

Abstract
Two papers published recently in Science exploit new transgenic mouse systems to explore the path that activated CD8+ T cells take on the way to memory differentiation.
Primary infection results in the activation and proliferation of rare naive CD8+ T cells that have been selected on the basis of their ability to respond specifically to the offending pathogen. At the population level, these expanded clones express effector molecules, including granzyme B, that aid in the elimination of infected host cells. After pathogen clearance, most pathogen-specific CD8+ T cells die, but a substantial fraction (5–10%) of the cells survive long-term and rapidly protect the host in the event of reinfection. Memory T cells differ from both naive and effector T cells and show a broad range of differentiation states defined by phenotype, function, anatomic distribution and contribution to protection from reinfection1. Thus, activated naive T cells differentiate into terminal effector T cells that die, and into a diverse array of memory T cells that persist in the host. Two recent papers in Science provide insight into the regulation of these fate ‘decisions’2,3.
How and when is it determined which pathogen-specific CD8+ T cells die after the resolution of infection and which become long-lived memory cells? It has long been debated whether transit through an effector stage is required for memory development to occur or even if effector cells can differentiate into memory cells rather than constituting a terminally differentiated lineage that is fated for death. These fundamental immunological questions have been difficult to answer, and the existing evidence supports a variety of models. One possibility is that asymmetric division after the activation of a naive T cell results in the formation of two daughter cells with polarized cell fates: a terminal effector lineage and a memory lineage4 (Fig. 1a). A related model dictates that naive T cells differentiate into two lineages of effectors: one terminal and one leading to memory cell generation5,6 (Fig. 1b). Alternatively, memory cells could arise from an expanded population of pluripotent effector cells and the fate ‘decision’ could be regulated by extrinsic developmental cues, including antigen stimulation and the cytokine milieu, or could be purely stochastic.
Figure 1.
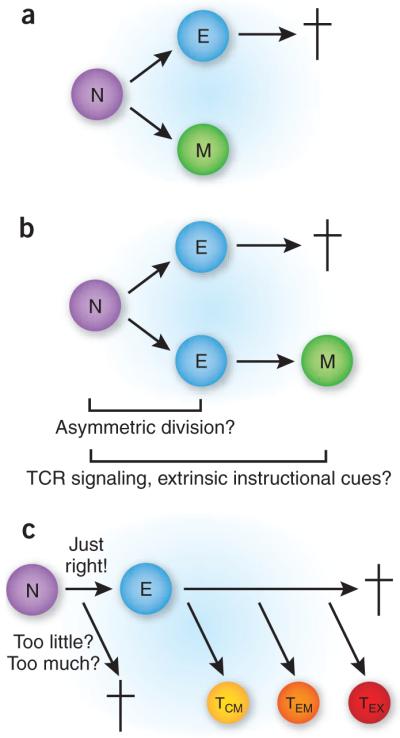
Potential pathways for the development of memory CD8+ T cells. (a) In the dual-lineage model, two separate pathways are followed immediately after initial activation of a naive CD8+ T cell (N), one leading to effector cells (e) that go on to die (†) and another leading to the generation of long-lived memory cells (M) without transit through an effector stage. This model is consistent with early asymmetric division that produces separate lineages. (b) In the dual-effector lineage model, two populations of effector cells are generated: one subset goes on to die, and the other is the eventual source of memory cells. In this scenario, it is also possible that asymmetric division early after activation generates the two effector cell lineages. Alternatively, the amount of TCR signaling or the extent of integration of all necessary signals (TCR, costimulation, cytokines) could influence whether one lineage is produced or both are produced. This model is compatible with the results of both papers discussed here. (c) In the ‘Goldilocks’ linear model, the strength of stimulation regulates the response outcome. Too little or too much signaling leads to a nonproductive response or perhaps to arrest at the effector stage, as described by Teixeiro and colleagues2. with optimal signal integration during the activation of naive T cells, effector cells are produced that develop into central memory cells (TCM) and effector memory cells (TeM) and, in some cases, as during chronic infection, exhausted memory cells (TeX). This model is supported by the results of Bannard and colleagues3, and is also in agreement with the findings of Teixeiro et al.
Defining the regulatory requirements for commitment to the long-lived protective memory lineage could provide the potential for rational manipulation of vaccination. Overlying the basic idea of memory cell development is the additional complexity of memory cell subset heterogeneity, which also may be designated early during T cell responses but could be further influenced by tissue-specific environmental cues late after priming7. In general, CD8+ memory T cell subsets can be broadly categorized as central memory or effector memory on the basis of location and function, but their origins are also a matter of speculation8. Like naive CD8+ T cells, cells of both of these memory subtypes may also be considered stem cells, as they undergo self-renewal, albeit at distinct rates1, and are able to respond to secondary or tertiary antigenic challenges, resulting in production of new effector and memory cells. Thus, this additional diversity must be accounted for when developing complete models of immune response cellular dynamics.
Bannard et al. seek a definitive answer to the question of whether effector cells can differentiate into memory cells or represent terminally differentiated cells incapable of surviving after the clearance of infection3. They developed an inducible system which indelibly labels (via Cre-mediated recombination) cells that have granzyme B promoter activated the (one ‘signature’ of effector cells) with a fluorescent protein (EYFP)3. Pulsing cells with the inducer tamoxifen during the initiation of the response results in EYFP expression in a subset (~20–30%) of the responding antigen-specific population because of inefficiency in the Cre recombinase–mediated recombination step. The authors exploit this elegant system to track the fate of CD8+ T cells that express granzyme B at various times during the course of the immune response to influenza virus infection. They find that effector cells that had upregulated granzyme B expression within the first few days after infection continue to proliferate and also form long-lived memory cells in both lymphoid and nonlymphoid tissues. Moreover, labeled memory cells that had expressed granzyme B during the response to infection differentiate into both central memory and effector memory CD8+ T cells and are fully able to proliferate in response to reinfection, fulfilling a hallmark property of T cell memory. Although these observations provide very strong evidence that effector cells can differentiate into memory cells, an important question is left unanswered. This model system cannot determine whether a subset of effector cells is committed to die while others have long-term survival potential. For example, the assay may not discriminate between cells with low expression of granzyme B and those with high expression of granzyme B. Alternatively, memory precursors could differ from terminally differentiated effectors in the expression of molecules other than granzyme B. Thus, the results do not refute the hypothesis that there could be two kinds of effector cells, one of which is terminally differentiated (Fig. 1b). However, this important study does show that effector differentiation, as defined by granzyme B expression, does not preclude differentiation into long-lived memory CD8+ T cells that populate lymphoid and nonlymphoid tissues.
Palmer and colleagues have also developed a new transgenic system to investigate the relationship between effector and memory CD8+ T cells2. In this study, they introduce point mutations in the sequence encoding the T cell antigen receptor-β (TCRβ) conserved antigen receptor transmembrane motif of CD8+ T cells. They find that after infection with Listeria monocytogenes, transferred mutant clonotypic TCR-transgenic CD8+ T cells undergo robust proliferation and differentiation into effector cells. However, the development of pathogen-specific memory CD8+ T cells is mostly abrogated by the mutation. This defect correlates with poor polarization of TCRs to the immunological synapse, although despite this, wild-type and mutant T cells are equally able to maintain stable interactions with peptide-coated antigen-presenting cells, at least in vitro. The data further demonstrate that there is less recruitment of PKC-θ to the synapse and less ‘downstream’ NF-κB induction. This may not be the whole story, as PKC-θ is not required for the generation of antiviral effector or memory CD8+ T cells9, which leaves the precise defect in the mutant CD8+ T cells that blocks antibacterial memory development unclear. Nonetheless, the results suggest that memory development is not an axiomatic consequence of the generation of an effector population; that is, apparently normal effector cells may not have memory potential. This study supports the possibility that two lineages, one effector and one memory, develop after the activation of naive CD8+ T cells but falls short of proof for the two-lineage model, as it does not demonstrate at which point mutant effector cells become terminally differentiated. However, the study does provide intriguing evidence that the quality of ‘signal 1’ derived from the TCR can be ‘translated’ into a transcriptional program that is dispensable for effector differentiation but is essential for memory development (Fig. 1c). This observation supports the idea that the memory pathway can derive from a distinct T cell-intrinsic fate ‘decision’.
Whether the circumstances produced by Teixeiro et al. are recapitulated during a normal immune response is unclear at present. For example, do wild-type endogenous polyclonal CD8+ T cells show heterogeneity in NF-κB localization that in turn dictates effector death versus survival? Regardless of this caveat, the ability to dissociate effector and memory generation provides an exceptional system with which to begin to define the molecular ‘fingerprint’ associated with memory development. Additionally, this system, as well as variations on the model of Bannard et al., could be used to determine whether and when the fate ‘decision’ becomes terminal. Could exogenous factors ‘rescue’ the defect early or late in the response? Or is the initial TCR signal deterministic for the eventual outcome? It should also be noted that the environmental milieu of a particular response to immunization or infection will also affect the outcome10, so defining universal rules governing memory T cell development will continue to pose considerable challenges.
Although the ‘holy grail’ of immune memory—that is, the identification of the precise factors that initially specify which cell lives or dies at the outset of an immune response—remains elusive, the prize draws closer. The ultimate challenge is to identify the true memory precursor at the single-cell level at the earliest moment possible during an immune response and to determine the signals required for the appearance of that cell type and the ‘downstream’ development of memory lineages. Additional creative experimental approaches akin to those discussed here will be needed to solve this enigma.
References
- 1.Lefrançois L. Immunol. Rev. 2006;211:93–103. doi: 10.1111/j.0105-2896.2006.00393.x. [DOI] [PubMed] [Google Scholar]
- 2.Teixeiro E, et al. Science. 2009;323:502–505. doi: 10.1126/science.1163612. [DOI] [PubMed] [Google Scholar]
- 3.Bannard O, Kraman M, Fearon DT. Science. 2009;323:505–509. doi: 10.1126/science.1166831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang JT, et al. Science. 2007;315:1687–1691. doi: 10.1126/science.1139393. [DOI] [PubMed] [Google Scholar]
- 5.Joshi NS, et al. Immunity. 2007;27:281–295. doi: 10.1016/j.immuni.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sarkar S, et al. J. Exp. Med. 2008;205:625–640. doi: 10.1084/jem.20071641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Masopust D, vezys v., wherry e.J., Barber DL, Ahmed R. J. Immunol. 2006;176:2079–2083. doi: 10.4049/jimmunol.176.4.2079. [DOI] [PubMed] [Google Scholar]
- 8.Masopust D, Lefrançois L. Microbes Infect. 2003;5:221–226. doi: 10.1016/s1286-4579(03)00014-5. [DOI] [PubMed] [Google Scholar]
- 9.Marsland BJ, et al. Proc. Natl. Acad. Sci. USA. 2005;102:14374–14379. doi: 10.1073/pnas.0506250102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harty JT, Badovinac v.P. Nat. Rev. Immunol. 2008;8:107–119. doi: 10.1038/nri2251. [DOI] [PubMed] [Google Scholar]