

Abstract
Isolated rings of hamster aorta produced an unstable substance which inhibited platelet aggregation in vitro and had the same characteristics as prostacyclin.
Prostacyclin inhibited adenosine diphosphate (ADP)-induced aggregation of hamster platelets in vitro.
The effects of prostacyclin on ADP-induced platelet thrombi in the microcirculation of the hamster cheek pouch were studied with a television microscope.
Prostacyclin caused a dose-dependent increase in the time of iontophoretic application of ADP which was required to induce platelet thrombi formation and embolization in venules (30 to 40 μm diameter).
Prostacyclin caused a dose-dependent reduction in the total time during which ADP-induced thrombi were observed following local electrical damage to arterioles (40 to 80 μm diameter).
Thrombus formation in venules and arterioles was abolished by 500 ng/ml prostacyclin in the Krebs solution superfusing the hamster cheek pouch.
Prostacyclin was approximately twenty times more potent than prostaglandin E1 in preventing thrombus formation in the microcirculation.
Introduction
Prostaglandin (PG) E1 (Kloeze, 1967) and PGD2 (Smith, Silver, Ingerman & Kocsis, 1974) are powerful inhibitors of platelet aggregation in vitro, while PGE2 has been reported to enhance aggregation in low concentrations and inhibit aggregation at higher ones. Prostaglandins from the A, B and F series are relatively inactive (for review, see Howie, 1976).
PGG2 and PGH2, the cyclic endoperoxide intermediates in prostaglandin biosynthesis are converted to thromboxane A2 (TXA2) (Hamberg, Svensson & Samuelsson, 1975) by an enzyme in platelet microsomes (Needleman, Moncada, Bunting, Vane, Hamberg & Samuelsson, 1976) or to prostacyclin (PGI2) by an enzyme located in blood vessel walls (Moncada, Gryglewski, Bunting & Vane, 1976a). TXA2 and PGI2 are unstable substances with opposite effects on platelet aggregation in vitro; TXA2 induces aggregation and PGI2 is the most potent known inhibitor of aggregation. Moncada, Gryglewski, Bunting & Vane (1976b) have suggested that a balance between formation of anti- and pro-aggregatory substances could contribute to the maintenance of the integrity of vascular endothelium and disruption of this balance may contribute to the formation of intravascular thrombi.
Prostacyclin inhibits adenosine diphosphate (ADP)-induced aggregation of human, rabbit, sheep, horse and rat platelets (Moncada, Vane & Whittle, 1978). Furthermore, prostacyclin is produced by vascular tissue from a number of different species including man (Moncada el al., 1976a; 1976b; Moncada, Higgs & Vane, 1977).
Emmons, Hampton, Harrison, Honour & Mitchell (1967), first demonstrated that PGE1 inhibits platelet thrombus formation in vivo in injured rabbit-brain arteries. Westwick (1977) has now shown that both PGE1 and PGG2 inhibit thrombus formation in hamster cheek pouch arterioles. In this paper we have investigated the production of prostacyclin-like material by hamster blood vessels, the sensitivity of hamster platelets to protacyclin and the effects of prostacyclin on the formation of platelet thrombi in venules and arterioles of the hamster cheek pouch. Some of these results have been communicated to the British Pharmacological Society (Higgs, Moncada & Vane, 1977).
Methods
Platelet aggregation in vitro
Male golden hamsters (100 to 150 g) were anaesthetized with sodium pentobarbitone (75 mg/kg i.p.) and 5 to 7 ml blood was withdrawn through a cannula in a carotid artery or the abdominal aorta. Human blood was taken from the anticubital vein of healthy volunteers who had not taken aspirin in the previous 2 weeks. Fresh hamster or human blood was citrated (0.1 volume of 3.15% (w/v) trisodium citrate) and centrifuged at 200 g for 15 min to obtain platelet-rich plasma (PRP). Platelet aggregation was induced by the addition of 0.5 to 10 μl of 10−3 m ADP to 0.3 ml hamster PRP or 0.5 ml human PRP in a Born-type aggregometer (Payton dual channel). Aggregation was measured as a change in light transmitted through the platelet suspension (Born, 1962).
Assay of prostacyclin activity
The sodium salt of prostacyclin (Johnson, Lincoln, Thompson, Nidy, Mizsak & Axen, 1977; Whittaker, 1977) was dissolved immediately before use in 50 Mm Tris buffer (pH 7.5) and different doses (100 pg to 2 ng in 0.1 to 2 μl volumes) were added to hamster or human PRP 1 min before the addition of the lowest dose of ADP which caused both phases of aggregation in untreated PRP. Aggregation was measured as the change in transmitted light 2.5 min after the addition of ADP. Inhibition was calculated as percentage reduction of the control response. The IC50 for inhibition of aggregation of hamster platelets by prostacyclin was calculated from a dose-response curve.
Prostacyclin production
Lengths of hamster aorta (1.5 to 2.0 cm) were cleaned of connective tissue, weighed and washed in cold 50 Mm Tris buffer (pH 7.5). The tissue (10 to 25 mg) was chopped into rings and incubated in 200 μl buffer for 3 min at 22°C. Aliquots (10 to 100 μl) of the supernatant of this incubation mixture were added to human PRP to test for anti-aggregating activity. The activity of supernatants was tested immediately, 30 min after standing at 22°C or after boiling for 15 seconds. The generation of anti-aggregating activity by hamster aorta was measured after incubation with PGH2 (1 μg/ml for 3 min at 22°C) in untreated rings or rings pretreated with 15-hydroperoxy arachidonic acid (15-HPAA; 10 μg/ml for 10 min at 22°C). Anti-aggregating activity was assayed against standard prostacyclin and expressed as ng/mg wet weight of tissue. In four experiments, hamsters were given aspirin orally (500 mg/kg) 1–2 h before the aortae were removed.
The hamster cheek pouch preparation
The microcirculation of the hamster cheek pouch was studied by the method of Duling, Berne & Born (1968). Male golden hamsters (approx. 100 g) were anaesthetized with sodium pentobarbitone (75 mg/kg, i.p.) and their everted cheek pouches were immersed in a well (9 ml) in a perspex microscope stage. The preparation was superfused at 5 ml/min with Krebs solution (composition g/l glass-distilled water: NaCl 7.01, NaHCO3 1.68, KCl 0.3503, MgCl 0.2437, CaCl2, 6H2O 0.2271 and d-glucose 1.80) at 36°C which was bubbled with 5% CO2 in O2. Blood vessels in the microcirculation of the cheek pouch were examined microscopically with long-working distance objectives (10 ×, 20 × or 32 ×) attached to a fixed stage microscope (Leitz, Laborlux). The preparation was viewed on a 10 inch television monitor (Hitachi, VM126AK) by attaching a light-weight closed-circuit television camera (Hitachi, HV62K) to the photo-tube of the microscope.
Thrombus formation in venules
Platelet thrombi were induced in venules (30 to 40 μm diameter) by the iontophoretic application of ADP to the blood vessel walls (Begent & Born, 1970). Glass electrodes (Jencons, ‘Vac-Seal’) were drawn out to form micropipettes (tip diameter = 2.5 to 5 μm) with a pipette puller (Scientific Research Instruments Ltd.). Micropipettes containing a solution of 10−2 m ADP were manipulated by means of a Leitz micromanipulator so that their tips were adjacent to the venules. ADP was applied to the blood vessel wall by passing a current (200–300 nA) between the micropipette and a reference electrode immersed in the superfusing Krebs solution. Current was supplied by a variable power supply (0 to 60 V) via a 100 MΩ current limiting resistor. The voltage drop across a 100 kω resistor, in series with the power supply was measured by a digital volt meter. The time of ADP application which was required to induce the formation and embolization of platelet thrombi as described by Begent, Born & Sharp (1972) was measured before, during and after infusions (0.05 ml/min) of different concentrations of PGI2, PGE1 or 6-oxo-PGF1α, dissolved in 50 Mm Tris buffer (pH 7.5), into the Krebs solution superfusing the microcirculation.
Thrombus formation in arterioles
Thrombus formation in arterioles (40 to 80 μm diameter) was induced by electrical damage to the blood vessel wall followed by an injection of 10 μl 10−3 m ADP into the superfusing Krebs solution (Westwick, 1977). Current (10 to 15 μA) was applied to arteriole walls for 30 s via a micropipette filled with 1 m KCl, using the circuit described above, but with the 100 MΩ current limiting resistor switched out. The blood vessel was observed for 5 min after stimulation and the total time that platelet thrombi were present (‘total thrombus time’) was recorded. The same vessel was stimulated 3 to 5 times before, during and after the infusion (0.05 ml/min) of different concentrations of PGI2, PGE1 or 6-oxo-PGF1α into the superfusing Krebs solution. The means of the total thrombus times in the presence of PGI2, PGE1 or 6-oxo-PGF1α were expressed as a percentage of the means of the control times.
Measurement of mean blood velocity
Mean blood velocity in venules and arterioles was measured by the method of Begent & Born (1970). The movement of embolized thrombi, downstream from their site of formation, was recorded with a video-tape recorder (National, NV-3030E). By playing back the video-tape in slow motion, the mean blood velocity along a measured length of blood vessel was calculated.
Results
Prostacyclin prevented the aggregation of hamster platelets in vitro (IC50 = 1.5 ± 0.8 ng/ml; mean ± s.e. mean, n = 3). Chopped rings of hamster aorta incubated for 3 min produced a substance which prevented the ADP-induced aggregation of human platelets. The anti-aggregating activity was equivalent to 0.08 ± 0.02 ng (n = 7) prostacyclin/mg wet weight of tissue and was not present after boiling the supernatant or standing it at room temperature for 30 minutes. PGH2 enhanced the production of anti-aggregating material, an effect which was inhibited by 15-HPAA. Aortae removed from hamsters 1 to 2 h after oral aspirin (500 mg/kg) produced only 0.0075 ± 0.001 ng (n = 4) prostacyclin-like activity/mg wet weight of tissue.
Thrombus formation in venules
After preparation of the hamster cheek pouch, a 30 to 45 min equilibrium period was allowed before venules with a diameter of 30 to 40 μm were selected for observation. Currents of up to 500 nA applied to pipettes filled with 0.9% w/v NaCl solution (saline) did not cause formation of platelet thrombi; nor did pipettes filled with 10−2 m ADP, in the absence of current. When currents of 200 to 300 nA were applied to ADP-filled pipettes, thrombi appeared on the vessel wall close to the tip and at sites downstream from the tip. During continuous application of ADP, thrombi adhered to vessel walls before they embolized and were swept away in the blood stream.
Measurements of mean blood velocity were made, and only venules in which the velocity was 1.0 to 2.0 mm/s were studied. When the velocity fell below 1.0 mm/s, the preparation was discarded.
The mean time of ADP application required to induce the formation and embolization of platelet thrombi in venules was 20.8 ± 3.2 s (n = 8). Repeated stimulation of the same venule gave a consistent response for up to 3 hours. Prostacyclin (1 to 200 ng/ml in superfusing Krebs solution) caused dose-dependent increases in this time and at concentrations of 0.5 to 1.0 μg/ml, continuous application of ADP for up to 5 min failed to induce thrombus formation (Figure 1). The maximum effects of prostacyclin infusions were seen after 10 minutes. At concentrations between 200 and 500 ng prostacyclin/ml, abolition of thrombus formation was seen in some, but not all, experiments. With all concentrations of PGI2 the response to ADP returned to control values after the infusion had stopped. After low doses, the normal response returned almost immediately and after higher doses (0.5 to 1 μg/ml) the recovery period was up to 30 minutes.
Figure 1.
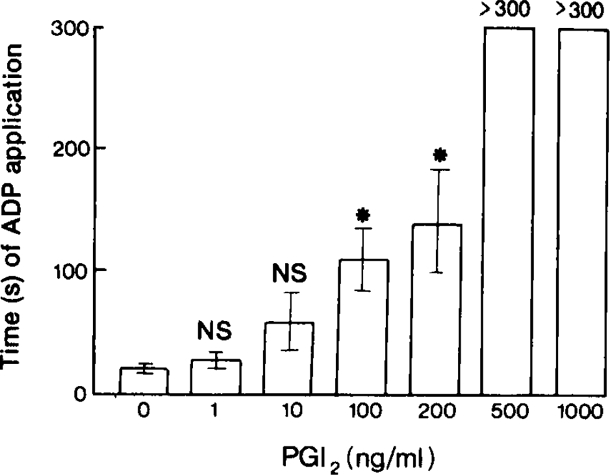
The time in seconds of iontophoretic application of adenosine diphosphate (ADP) to venules (30 to 40 μm) necessary to induce the formation and embolization of platelet thrombi in the presence of increasing concentrations of prostacyclin (PGI2) infused into the Krebs solution superfusing the hamster cheek pouch microcirculation. Each column represents the mean of 4–8 experiments and the bars are ± 1 s.e. mean. NS, P > 0.01; *P < 0.01.
6-oxo-PGF1α, the stable product of prostacyclin degradation (Johnson, Morton, Kinner, Gorman, McGuire, Sun, Whittaker, Bunting, Salmon, Moncada & Vane, 1976) did not affect thrombus formation in venules at concentrations up to 10 μg/ml. PGE1 (1 μg/ml) increased the time of ADP application required to induce thrombus formation and embolization to 61.2 ± 10.0 s (n = 5) and PGE1 at 10 μg/ml abolished thrombus formation.
PGI2 (1 μg/ml), PGE1 (10 μg/ml) or 6-oxo-PGF1α (10 μg/ml) did not cause a change in the mean diameter of the venules observed. There was also no significant change in mean blood flow at the highest concentrations of PGI2, PGE1 or 6-oxo-PGF1α at which embolization of thrombi could still be used as a measure of blood flow.
Thrombus formation in arterioles
About half the pre-capillary arterioles seen in the hamster cheek pouch had inherent tone and the others were assumed to be maximally dilated, for they were tested with PGE1 (100 ng) and PGI2 (10 ng) and did not respond by vasodilatation (Higgs, Moncada & Vane, 1978). Thrombus formation was always studied in maximally dilated vessels (40 to 80 μm diameter). Application of 10 to 15 μA for 30 s to arteriole walls did not induce thrombus formation, although localized constriction of the vessel was often seen. When 10 μl 10−3 m ADP was injected into the Krebs solution proximal to the vessels being observed, immediately after the application of current, platelet thrombi formed at the stimulated site. Thrombi embolized and continued to form at the damaged site for up to 5 min after stimulation. Mean blood velocity in the vessels studied was 2.0 to 7.0 mm/s and total thrombus time varied from 90 s in arterioles with high blood velocity to 290 s in vessels with slower velocity. Because of this variation in control response, the effects of PGI2, PGE1 or 6-oxo-PGF1α on total thrombus time were expressed as a percentage of the control response in the same blood vessel.
Prostacyclin (25 to 200 ng/ml) caused a dose-dependent reduction in the total thrombus time (Figure 2) and 500 ng/ml abolished arteriolar thrombus formation. Total thrombus time returned to control values 30 min after the prostacyclin infusions had stopped. Thrombus time was reduced to 49 ± 10% (n = 5) of control values by PGE1 at 1 μg/ml and abolished at 10 μg/ml. 6-oxo-PGF1α (1 to 10 μg/ml) did not affect thrombus formation. All concentrations of PGI2, PGE1 and 6-oxo-PGF1α tested produced maximal dilatation in pre-capillary vessels with inherent tone, but there were no significant changes in mean blood velocity in the maximally dilated vessels used for studying thrombus formation.
Figure 2.
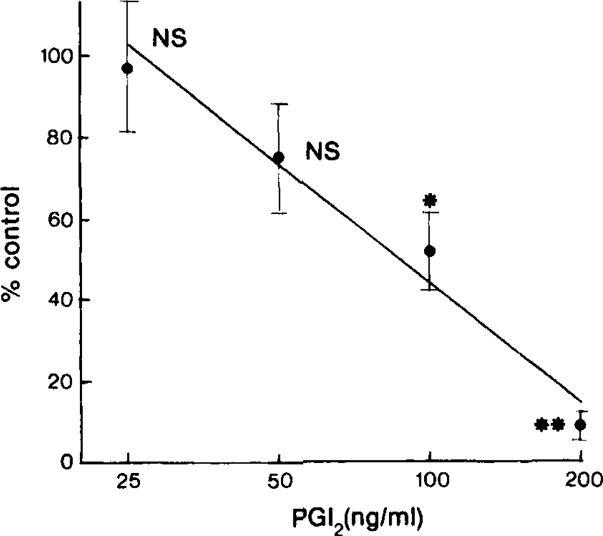
Inhibition of adenosine diphosphate (ADP)-induced thrombus formation in electrically damaged arterioles (40 to 80 μm) by increasing concentrations of prostacyclin (PGI2) infused into the Krebs solution superfusing the hamster cheek pouch microcirculation. Inhibition is measured as a reduction in total thrombus time (see text) compared with control values. Each point is the mean of 4–7 experiments and the bars represent ± 1 s.e. mean. The straight line was fitted to the log dose-response curve by regression analysis. There is a highly significant linear regression (P > 0.001) and there is no significant deviation from linearity. NS, P > 0.01; *P < 0.0025, **P < 0.0005.
Discussion
These results demonstrate that prostacyclin inhibits ADP-induced aggregation of hamster platelets. The inhibitory activity of prostacyclin on hamster platelets is similar to that reported for rat, rabbit and human (Moncada el al., 1978).
The anti-aggregating material generated by rings of hamster aorta has similar characteristics to authentic prostacyclin. It is unstable, disappearing after 30 min at room temperature, and heat labile, being destroyed by boiling for 15 seconds. Also, production of anti-aggregating material is enhanced by incubation of the tissue with PGH2 and inhibited by incubation with 15-HPAA, which inhibits prostacyclin generation (Moncada et al., 1976b). Furthermore, production of prostacyclin-like material by aortae from hamsters treated with aspirin is reduced by over 90%. The basal production of activity by hamster tissue is comparable to production by human arterial tissue (Moncada et al., 1977).
The observations of thrombus formation in the hamster microcirculation show that prostacyclin is a powerful inhibitor of platelet aggregation in vivo, being approximately twenty times more potent than PGE1. Reduction in thrombus formation is independent of changes in mean blood velocity, a factor which is known to influence the growth rate of thrombi in this preparation (Begent & Born, 1970).
Westwick (1977) has shown that PGE1 and PGG2 are equipotent in inhibiting thrombus formation in arterioles in the hamster check pouch, whereas PGE2 or PGD2 are inactive. PGG2 induces platelet aggregation in vitro (Hamberg, Svensson, Wakabayashi & Samuelsson, 1974) and the only metabolite of PGG2 which is anti-aggregatory in hamster arterioles is prostacyclin. The anti-thrombotic activity of PGG2 must, therefore, be interpreted as being due to partial conversion to prostacyclin in the blood vessel wall.
These results support the theory that generation of prostacyclin is an important natural defence against intra-vascular thrombus formation (see Moncada & Vane, 1977).
Acknowledgments
We wish to thank Dr J. Westwick for help and advice with the hamster cheek pouch preparation and Mr D. C. Cardinal for help with the electronics.
References
- Begent NA, Born GVR. Growth rate in vivo of platelet thrombi, produced by iontophoresis of ADP, as a function of mean blood flow velocity. Nature. 1970;227:926–930. doi: 10.1038/227926a0. [DOI] [PubMed] [Google Scholar]
- Begent NA, Born GVR, Sharp DE. The initiation of platelet thrombi in normal venules and its acceleration by histamine. J. Physiol. 1972;223:229–242. doi: 10.1113/jphysiol.1972.sp009843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Born GVR. Aggregation of blood platelets by adenosine diphosphate and its reversal. Nature, Lond. 1962;194:927–929. doi: 10.1038/194927b0. [DOI] [PubMed] [Google Scholar]
- Duling BR, Berne RM, Born GVR. Microiontophoretic application of vasoactive agents to the micro-circulation of the hamster cheek pouch. Microvascular Res. 1968;1:158–173. [Google Scholar]
- Emmons PR, Hampton JR, Harrison MJC, Honour AJ, Mitchell JRA. Effect of prostaglandin E1 on platelet behaviour in vitro and in vivo. Br. med. J. 1967;ii:468–472. doi: 10.1136/bmj.2.5550.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamberg M, Svensson J, Samuelsson B. Thromboxanes: A new group of biologically active compounds derived from prostaglandin endoperoxides. Proc. natn Acad. Sci., U.S.A. 1975;72:2994–2998. doi: 10.1073/pnas.72.8.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamberg M, Svensson J, Wakabayashi T, Samuelsson B. Isolation and structure of two prostaglandin endoperoxides that cause platelet aggregation. Proc. natn Acad. Sci., U.S.A. 1974;71:345–349. doi: 10.1073/pnas.71.2.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgs GA, Moncada S, Vane JR. Prostacyclin (PGI2) inhibits the formation of platelet thrombi induced by adenosine diphosphate (ADP) in vivo. Br. J. Pharmac. 1977;61:137P. [PMC free article] [PubMed] [Google Scholar]
- Higgs GA, Moncada S, Vane JR. Prostacyclin as a potent dilator of arterioles in the hamster cheek pouch. J. Physiol. 1978;275:30–31P. [PubMed] [Google Scholar]
- Howie PW. Prostaglandins and blood coagulation. In: Karim SMM, editor. Advances in Prostaglandin Research. Prostaglandins: Physiological, Pharmacological and Pathological Aspects. Lancaster: MTP Press Ltd; 1976. pp. 277–291. No. 3. [Google Scholar]
- Johnson RA, Lincoln FH, Thompson JL, Nidy EG, Mizsak SA, Axen U. Synthesis and stereochemistry of prostacyclin and synthesis of 6-oxo-prostaglandin F1α. J. Am. Chem. Soc. 1977;99:4182–4184. doi: 10.1021/ja00454a060. [DOI] [PubMed] [Google Scholar]
- Johnson RA, Morton DR, Kinner JH, Gorman RR, McGuire JR, Sun FF, Whittaker N, Bunting S, Salmon JA, Moncada S, Vane JR. The chemical characterization of prostaglandin X (prostacyclin) Prostaglandins. 1976;12:915–928. doi: 10.1016/0090-6980(76)90126-x. [DOI] [PubMed] [Google Scholar]
- Kloeze J. Influence of prostaglandins on platelet adhesiveness and platelet aggregation. In: Bergström S, Samuelsson B, editors. Prostaglandins. Vol. 2. Stockholm: Almquist and Wiksell; 1967. pp. 241–252. Nobel Symposium. [Google Scholar]
- Moncada S, Gryglewski RJ, Bunting S, Vane JR. An enzyme isolated from arteries transforms prostaglandin endoperoxides to an unstable substance that inhibits platelet aggregation. Nature. 1976a;263:663–665. doi: 10.1038/263663a0. [DOI] [PubMed] [Google Scholar]
- Moncada S, Gryglewski RJ, Bunting S, Vane JR. A lipid peroxide inhibits the enzyme in blood vessel microsomes that generates from prostaglandin endoperoxides the substance (prostaglandin X) which prevents platelet aggregation. Prostaglandins. 1976b;12:715–737. doi: 10.1016/0090-6980(76)90048-4. [DOI] [PubMed] [Google Scholar]
- Moncada S, Higgs EA, Vane JR. Human arterial and venous tissues generate prostacyclin (prostaglandin X), a potent inhibitor of platelet aggregation. Lancet. 1977;i:18–20. doi: 10.1016/s0140-6736(77)91655-5. [DOI] [PubMed] [Google Scholar]
- Moncada S, Vane JR. The discovery of prostacyclin. A fresh insight into arachidonic acid metabolism. In: Kharasch N, Fried J, editors. Biochemical Aspects of Prostaglandins and Thromboxanes. London: Academic Press; 1977. pp. 155–177. [Google Scholar]
- Moncada S, Vane JR, Whittle BJR. Relative potency of prostacyclin (PGI2), prostaglandin E1 and prostaglandin D2 as inhibitors of platelet aggregation in several species. J. Physiol. 1978;273:2–4P. [PubMed] [Google Scholar]
- Needleman P, Moncada S, Bunting S, Vane JR, Hamberg M, Samuelsson B. Identification of an enzyme in platelet microsomes which generates thromboxane A2 from prostaglandin endoperoxides. Nature. 1976;261:558–560. doi: 10.1038/261558a0. [DOI] [PubMed] [Google Scholar]
- Smith JB, Silver MJ, Ingerman CM, Kocsis JJ. PGD2 inhibits aggregation of human platelets. Thromb. Res. 1974;5:291–299. doi: 10.1016/0049-3848(74)90168-6. [DOI] [PubMed] [Google Scholar]
- Westwick J. Modulation of thrombus formation in vivo by prostaglandins. Br. J. Pharmac. 1977;61:138P–139P. [PMC free article] [PubMed] [Google Scholar]
- Whittaker N. A synthesis of prostacyclin sodium salt. Tetrahedron Lett. 1977;32:2805–2808. [Google Scholar]