

Abstract
Resting splenic venous outflow from anaesthetized dogs contains prostaglandin-like material: the concentration increases after intra-arterial injections of bradykinin into the spleen, and is abolished by treatment with indomethacin.
Intra-arterial injections of bradykinin into the spleen of lightly anaesthetized dogs elicit a dose-dependent reflex increase in the blood pressure, which is reduced but not abolished by treatment with indomethacin.
Addition of prostaglandin E1 or E2 either by injections or by infusions restores the reflex increase in the blood pressure due to bradykinin injections after indomethacin treatment.
The sensitizing action of endogenously released prostaglandins at or near the afferent nerve endings is discussed.
The analgesic activity of aspirin-like drugs is explained in terms of the removal of the sensitizing activity of prostaglandins.
Introduction
Inhibition of prostaglandin biosynthesis has been proposed as the mechanism of action of aspirin-like drugs (Vane, 1971, 1972a, b). Whereas the anti-inflammatory and anti-pyretic aspects of their activity could be adequately explained by this mechanism, the analgesic action was more difficult to account for, because the available data concerning the ‘algesic’ activity of prostaglandins were, until very recently, contradictory. On the one hand there were reports that prostaglandins applied to the blister base (Horton, 1963) or given by intradermal injection (Crunkhorn & Willis, 1971) did not produce pain; on the other, intravenous or intramuscular injections caused pain (Karim, 1971; Collier, Karim, Robinson & Somers, 1972; Gillespie, 1972).
Recently Collier & Schneider (1972) found that prostaglandins injected into the peritoneal cavity of mice elicited a writhing response which, in contrast to that produced by other substances like bradykinin, could not be blocked by aspirinlike drugs. They suggested that prostaglandins could be one of the pain mediators released by a noxious stimulus. Subsequently, Moncada, Ferreira & Vane (1972) showed that bradykinin, when injected intra-arterially into dog isolated spleens in doses which produce pain in the intact animal, released prostaglandin-like material into the splenic venous outflow. This release was blocked by infusions of indomethacin.
Ferreira (1972) demonstrated that prostaglandins produced pain in human volunteers when injected subdermally in high doses, but in concentrations likely to be found in inflammation, the main feature of their action was to produce a long-lasting hyperalgesia i.e. a sensitization to mechanical or to other chemical stimuli. He postulated that the analgesic activity of aspirin-like drugs was indeed due to prevention of biosynthesis of prostaglandins, thus removing this sensitizing effect.
The present results reinforce this hypothesis. In one series of experiments we measured the release of prostaglandin-like material into the splenic venous outflow induced by intra-arterial injections of bradykinin. In another series, we used the technique described by Guzman, Braun & Lim (1962) and Guzman, Braun, Lim, Potter & Rodgers (1964) to study the effects of endogenously released and exogenously added prostaglandins on the nociceptive activity of bradykinin.
Methods
Blood-bathed organ experiments
The blood-bathed organ technique (Vane, 1964, 1969) was used to measure release of prostaglandin-like material from dog spleen in vivo. Seventeen dogs of either sex weighing 11·5–24 kg were anaesthetized with pentobarbitone sodium (30 mg/kg intravenously). The spleen was exposed through an incision in the abdominal midline and completely isolated from its gastric, pancreatic and colonic circulation as previously described (Guzman et al., 1964). A small branch of the splenic artery was cannulated retrogradely for injections into the spleen. The splenic vein was cannulated and the outflow was led through polyethylene tubing into a cannula in the femoral vein. A portion (10 ml/min) of the splenic venous blood was withdrawn through a side arm by a pump and was used to superfuse a rat stomach strip (Vane, 1957), a chick rectum (Mann & West, 1950), and a rat colon (Regoli & Vane, 1964) for the detection of prostaglandins (Ferreira & Vane, 1967) and a cat terminal ileum (Ferreira, Ng & Vane, 1973) for the detection of bradykinin. They were rendered more specific to these substances by pretreatment for two to three hours (in Krebs solution) with a mixture of antagonists (Gilmore, Vane & Wyllie, 1968) and indomethacin 1 μg/ml. After superfusing the assay tissues, the blood was returned to the animal by gravity via an external jugular vein.
In three experiments, the femoral vein was cannulated so that blood could be withdrawn from the hind leg and used as an alternative bathing fluid for the assay tissues; in these experiments injections into the hind leg were made through a catheter inserted retrogradely into a branch of the femoral artery. Heparin (1000 i.u./kg) was injected intravenously before the external circulation of blood was started. Artificial ventilation was maintained by a pump through an intra-tracheal cannula.
The amounts of prostaglandin-like material released by intra-arterial injections of bradykinin were determined by comparison of the effects of the released substances on the blood-bathed tissues with the effects of prostaglandins infused directly into the blood as it left the vascular bed, on the way to the assay tissues.
Pressor reflex experiments
The stimulation of sensory nerves by bradykinin was studied by measuring the reflex increase in the blood pressure (Guzman et al., 1962; Hashimoto, Kumukura & Taira, 1964). Thirty-five dogs of either sex weighing 7–5–15 kg were anaesthetized with thiopentone (30 mg/kg) and chloralose (30–50 mg/kg) intravenously. Additional doses of chloralose (15 mg/kg) were given as required. The spleen was exposed and isolated as in the first group of experiments and a branch of the splenic artery was cannulated retrogradely for intra-arterial injections. One femoral artery was cannulated for measuring blood pressure with a Statham transducer (P23dB), the output of which was displayed on a potentiometric pen recorder (Servoscribe). The success of these experiments depended upon the depth of anaesthesia. If it was too deep (corneal and patellar reflexes absent), bradykinin had no hypertensive effect; if it was too light, different doses of bradykinin did not give graded responses. Because of this, pressor effects of bradykinin were only compared for 45–60 min periods, during which the depth of anaesthesia remained fairly constant. In some dogs, graded pressor effects could not be obtained after intra-arterial bradykinin and in others, changes in respiration interfered too much with basal blood pressure; all these (10 out of 35) have been excluded from the results. Atropine (1 mg/kg) was given intravenously to those animals in which there was bronchial hypersecretion.
Drugs used were: adrenaline hydrogen tartrate (BDH Biochemicals Ltd.), atropine sulphate (BDH Biochemicals Ltd.), bradykinin (Sandoz), α-chloralose (Sigma), eledoisin (ELD 950; Sandoz), 5-hydroxytryptamine creatinine sulphate (May & Baker), indomethacin (Merck Sharp & Dohme), nitroglycerine (1% v/v in ethanol), pentobarbitone sodium (Nembutal; Abbot), phenoxybenzamine (Dibenyline; S.K.F.), prostaglandins E1 and E2 (Upjohn) and thiopentone sodium (Pentothal; Abbot).
Results
Basal and stimulated outputs of prostaglandin-like substances
Splenic venous blood contained a basal concentration of prostaglandin-like material, as shown by the contractions of the assay tissues it produced when compared with femoral blood. Figure 1 shows an experiment in which the basal release was equivalent to a concentration of prostaglandin E2 of 1 ng/ml. In two other experiments, the basal release was estimated as 1·5 and 1 ng/ml. Indomethacin (2–4 mg/kg intravenously) abolished the basal output of prostaglandin-like material in splenic venous blood. When the assay tissues were bathed in femoral venous blood, their tone was unchanged by indomethacin, showing the absence of detectable prostaglandins in femoral venous blood (Figure 1).
Fig. 1.
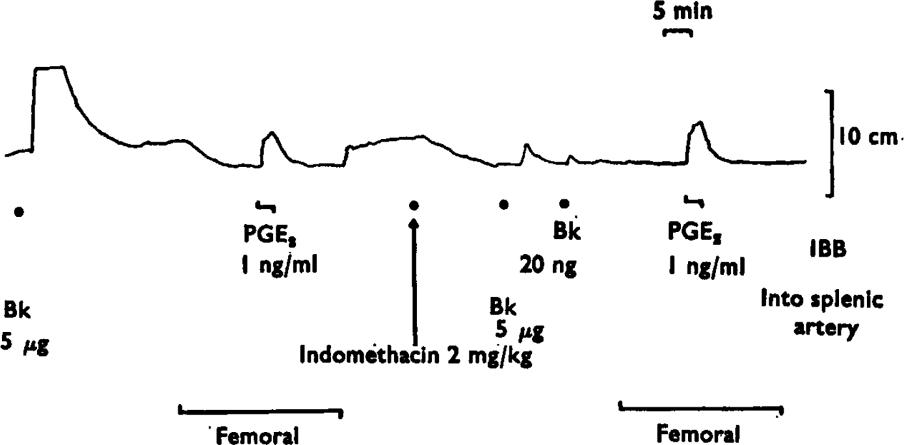
Basal release of prostaglandin-like material from the spleen. A rat stomach strip was bathed in splenic venous blood, except during the periods marked ‘femoral’, when it was bathed in femoral venous blood. The tracing shows the contractions of the stomach strip to prostaglandin-like material released by bradykinin (Bk, 5 μg) injected into the spleen or to direct infusions of prostaglandin E2 (PGE2) into the blood bathing the tissue (IBB). A reduction in tone is produced by bathing the stomach strip with femoral venous blood. Indomethacin (2 mg/kg i.v.) abolished the release of prostaglandin-like material by 5 μg of bradykinin and the contractions of the stomach strip induced by splenic rather than femoral venous blood. Note that the tone of the stomach strip in femoral blood was the same before and after indomethacin, showing that there was no prostaglandin-like material in femoral venous blood. Time 5 min; vertical scale 10 cm.
The release of prostaglandin-like material by intra-splenic injections of bradykinin was demonstrated in 15 dogs. The assay tissues were bathed in splenic venous blood and were sensitive to infused prostaglandins in concentrations as low as 0·5–2 ng/ml. Bradykinin released prostaglandin-like material in amounts up to a peak concentration of 10 ng/ml (assayed as prostaglandin E2). The relative contractions of the assay tissues showed that the prostaglandin-like activity was mostly E type. The amounts released varied from experiment to experiment, but the higher dose (10 μg) of bradykinin always released greater amounts of prostaglandin-like material than the lower dose (5 μg). With 5 μg bradykinin the average peak release was 2·5 ng E2-like material (7 experiments) and with 10 μg bradykinin it was 3·4 ng (12 experiments). Figure 2 shows the release induced by intrasplenic injection of 10 μg bradykinin.
Fig. 2.
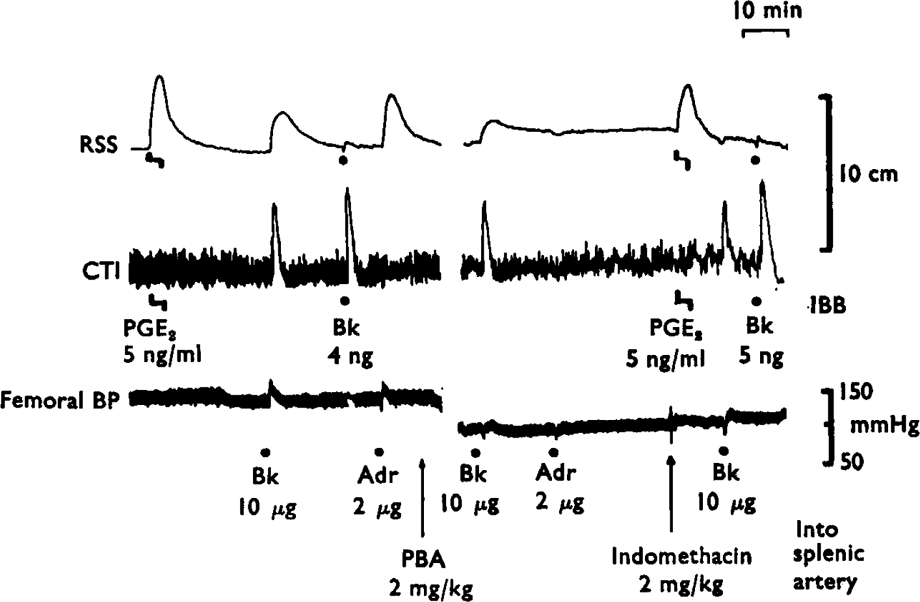
Effects of phenoxybenzamine and indomethacin on the release of prostaglandin-like material by bradykinin and adrenaline. The lower tracing shows the femoral blood pressure of the animal. The upper two tracings show the contractions of the rat stomach strip (RSS) the cat terminal ileum (CTI) bathed in splenic venous blood. Infusions directly into the bathing blood (IBB) of prostaglandin E2 (5 ng/ml) or injections of bradykinin (4 ng and 5 ng) gave selective responses on RSS and CTI. When bradykinin (Bk, 10 μg) or adrenaline (Adr, 2 μg) was injected into the splenic artery, contractions of the RSS showed the release of prostaglandin-like material. Phenoxybenzamine (PBA, 2 mg/kg i.v.) abolished the release due to adrenaline without changing the release due to bradykinin. Indomethacin (2 mg/kg i.v.) abolished the release due to bradykinin. Time 10 min; vertical scales 10 cm and mmHg.
Adrenaline (1–5 μg) also released prostaglandin-like material (2–10 ng/ml) into the splenic venous outflow. On a molar basis, adrenaline was more potent than bradykinin in five experiments, equal in two and less potent in three. Phenoxy-benzamine (2 mg/kg i.v.) blocked the release induced by adrenaline but not that due to bradykinin (Figure 2). Similar results were obtained in four other experiments. Indomethacin (2–4 mg/kg i.v.) abolished the release of prostaglandin-like material by bradykinin in 5 experiments and reduced it by 50–90% in 4 experiments. Partial blockade of release (50–70%) was seen in two experiments in which only 50–100 μg/kg of indomethacin was given; a further dose of indomethacin (4 mg/kg) then abolished the release.
Pressor reflex experiments
Bradykinin injected intra-arterially into the spleen produced a reflex rise in blood pressure which was proportional to the dose (Figures 3–7).
Fig. 3.
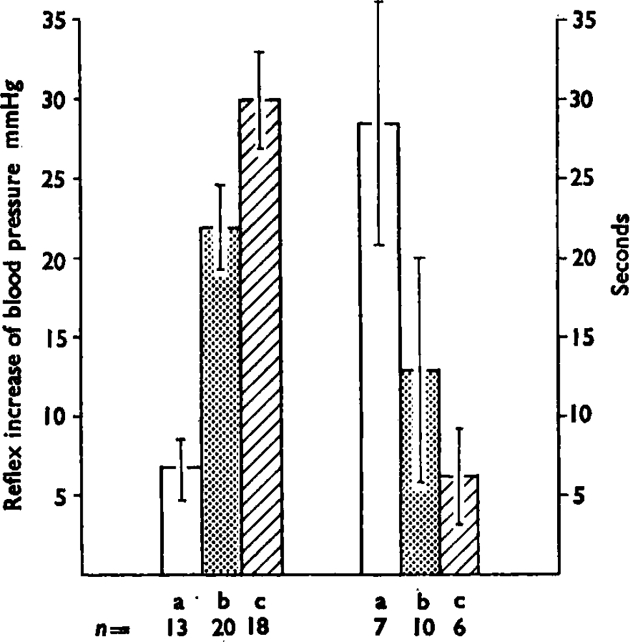
The histogram shows the mean (± s.e.m.) maximum increase in blood pressure (left hand group) and the mean (+ s.e.m.) delay time in seconds to the start of the response (right hand group) induced by intrasplenic injection of three dose ranges of bradykinin; (a) is 0·05–0·1 μg, (b) is 0·2–1 μg and (c) is 2–10 μg. Numbers under the bars (n) are number of experiments. As the doses increase, the pressor effect increases and the delay decreases. The mean values are calculated using the reciprocals of the delays to overcome the difficulty of infinite delays when there was no response to a given dose. Lines show standard errors of the means.
Fig. 7.
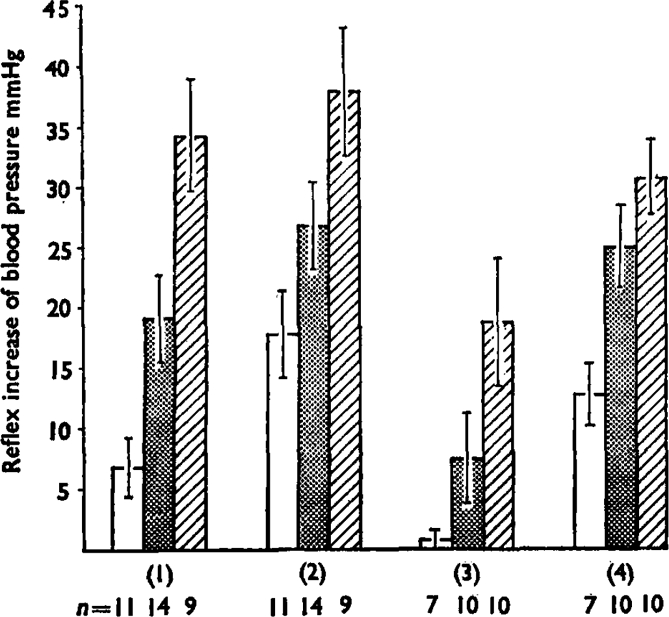
This summarizes the reflex pressor increases in blood pressure produced by bradykinin injections at 0·1 μg (open bars), 0·5 μg (stippled bars) and 2 μg (hatched bars). Numbers of experiments (n) are shown below. Group 1 are the mean increases (+ s.e.m.) for control injections and group 2 shows the potentiation produced by simultaneous injection (5–20 μg) or infusion (50–200 ng/min) of prostaglandin E1. Differences between the effects of bradykinin (by paired t tests) had P values of <0·01 for 0·1 μg, <0·02 for 0·5 μg, and <0·l for 2 μg. Group 3 shows the reduced effects of bradykinin after dosage of the does with indomethacin (1–5 mg/kg i.v.) and group 4 shows the potentiation of these effects by prostaglandin E1. The differences were all significant at P<0·01. Groups 1 and 2 were paired observations in the same animals, as were groups 3 and 4.
A rise in blood pressure of more than 5 mmHg was considered to be a response. This was seen in 7 out of 13 dogs with 0·1 μg bradykinin and in all dogs with 0·5 μg bradykinin. Increasing the depth of anaesthesia abolished the pressor response.
The reflex pressor effect appeared within 3–20 s after the injection of bradykinin and the delay time decreased as the dose increased (Figures 3 and 4). Adrenaline (10–20 μg) injected into the spleen also caused a rise in blood pressure but this was not affected by increasing the depth of anaesthesia.
Fig. 4.
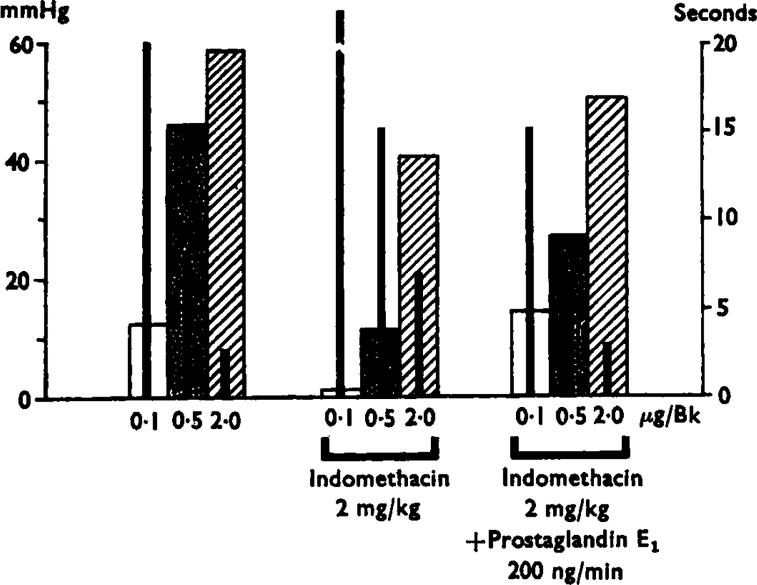
Results of a single experiment showing variations in the magnitude and relay of the responses to bradykinin injections of 0·1, 0·5 and 2·0 μg by indomethacin treatment (left hand group) and the mean (+s.e.m.) delay time in seconds to the start of the response in mmHg (left hand scale) and the thin black bars inside the broad ones represent the delays in seconds (right hand scale). Treatment with indomethacin (2 mg/kg i.v.) reduced the pressor effects and increased the delays. Infusion into the spleen of prostaglandin E1 (200 ng/min) after indomethacin increased the values towards pretreatment levels.
Inhibition of the pressor reflex effect by indomethacin
Within 15 min of an intravenous injection of indomethacin (1–5 mg/kg; 16 experiments), the reflex pressor response induced by bradykinin was reduced and the delay to its appearance was increased. Figure 4 shows that the responses to the largest dose of bradykinin were least affected. Indomethacin gave an overall reduction of 72% of the pressor effects of the 0·5 μg dose of bradykinin and 47% of those of the 2 μg dose. The pressor effects of adrenaline injected into the spleen were not reduced by indomethacin.
Potentiation of the pressor reflex by prostaglandin E1 and E2
Prostaglandin E1 given either by infusion (50–200 ng/min in 8 experiments) or injection (5–20 μg in 9 experiments) intra-arterially to the spleen potentiated the reflex pressor activity of bradykinin (0·5–10 μg) and shortened the delay time. The same injections of prostaglandin E1 or E2 by themselves produced either no response or a slight hypotension (5–15 mmHg); infusions had no effect. Figure 5 shows the results of all experiments in which prostaglandin E1 was injected with bradykinin. The effects of the smaller doses of bradykinin were more potentiated than those of the higher doses.
Fig. 5.
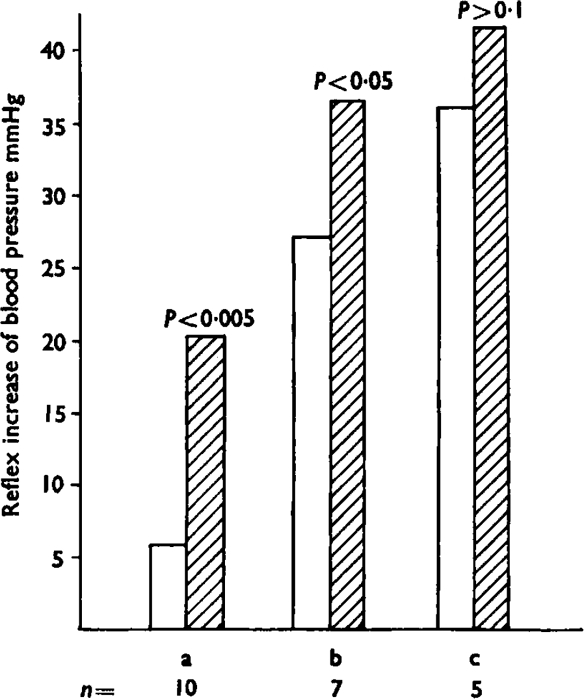
Potentiation by addition of prostaglandin E1 of the reflex increase of blood pressure induced by bradykinin. The open bars show the pressor effects of three dose ranges of bradykinin; (a) is 0·05–0·1 μg, (b) is 0·2–1 μg and (c) is 2–10 μg. The hatched bars show the effects of the same doses, but with the simultaneous injection of prostaglandin E1 (5–20 μg). This produced a significant increase in the pressor response in the first group (P<0·005) and the second group (P<0·05) but not in the third (P>0·1). Numbers of observations (n) are shown below the bars.
Figure 6 is of a blood pressure tracing from a dog. Bradykinin injection into the spleen gave graded increases in blood pressure which were greatly augmented during an infusion of prostaglandin E1 (200 ng/minute). The potentiation increased as the infusion was maintained and only slowly declined when the prostaglandin E1 infusion was stopped. A similar potentiation of the reflex pressor effects of bradykinin was induced by prostaglandin E2, but 4–8 times more had to be used.
Fig. 6.
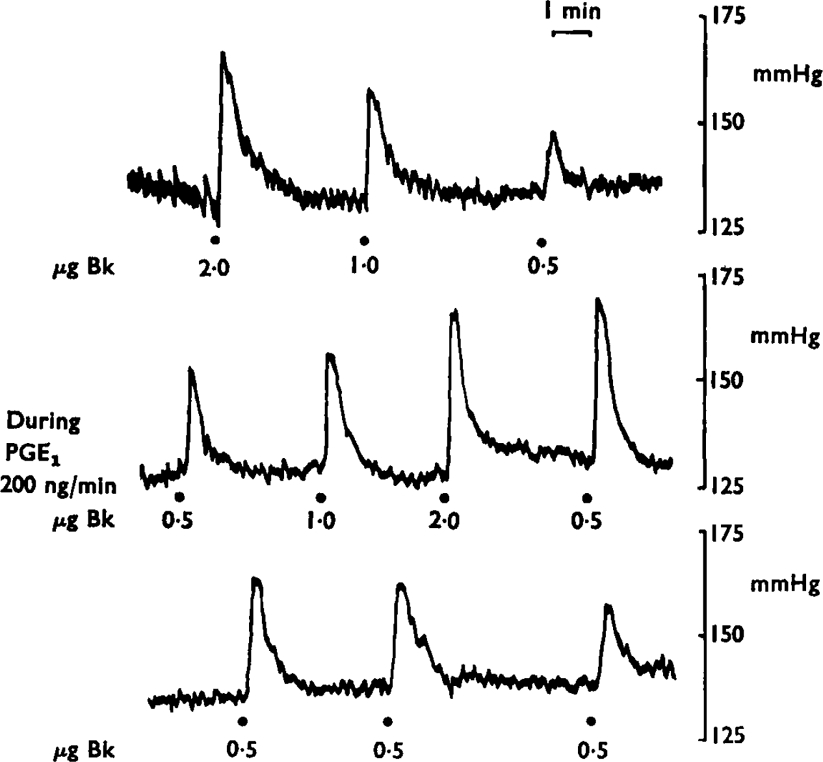
Prostaglandin E1 (PGE1) potentiates the reflex effect of bradykinin. A continuous tracing of the blood pressure of a dog is shown arranged in three panels. In the upper one, the hypertensive responses to 2·0, 1·0 and 0·5 μg of bradykinin are shown; in the second tracing the same injections are repeated during infusion of prostaglandin E1 (200 ng/minute). After the infusion (third tracing) the pressor effects of 0·5 μg of bradykinin gradually declined towards pre-treatment levels. Time 1 min; vertical scales mmHg.
After indomethacin administration had reduced the reflex pressor response induced by bradykinin, addition of prostaglandin E1 or E2 tended to restore the rise in pressure back to control levels. The results of a single experiment have already been presented in Fig. 4: the results of all the experiments are shown in Figure 7.
In contrast to the potentiation of bradykinin responses, prostaglandin E1 given either by injection or infusion reduced the pressor effects of adrenaline injected intra-arterially into the spleen. To test whether the potentiation of the bradykinin response was linked with the vasodilatation induced by prostaglandin E1, other vasodilator substances were used. There was no potentiation of the bradykinin response by simultaneous injections of nitroglycerine (1 μg/kg; 1 experiment) or infusions of eledoisin (0·5–1 μg/min; 2 experiments) or even bradykinin (0·5–1 μg/min; 2 experiments). However, 5-hydroxytryptamine (0·2–1 μg/min; 3 experiments) potentiated the bradykinin response.
Discussion
The discovery that aspirin-like drugs inhibit the biosynthesis of prostaglandins (Vane, 1971; Smith & Willis, 1971; Ferreira, Moncada & Vane. 1971) suggested that prostaglandins were involved in the mediation of pain. This possibility was further strengthened by the work of Collier & Schneider (1972) who found that prostaglandins were the most powerful substances known for induction of the writhing reflex in mice. This effect, in contrast to that produced by other noxious stimuli, was not antagonized by aspirin-like drugs. Recently, on the basis of the effects of prostaglandin infusions into man, Ferreira (1972) suggested that their main contribution to the pain of inflammation was their ability to sensitize the sensory nerve endings to mechanical or chemical stimulation. Our present results reinforce and extend this conclusion by using the same system by which Lim, Guzman, Rodgers, Goto, Braun, Dickerson & Engle (1964) demonstrated that aspirin-like drugs exert their analgesic action peripherally, near to or at the site of generation of pain.
Bradykinin in doses (1–10 μg) which produced pain when injected intra-arterially into the spleen of lightly anaesthetized dogs (Guzman et al., 1964) caused a release of prostaglandin-like material from the isolated prefused spleen of the dog (Moncada et al., 1972). This release was blocked by indomethacin. We have now shown that there is a basal output of prostaglandin-like material from the spleen of the dog in vivo and this output is increased by injections of bradykinin. The release induced by bradykinin is of the same order as that observed with equimolar doses of adrenaline and is abolished by indomethacin. However, phenoxybenzamine abolished the release by adrenaline but not that by bradykinin. We have not attempted to characterize further the prostaglandin(s) released, but in other dog and cat spleen perfusions the release was predominantly of prostaglandin E2 (Davies, Horton & Withrington, 1968; Gilmore et al., 1968; Ferreira, Moncada & Vane, 1973). Bradykinin also causes release of prostaglandin E2 from dog kidney (McGiff, Terragno, Malik & Lonigro, 1972). The relative effects of the prostaglandin-like material on the assay tissues we used, together with the fact that a specific inhibitor of prostaglandin biosynthesis prevented its release, suggest that here, also, it was mainly a prostaglandin of the E series.
Two conclusions can be inferred from these results. First, the mechanism of prostaglandin release by bradykinin, which does not contract the dog isolated spleen (Moncada et al., 1972) is different from that induced by adrenaline, which is associated with splenic contraction (Ferreira & Vane, 1967). Secondly, it seems unlikely that prostaglandins directly mediate the pain-producing activity of bradykinin, for adrenaline also releases prostaglandins but does not cause pain (Guzman et al., 1964).
To test the possibility that prostaglandin release facilitated production of pain, we used the reflex pressor effect of intrasplenic injections of bradykinin. This reflex has already been fully characterized as an autonomic response due to the stimulation of afferent (pain) nerve endings (Guzman et al., 1962; 1964; Hashimoto et al., 1964) and is reduced by aspirin-like drugs. Moreover, the reflex depends on the degree of anaesthesia of the animal, thus distinguishing it from the pressor effects of adrenaline which remain unaffected when anaesthesia is deepened.
Indomethacin, in doses which completely blocked the resting output of prostaglandins and the increased release by intrasplenic injections of bradykinin, reduced but did not abolish the pressor reflex induced by bradykinin. This inhibition was much more evident for the lower doses of bradykinin. These results further support the idea that prostaglandin release does not mediate the pain-producing action of bradykinin, but in some way sensitizes the pain receptors. Certainly, the reflex was potentiated when prostaglandin E1 and E2 was administered to the spleen simultaneously with the bradykinin. This sensitization was greater with the small doses of bradykinin, possibly because the higher doses gave an almost maximal effect.
The potentiation by prostaglandins of the E series of the pressor reflex induced by intrasplenic injection of bradykinin might have been the consequence of their vasodilator action. This possibility was ruled out by the fact that infusions of other vasodilator drugs had no potentiating action. Moreover, 5-hydroxytryptamine, which causes vasoconstriction in the dog spleen (Riva Sanseverino & Urbano, 1962) was as effective as prostaglandin E2 (but less potent than prostaglandin E1) in potentiating the pressor effects of bradykinin. This result confirms that of Sicuteri (1966) who found that 5-hydroxytryptamine potentiated the pain-producing activity of bradykinin in the dorsal vein of the hand.
We propose, therefore, that prostaglandins accentuate rather than mediate the pain-producing activity of bradykinin. There is a basal output of prostaglandins from the spleen and this is increased by injections of bradykinin. The analgesic action of aspirin-like drugs can thus be explained by the abolition of the generation of prostaglandins which normally sensitize the pain receptors to the action of bradykinin. A similar potentiation of the effects of bradykinin by a released prostaglandin may explain those other effects of bradykinin (see Collier, 1969) which are partially blocked by aspirin-like drugs. In other tissues such as the skin, no basal prostaglandin release has been shown. However, prostaglandins are found in inflammation (Willis, 1969; Greaves, Søndergaard & McDonald-Gibson, 1971). This difference could, therefore, explain why the aspirin-like drugs fail to block pain produced in normal skin (Collier, Hammond, Horwood-Barrett & Schneider, 1964; Winter & Flataker, 1965) but do so when the skin is inflamed (Randall & Selitto, 1957).
Aspirin-like drugs do not increase pain threshold by more than 30% (Wolff, Hardy & Goodell, 1941; Winder, Pfeiffer & Maison, 1946; Winder, 1947). a fact which also fits with the suggestion that their analgesic action is exerted by removal of a prostaglandin sensitization (Ferreira, 1972). When there is no previous inflammation, aspirin-like drugs are probably effective analgesics when the stimulus used leads to an immediate release of prostaglandins. In such instances there would be a delay between the application of a noxious stimulus and the appearance of the response. For instance, there is a long delay between injection of phenylbenzoquinone and the production of the writhing response (Keith, 1960). Aspirin blocks this delayed writhing response but has low activity against the almost immediate writhing response induced by hypertonic saline (Collier & Schneider, 1969). Aspirin-like drugs have a low activity against the early writhing response induced by bradykinin (Collier, Dinneen, Johnson & Schneider. 1968) but are very active against the delayed response (Emele & Shanaman, 1963). In our experiments prostaglandins also shorten the delay in the appearance of the pressor response to bradykinin, especially for the low doses of bradykinin.
Our experiments do not rule out the possibility that part of the analgesic action of aspirin-like drugs is mediated by a central effect. If it is, this could also involve the prostaglandin system, for electrical stimulation of the hind paw of frogs (Ramwell, Shaw & Jessup, 1966) or the radial nerves of cats (Ramwell & Shaw, 1966) released prostaglandins in the spinal cord and in the sensory cortex. Some prostaglandins also can facilitate polysynaptic reflexes (Siggins, Hoffer & Bloom, 1971). There is indeed evidence that there is a central component to the action of paracetamol. This substance crosses the blood brain barrier more easily than aspirin (Davison, Guy, Levitt & Smith, 1961) and, unlike aspirin, increases the pain threshold in both inflamed and normal paws (Randall & Selitto, 1958). Thus, it could act by removing the facilitation produced by centrally-released prostaglandins.
In conclusion our experiments show that the analgesia produced by aspirinlike drugs can be explained by the removal of the facilitation produced by endogenously released prostaglandins, which normally lead to hyperalgesia to mechanical or chemical stimulation. Such an action explains why aspirin and its congeners are only weak analgesics.
Acknowledgments
We thank Mr. M. J. Parnham and Miss V. Pontieri for technical assistance; Upjohn for the gift of prostaglandins, Merck, Sharp & Dohme for the gift of indomethacin and the Wellcome Trust and the University of Honduras for grants.
REFERENCES
- Collier HOJ. A pharmacological analysis of aspirin. Adv. Pharmac. Chemother. 1969;7:333–405. [PubMed] [Google Scholar]
- Collier HOJ, Hammond AR, Horwood-Barrett S, Schneider C. Rapid induction by acetylcholine, bradykinin and potassium of a nociceptive response in mice and its selective antagonism by aspirin. Nature, Lond. 1964;204:1316–1318. doi: 10.1038/2041316b0. [DOI] [PubMed] [Google Scholar]
- Collier HOJ, Dinneen LC, Johnson CA, Schneider C. The abdominal constriction response and its suppression by analgesic drugs in the mouse. Br. J. Pharmac. Chemother. 1968;32:295–310. doi: 10.1111/j.1476-5381.1968.tb00973.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier HOJ, Schneider C. Profile of activity in rodents of some narcotics and narcotic antagonistic drugs. Nature, Lond. 1969;224:610–612. doi: 10.1038/224610a0. [DOI] [PubMed] [Google Scholar]
- Collier HOJ, Schneider C. Nociceptive response to prostaglandins and analgesic actions of aspirin and morphine. Nature New Biol. 1972;236:141–143. doi: 10.1038/newbio236141a0. [DOI] [PubMed] [Google Scholar]
- Collier JG, Karim SMM, Robinson B, Somers K. Action of prostaglandins A2, B1, E2 and F2α on superficial hand veins of man. Br. J. Pharmac. 1972;44:374–375P. [PMC free article] [PubMed] [Google Scholar]
- Crunkhorn P, Willis AL. Cutaneous reactions to intradermal prostaglandins. Br. J. Pharmac. 1971;41:57–64. doi: 10.1111/j.1476-5381.1971.tb09934.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies BN, Horton EW, Withrington PG. The occurrence of prostaglandin E2 in splenic venous blood of the dog following splenic nerve stimulation. Br. J. Pharmac. Chemother. 1968;32:127–135. doi: 10.1111/j.1476-5381.1968.tb00436.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davison C, Guy JL, Levitt M, Smith PK. The distribution of certain non-narcotic analytic agents in the CNS of several species. J. Pharmac. exp. Ther. 1961;134:176–183. [PubMed] [Google Scholar]
- Emele JF, Shanaman J. Bradykinin writhing: method for measuring analgesia. Proc. Soc. exp. Biol. 1963;114:680–681. doi: 10.3181/00379727-114-28768. [DOI] [PubMed] [Google Scholar]
- Ferreira SH. Prostaglandins, aspirin-like drugs and analgesia. Nature New Biol. 1972;240:200–203. doi: 10.1038/newbio240200a0. [DOI] [PubMed] [Google Scholar]
- Ferreira SH, Moncada S, Vane JR. Indomethacin and aspirin abolish prostaglandin release from the spleen. Nature New Biol. 1971;231:237–239. doi: 10.1038/newbio231237a0. [DOI] [PubMed] [Google Scholar]
- Ferreira SH, Moncada S, Vane JR. Some effects of inhibiting endogenous prostaglandin formation on the responses of the cat spleen. Br. J. Pharmac. 1973;47:48–58. doi: 10.1111/j.1476-5381.1973.tb08157.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira SH, Ng KK, Vane JR. The continuous bioassay of the release and disappearance of histamine in the circulation. Br. J. Pharmac. 1973 doi: 10.1111/j.1476-5381.1973.tb17265.x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira SH, Vane JR. Prostaglandins: their disappearance from and release into the circulation. Nature. 1967;216:868–873. doi: 10.1038/216868a0. [DOI] [PubMed] [Google Scholar]
- Gillespie A. Prostaglandin-oxytocin enhancement and potentiation and clinical application. Br. Med. J. 1972;1:150–152. doi: 10.1136/bmj.1.5793.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore N, Vane JR, Wyllie JH. Prostaglandins released by the spleen. Nature. 1968;218:1135–1140. doi: 10.1038/2181135a0. [DOI] [PubMed] [Google Scholar]
- Greaves MW, Søndergaard J, McDonald-Gibson W. Recovery of prostaglandins in human cutaneous inflammation. Br. Med. J. 1971;2:258–260. doi: 10.1136/bmj.2.5756.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman F, Braun C, Lim RKS. Visceral pain and the pseudoaffective response to intra-arterial injections of bradykinin and other algesic agents. Arch Int. Pharmacodyn. 1962;136:353–384. [PubMed] [Google Scholar]
- Guzman F, Braun C, Lim RKS, Potter GD, Rodgers DW. Narcotic and nonnarcotic analgesics which block visceral pain evoked by intra-arterial injections of bradykinin and other algesic agents. Arch. Int. Pharmacodyn. 1964;149:571–588. [PubMed] [Google Scholar]
- Hashimoto K, Kumakura S, Taira N. Vascular reflex responses induced by an intraarterial injection of aza-azepinophenothiazine, andromidotoxin, veratridine, bradykinin and kallikrein and blocking action of sodium salicylate. Jap. J. Physiol. 1964;14:299–308. doi: 10.2170/jjphysiol.14.299. [DOI] [PubMed] [Google Scholar]
- Horton EW. Action of prostaglandin E1 on tissues which respond to bradykinin. Nature. 1963;200:892–893. doi: 10.1038/200892b0. [DOI] [PubMed] [Google Scholar]
- Karim SMM. Action of prostaglandin in the pregnant woman. Ann. N. Y. Acad. Sci. 1971;180:483–498. doi: 10.1111/j.1749-6632.1971.tb53216.x. [DOI] [PubMed] [Google Scholar]
- Keith EF. Evaluation of analgesic substances. Am. J. Pharm. 1960;132:202–230. [PubMed] [Google Scholar]
- Lim RKS, Guzman F, Rodgers DW, Goto K, Braun C, Dickerson GD, Engle RJ. Site of action of narcotic and non-narcotic analgesics determined by blocking bradykinin-evoked visceral pain. Arch. int. Pharmacodyn. 1964;152:25–57. [PubMed] [Google Scholar]
- Mann M, West GB. The nature of hepatic and splenic sympathin. Br. J. Pharmac. Chemother. 1950;5:173–177. doi: 10.1111/j.1476-5381.1950.tb01004.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGiff JC, Terragno NA, Malik KU, Lonigro AJ. Release of a prostaglandin E-like substance from canine kidney by bradykinin. Circ. Res. 1972;31:36–43. doi: 10.1161/01.res.31.1.36. [DOI] [PubMed] [Google Scholar]
- Moncada SH, Ferreira SH, Vane JR. V. Int. Congress. Pharmacol. San Francisco: 1972. Does bradykinin cause pain through prostaglandin production. Abstracts of Volunteer Papers; p. 160. [Google Scholar]
- Ramwell PW, Shaw JE. Spontaneous and evoked release of prostaglandins from cerebral cortex of anaesthetized cats. Am. J. Physiol. 1966;211:125–134. doi: 10.1152/ajplegacy.1966.211.1.125. [DOI] [PubMed] [Google Scholar]
- Ramwell PW, Shaw JE, Jessup R. Spontaneous and evoked release of prostaglandins from frog spinal cord. Am. J. Physiol. 1966;211:998–1004. doi: 10.1152/ajplegacy.1966.211.4.998. [DOI] [PubMed] [Google Scholar]
- Randall LO, Selitto JJ. A method for measurement of analgesic activity on inflamed tissue. Arch Int. Pharmacodyn. 1957;111:409–419. [PubMed] [Google Scholar]
- Randall LO, Selitto JJ. Anti-inflammatory effects of Romilar CF. J. Amer. Pharmac. Assoc. 1958;47:313–314. [PubMed] [Google Scholar]
- Regoli D, Vane JR. A sensitive method for the assay of angiotensin. Br. J. Pharmac. Chemother. 1964;23:351–359. doi: 10.1111/j.1476-5381.1964.tb01591.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riva Sanseverino E, Urbano A. Azione della 5-idrossitriptamina sulla milza di cane. Arch. Sci. Biol. (Napoli) 1962;46:419–431. [PubMed] [Google Scholar]
- Sicuteri F. Sensitization of nociceptors by 5-hydroxytryptamine in man. In: Lim RKS, Armstrong D, Pardo EG, editors. Pharmacology of pain. London, N.Y.: Pergamon Press; 1966. pp. 57–86. [Google Scholar]
- Siggins G, Hoffer B, Bloom F. Prostaglandin-norepinephrine interactions in brain: microelectrophoretic and histochemical correlates. Ann. N.Y. Acad. Sci. 1971;180:302–323. doi: 10.1111/j.1749-6632.1971.tb53199.x. [DOI] [PubMed] [Google Scholar]
- Smith JB, Willis AL. Aspirin selectively inhibits prostaglandin production in human platelets. Nature New Biol. 1971;231:235–237. doi: 10.1038/newbio231235a0. [DOI] [PubMed] [Google Scholar]
- Vane JR. A sensitive method for the assay of 5-hydroxytryptamine. Br. J. Pharmac. Chemother. 1957;12:344–349. doi: 10.1111/j.1476-5381.1957.tb00146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vane JR. The use of isolated organs for detecting active substances in the circulating blood. Br. J. Pharmac. Chemother. 1964;23:360–373. doi: 10.1111/j.1476-5381.1964.tb01592.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vane JR. The release and fate of vaso-active hormones in the circulation. Br. J. Pharmac. 1969;35:209–242. doi: 10.1111/j.1476-5381.1969.tb07982.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nature New Biol. 1971;231:232–235. doi: 10.1038/newbio231232a0. [DOI] [PubMed] [Google Scholar]
- Vane JR. Prostaglandins in the inflammatory response. In: Lepow IH, Ward PA, editors. Inflammation mechanisms and control. New York and London: 1972a. pp. 261–279. [Google Scholar]
- Vane JR. Prostaglandins and the aspirin like drugs. Hospital Practice. 1972b;7:62–71. [Google Scholar]
- Willis AL. Release of histamine, kinin and prostaglandin during carrageenin-induced inflammation in the rat. In: Mantegazza P, Horton EW, editors. Prostaglandins, Peptides and Amines. London and New York: Acad. Press; 1969. pp. 31–38. [Google Scholar]
- Winder CV. A preliminary test for analgesic action in guinea-pigs. Arch. Int. Pharmacodyn. 1947;74:176–192. [PubMed] [Google Scholar]
- Winder CV, Pfeiffer CC, Maison GL. The nociceptive contraction of the cutaneous muscle of the guinea-pig as elicited by radiant heat. Arch. Int. Pharmacodyn. 1946;72:329–359. [PubMed] [Google Scholar]
- Winter CA, Flataker L. Nociceptive thresholds as affected by parental administration of irritants and of various anti-nociceptive drugs. J. Pharmac. exp. Ther. 1965;148:373–379. [PubMed] [Google Scholar]
- Wolff HG, Hardy JD, Goodell H. Measurement of the effect on the pain threshold of acetylsalicylic acid, acetanilid, acetophenetidin. aminopyrine, ethyl-alcohol, trichlorethylene, a barbiturate, quinine, ergotamine tartrate and caffeine: an analysis of their relations to the pain experience. J. Clin. Invest. 1941;20:63–80. doi: 10.1172/JCI101196. [DOI] [PMC free article] [PubMed] [Google Scholar]