological Engineering under the supervision of Prof. Taeghwan Hyeon at Seoul National University. He worked as a visiting postdoctoral fellow (2008) in the Department of Radiology and Institute for Cell Engineering at the Johns Hopkins School of Medicine, and currently is working as a postdoctoral researcher at Harvard University and Wyss Institute under the direction of Prof. David Mooney. His current research is focused on cancer vaccine, therapeutic angiogenesis, and nano- and bio-materials for tissue engineering.
Jaeyun Kim received his BS (2001), MS (2003), and PhD (2007) in School of Chemical and Biological Engineering under the supervision of Prof. Taeghwan Hyeon at Seoul National University. He worked as a visiting postdoctoral fellow (2008) in the Department of Radiology and Institute for Cell Engineering at the Johns Hopkins School of Medicine, and currently is working as a postdoctoral researcher at Harvard University and Wyss Institute under the direction of Prof. David Mooney. His current research is focused on cancer vaccine, therapeutic angiogenesis, and nano- and bio-materials for tissue engineering.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schuler G, Schuler-Thurner B, Steinman RM. Curr Opin Immunol. 2003;15:138–147. doi: 10.1016/s0952-7915(03)00015-3. [DOI] [PubMed] [Google Scholar]
- 2.Gilboa E. J Clin Invest. 2007;117:1195–1203. doi: 10.1172/JCI31205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Steinman RM, Banchereau J. Nature. 2007;449:419–426. doi: 10.1038/nature06175. [DOI] [PubMed] [Google Scholar]
- 4.Banchereau J, Steinman RM. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 5.Steinman RM. Nat Med. 2007;13:1155–1159. doi: 10.1038/nm1643. [DOI] [PubMed] [Google Scholar]
- 6.Steinman RM. Immunity. 2008;29:319–324. doi: 10.1016/j.immuni.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 7.Steinman RM. Eur J Immunol. 2007;37(Suppl 1):S53–60. doi: 10.1002/eji.200737400. [DOI] [PubMed] [Google Scholar]
- 8.Jahnisch H, Fussel S, Kiessling A, Wehner R, Zastrow S, Bachmann M, et al. Clin Dev Immunol. 2010;2010:517493. doi: 10.1155/2010/517493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gao J, Gu H, Xu B. Acc Chem Res. 2009;42:1097–1107. doi: 10.1021/ar9000026. [DOI] [PubMed] [Google Scholar]
- 10.Kim J, Piao Y, Hyeon T. Chem Soc Rev. 2009;38:372–390. doi: 10.1039/b709883a. [DOI] [PubMed] [Google Scholar]
- 11.Zrazhevskiy P, Sena M, Gao X. Chem Soc Rev. 2010;39:4326–4354. doi: 10.1039/b915139g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Torchilin VP. Nat Rev Drug Discov. 2005;4:145–160. doi: 10.1038/nrd1632. [DOI] [PubMed] [Google Scholar]
- 13.Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nat Nanotechnol. 2007;2:751–760. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 14.Langer R, Vacanti JP. Science. 1993;260:920–926. doi: 10.1126/science.8493529. [DOI] [PubMed] [Google Scholar]
- 15.Lee KY, Mooney DJ. Chem Rev. 2001;101:1869–1879. doi: 10.1021/cr000108x. [DOI] [PubMed] [Google Scholar]
- 16.Lutolf MP, Hubbell JA. Nat Biotechnol. 2005;23:47–55. doi: 10.1038/nbt1055. [DOI] [PubMed] [Google Scholar]
- 17.Freed LE, Guilak F, Guo XE, Gray ML, Tranquillo R, Holmes JW, Radisic M, Sefton MV, Kaplan D, Vunjak-Novakovic G. Tissue Eng. 2006;12:3285–3305. doi: 10.1089/ten.2006.12.3285. [DOI] [PubMed] [Google Scholar]
- 18.Discher DE, Mooney DJ, Zandstra PW. Science. 2009;324:1673–1677. doi: 10.1126/science.1171643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huebsch N, Mooney DJ. Nature. 2009;462:426–432. doi: 10.1038/nature08601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Place ES, Evans ND, Stevens MM. Nat Mater. 2009;8:457–470. doi: 10.1038/nmat2441. [DOI] [PubMed] [Google Scholar]
- 21.Tsang KY, Cheung MC, Chan D, Cheah KS. Cell Tissue Res. 2010;339:93–110. doi: 10.1007/s00441-009-0893-8. [DOI] [PubMed] [Google Scholar]
- 22.Dvir T, Timko BP, Kohane DS, Langer R. Nat Nanotechnol. 2011;6:13–22. doi: 10.1038/nnano.2010.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Steinman RM, Cohn ZA. J Exp Med. 1973;137:1142–1162. doi: 10.1084/jem.137.5.1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Forster R, Davalos-Misslitz AC, Rot A. Nat Rev Immunol. 2008;8:362–371. doi: 10.1038/nri2297. [DOI] [PubMed] [Google Scholar]
- 25.Akira S. Curr Opin Immunol. 2003;15:5–11. doi: 10.1016/s0952-7915(02)00013-4. [DOI] [PubMed] [Google Scholar]
- 26.Takeda K, Kaisho T, Akira S. Annu Rev Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 27.Jenner E. An inquiry into the causes and effects of the variolae vaccinae, a disease discovered in some of the western counties of England, particularly Gloucestershire and known by the name of the cow pox, Law, London. 1798 [Google Scholar]
- 28.Meissner M, Reichert TE, Kunkel M, Gooding W, Whiteside TL, Ferrone S, et al. Clin Cancer Res. 2005;11:2552–2560. doi: 10.1158/1078-0432.CCR-04-2146. [DOI] [PubMed] [Google Scholar]
- 29.Shevach EM. Nat Med. 2004;10:900–901. doi: 10.1038/nm0904-900. [DOI] [PubMed] [Google Scholar]
- 30.Mbeunkui F, Johann DJ., Jr Cancer Chemother Pharmacol. 2009;63:571–582. doi: 10.1007/s00280-008-0881-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Du C, Wang Y. J Exp Clin Cancer Res. 2011;30:12. doi: 10.1186/1756-9966-30-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Steinman RM, Dhodapkar M. Int J Cancer. 2001;94:459–473. doi: 10.1002/ijc.1503. [DOI] [PubMed] [Google Scholar]
- 33.Hsu FJ, Benike C, Fagnoni F, Liles TM, Czerwinski D, Taidi B, et al. Nat Med. 1996;2:52–58. doi: 10.1038/nm0196-52. [DOI] [PubMed] [Google Scholar]
- 34.Kleindienst P, Brocker T. J Immunol. 2003;170:2817–2823. doi: 10.4049/jimmunol.170.6.2817. [DOI] [PubMed] [Google Scholar]
- 35.Berzofsky JA, Terabe M, Oh S, Belyakov IM, Ahlers JD, Janik JE, et al. J Clin Invest. 2004;113:1515–1525. doi: 10.1172/JCI21926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rosenberg SA, Yang JC, Restifo NP. Nat Med. 2004;10:909–915. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Purcell AW, McCluskey J, Rossjohn J. Nat Rev Drug Discov. 2007;6:404–414. doi: 10.1038/nrd2224. [DOI] [PubMed] [Google Scholar]
- 38.Steinman RM, Hawiger D, Nussenzweig MC. Annu Rev Immunol. 2003;21:685–711. doi: 10.1146/annurev.immunol.21.120601.141040. [DOI] [PubMed] [Google Scholar]
- 39.Vasir JK, Labhasetwar V. Adv Drug Deliv Rev. 2007;59:718–728. doi: 10.1016/j.addr.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pic E, Pons T, Bezdetnaya L, Leroux A, Guillemin F, Dubertret B, et al. Mol Imaging Biol. 2010;12:394–405. doi: 10.1007/s11307-009-0288-y. [DOI] [PubMed] [Google Scholar]
- 41.Collins DS, Findlay K, Harding CV. J Immunol. 1992;148:3336–3341. [PubMed] [Google Scholar]
- 42.Reddy R, Zhou F, Nair S, Huang L, Rouse BT. J Immunol. 1992;148:1585–1589. [PubMed] [Google Scholar]
- 43.Suzuki Y, Wakita D, Chamoto K, Narita Y, Tsuji T, Takeshima T, et al. Cancer Res. 2004;64:8754–8760. doi: 10.1158/0008-5472.CAN-04-1691. [DOI] [PubMed] [Google Scholar]
- 44.van Broekhoven CL, Parish CR, Demangel C, Britton WJ, Altin JG. Cancer Res. 2004;64:4357–4365. doi: 10.1158/0008-5472.CAN-04-0138. [DOI] [PubMed] [Google Scholar]
- 45.Popescu MC, Robb RJ, Batenjany MM, Boni LT, Neville ME, Pennington RW, et al. Blood. 2007;109:5407–5410. doi: 10.1182/blood-2006-08-039446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moon JJ, Suh H, Bershteyn A, Stephan MT, Liu H, Huang B, et al. Nat Mater. 2011;10:243–251. doi: 10.1038/nmat2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Z, Tongchusak S, Mizukami Y, Kang YJ, Ioji T, Touma M, et al. Biomaterials. 2011;32:3666–3678. doi: 10.1016/j.biomaterials.2011.01.067. [DOI] [PubMed] [Google Scholar]
- 48.Waeckerle-Men Y, Groettrup M. Adv Drug Deliv Rev. 2005;57:475–482. doi: 10.1016/j.addr.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 49.Shen H, Ackerman AL, Cody V, Giodini A, Hinson ER, Cresswell P, et al. Immunology. 2006;117:78–88. doi: 10.1111/j.1365-2567.2005.02268.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Waeckerle-Men Y, Allmen EU, Gander B, Scandella E, Schlosser E, Schmidtke G, et al. Vaccine. 2006;24:1847–1857. doi: 10.1016/j.vaccine.2005.10.032. [DOI] [PubMed] [Google Scholar]
- 51.Heit A, Schmitz F, Haas T, Busch DH, Wagner H. Eur J Immunol. 2007;37:2063–2074. doi: 10.1002/eji.200737169. [DOI] [PubMed] [Google Scholar]
- 52.Solbrig CM, Saucier-Sawyer JK, Cody V, Saltzman WM, Hanlon DJ. Mol Pharm. 2007;4:47–57. doi: 10.1021/mp060107e. [DOI] [PubMed] [Google Scholar]
- 53.Schlosser E, Mueller M, Fischer S, Basta S, Busch DH, Gander B, et al. Vaccine. 2008;26:1626–1637. doi: 10.1016/j.vaccine.2008.01.030. [DOI] [PubMed] [Google Scholar]
- 54.Alshamsan A, Haddadi A, Hamdy S, Samuel J, El-Kadi AO, Uludag H, et al. Mol Pharm. 2010;7:1643–1654. doi: 10.1021/mp100067u. [DOI] [PubMed] [Google Scholar]
- 55.Cruz LJ, Tacken PJ, Fokkink R, Joosten B, Stuart MC, Albericio F, et al. J Control Release. 2010;144:118–126. doi: 10.1016/j.jconrel.2010.02.013. [DOI] [PubMed] [Google Scholar]
- 56.Molavi O, Mahmud A, Hamdy S, Hung RW, Lai R, Samuel J, et al. Mol Pharm. 2010;7:364–374. doi: 10.1021/mp900145g. [DOI] [PubMed] [Google Scholar]
- 57.Prasad S, Cody V, Saucier-Sawyer JK, Saltzman WM, Sasaki CT, Edelson RL, et al. Nanomedicine. 2011;7:1–10. doi: 10.1016/j.nano.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Akagi T, Wang X, Uto T, Baba M, Akashi M. Biomaterials. 2007;28:3427–3436. doi: 10.1016/j.biomaterials.2007.04.023. [DOI] [PubMed] [Google Scholar]
- 59.Uto T, Wang X, Sato K, Haraguchi M, Akagi T, Akashi M, et al. J Immunol. 2007;178:2979–2986. doi: 10.4049/jimmunol.178.5.2979. [DOI] [PubMed] [Google Scholar]
- 60.Uto T, Akagi T, Hamasaki T, Akashi M, Baba M. Immunol Lett. 2009;125:46–52. doi: 10.1016/j.imlet.2009.05.008. [DOI] [PubMed] [Google Scholar]
- 61.Uto T, Wang X, Akagi T, Zenkyu R, Akashi M, Baba M. Biochem Biophys Res Commun. 2009;379:600–604. doi: 10.1016/j.bbrc.2008.12.122. [DOI] [PubMed] [Google Scholar]
- 62.Hamasaki T, Uto T, Akagi T, Akashi M, Baba M. Clin Vaccine Immunol. 2010;17:748–756. doi: 10.1128/CVI.00505-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Matsuo K, Ishii Y, Yoshinaga T, Akashi M, Mukai Y, Yoshioka Y, et al. Biol Pharm Bull. 2010;33:2003–2007. doi: 10.1248/bpb.33.2003. [DOI] [PubMed] [Google Scholar]
- 64.Minigo G, Scholzen A, Tang CK, Hanley JC, Kalkanidis M, Pietersz GA, et al. Vaccine. 2007;25:1316–1327. doi: 10.1016/j.vaccine.2006.09.086. [DOI] [PubMed] [Google Scholar]
- 65.Bourquin C, Anz D, Zwiorek K, Lanz AL, Fuchs S, Weigel S, et al. J Immunol. 2008;181:2990–2998. doi: 10.4049/jimmunol.181.5.2990. [DOI] [PubMed] [Google Scholar]
- 66.Zwiorek K, Bourquin C, Battiany J, Winter G, Endres S, Hartmann G, et al. Pharm Res. 2008;25:551–562. doi: 10.1007/s11095-007-9410-5. [DOI] [PubMed] [Google Scholar]
- 67.Kwon YJ, James E, Shastri N, Frechet JM. Proc Natl Acad Sci U S A. 2005;102:18264–18268. doi: 10.1073/pnas.0509541102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nguyen DN, Raghavan SS, Tashima LM, Lin EC, Fredette SJ, Langer RS, et al. Biomaterials. 2008;29:2783–2793. doi: 10.1016/j.biomaterials.2008.03.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cheung WH, Chan VS, Pang HW, Wong MK, Guo ZH, Tam PK, et al. Bioconjug Chem. 2009;20:24–31. doi: 10.1021/bc800167q. [DOI] [PubMed] [Google Scholar]
- 70.Zhou XF, Liu B, Yu XH, Zha X, Zhang XZ, Wang XY, et al. Small. 2007;3:1707–1713. doi: 10.1002/smll.200700151. [DOI] [PubMed] [Google Scholar]
- 71.Wilson NS, El-Sukkari D, Belz GT, Smith CM, Steptoe RJ, Heath WR, et al. Blood. 2003;102:2187–2194. doi: 10.1182/blood-2003-02-0513. [DOI] [PubMed] [Google Scholar]
- 72.Sixt M, Kanazawa N, Selg M, Samson T, Roos G, Reinhardt DP, et al. Immunity. 2005;22:19–29. doi: 10.1016/j.immuni.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 73.Erogbogbo F, Yong KT, Roy I, Hu R, Law WC, Zhao W, et al. ACS Nano. 2011;5:413–423. doi: 10.1021/nn1018945. [DOI] [PubMed] [Google Scholar]
- 74.Jeon YH, Kim YH, Choi K, Piao JY, Quan B, Lee YS, et al. Mol Imaging Biol. 2010;12:155–162. doi: 10.1007/s11307-009-0262-8. [DOI] [PubMed] [Google Scholar]
- 75.Jain R, Dandekar P, Patravale V. J Control Release. 2009;138:90–102. doi: 10.1016/j.jconrel.2009.05.010. [DOI] [PubMed] [Google Scholar]
- 76.Kim S, Lim YT, Soltesz EG, De Grand AM, Lee J, Nakayama A, et al. Nat Biotechnol. 2004;22:93–97. doi: 10.1038/nbt920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Anzai Y, Blackwell KE, Hirschowitz SL, Rogers JW, Sato Y, Yuh WT, et al. Radiology. 1994;192:709–715. doi: 10.1148/radiology.192.3.7520182. [DOI] [PubMed] [Google Scholar]
- 78.Swartz MA. Adv Drug Deliv Rev. 2001;50:3–20. doi: 10.1016/s0169-409x(01)00150-8. [DOI] [PubMed] [Google Scholar]
- 79.Reddy ST, Rehor A, Schmoekel HG, Hubbell JA, Swartz MA. J Control Release. 2006;112:26–34. doi: 10.1016/j.jconrel.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 80.Reddy ST, van der Vlies AJ, Simeoni E, Angeli V, Randolph GJ, O’Neil CP, et al. Nat Biotechnol. 2007;25:1159–1164. doi: 10.1038/nbt1332. [DOI] [PubMed] [Google Scholar]
- 81.Manolova V, Flace A, Bauer M, Schwarz K, Saudan P, Bachmann MF. Eur J Immunol. 2008;38:1404–1413. doi: 10.1002/eji.200737984. [DOI] [PubMed] [Google Scholar]
- 82.Kumamoto T, Huang EK, Paek HJ, Morita A, Matsue H, Valentini RF, et al. Nat Biotechnol. 2002;20:64–69. doi: 10.1038/nbt0102-64. [DOI] [PubMed] [Google Scholar]
- 83.Hori Y, Winans AM, Huang CC, Horrigan EM, Irvine DJ. Biomaterials. 2008;29:3671–3682. doi: 10.1016/j.biomaterials.2008.05.033. [DOI] [PubMed] [Google Scholar]
- 84.Hori Y, Stern PJ, Hynes RO, Irvine DJ. Biomaterials. 2009;30:6757–6767. doi: 10.1016/j.biomaterials.2009.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hori Y, Winans AM, Irvine DJ. Acta Biomater. 2009;5:969–982. doi: 10.1016/j.actbio.2008.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ali OA, Huebsch N, Cao L, Dranoff G, Mooney DJ. Nat Mater. 2009;8:151–158. doi: 10.1038/nmat2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ali OA, Emerich D, Dranoff G, Mooney DJ. Sci Transl Med. 2009;1:8ra19. doi: 10.1126/scitranslmed.3000359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ali OA, Doherty E, Bell WJ, Fradet T, Hudak J, Laliberte MT, et al. Pharm Res. 2011 doi: 10.1007/s11095-010-0361-x. [DOI] [PubMed] [Google Scholar]
- 89.Epardaud M, Elpek KG, Rubinstein MP, Yonekura AR, Bellemare-Pelletier A, Bronson R, et al. Cancer Res. 2008;68:2972–2983. doi: 10.1158/0008-5472.CAN-08-0045. [DOI] [PubMed] [Google Scholar]