

Abstract
Pulmonary arterial hypertension (PAH) is a rapidly progressive and fatal disease for which there is an ever-expanding body of genetic and related pathophysiological information on disease pathogenesis. The most common single culprit gene known is BMPR2, and animal models of the disease in several forms exist. There is a wealth of genetic data regarding modifiers of disease expression, penetrance, and severity. Despite the rapid accumulation of data in the last decade, a complete picture of the molecular pathogenesis of PAH leading to novel therapies is lacking. In this review, we attempt to summarize the current understanding of PAH from the genetic perspective. The most recent PAH demographics are discussed. Heritable PAH in the post-BMPR2 era is examined in detail as the most robust model of PAH genetics in both animal models and human pedigrees. Important downstream molecular pathways and modifiers of disease expression are reviewed in light of what is known about PAH pathogenesis. Current and emerging therapies are examined in light of genetic data. The role of genetic testing in PAH in the post-BMPR2 era is discussed. Finally, directions for future investigations that ideally will fulfill the promise of novel therapeutic or preventive strategies are discussed.
Keywords: BMPR2, heritable pulmonary arterial hypertension, idiopathic pulmonary arterial hypertension, pulmonary arterial hypertension, right ventricle
INTRODUCTION
Pulmonary arterial hypertension (PAH) is a disease of the pulmonary vasculature that is pathologically characterized by progressive neointimal proliferation leading to vaso-occlusive lesions as well as to dropout and pruning of the smallest pulmonary arteries.[1] This drives the clinically apparent disease, characterized by progressive dyspnea, exercise intolerance, increasing pulmonary vascular resistance, and ultimately right ventricular failure and death.[2] Under the current classification system, WHO Group 1 PAH is divided into disease subgroups that include heritable (HPAH, formerly familial PAH or FPAH), idiopathic (IPAH), and PAH associated (APAH) with a variety of other systemic diseases or drug/toxin exposures.[3]
Untreated, PAH results in death from right heart failure in less than 3 years for most patients.[4] Despite advancements in therapies, as evidenced by FDA approval of 7 drugs specifically for the treatment of PAH, the currently available therapies are, for most patients, only partially or temporarily effective. No therapies tested to date have shown any significant ability to reverse the disease, and despite suggestions in epidemiologic studies of improvement there has been no conclusive demonstration in large clinical trials of the ability of currently available therapies to prolong survival, although patients who have a good initial response to therapy may derive some survival benefit.[5–10]
Much has been learned about the genetic underpinnings of PAH since its initial descriptions as primary pulmonary hypertension by Dresdale and colleagues in 1951.[11] This line of inquiry expanded to include the recognition of PAH as a familial disease in some cases, with the initial description in 1954 of a family in which PAH was identified in a mother and in her son and sister.[12] Intense study of the inheritance and genetic patterns of familial PAH ultimately led to the identification of altered transforming growth factor beta (TGF-β) signaling via the bone morphogenetic protein receptor Type 2 (BMPR2) as being the key heritable risk factor for development of PAH,[13,14] with other primary (e.g., mutations in ALK1 or ENG) and modifier genetic risk factors having been identified as well. The evolution of the current understanding of the genetics of PAH, as well as existing questions, is the topic of this review.
DISCUSSION
PAH: Definition, incidence, and demographics
For the purposes of clinical and research definition, PAH is defined as a mean pulmonary artery pressure > 25 mmHg at rest,[15] confirmed by right heart catheterization, and in the absence of other conditions known to elevate pulmonary vascular pressures, such as pulmonary embolism, left-sided heart disease, other lung diseases, and various conditions associated with PAH.[16] HPAH is a subset of PAH in which patients either belong to a pedigree known to be affected by PAH in multiple family members or are found to have a mutation in a gene known to associate with PAH (most commonly BMPR2) in what was previously thought to be a case of sporadic or IPAH. It was this latter group of patients that prompted the official change in classification from familial PAH (FPAH) to HPAH.[3] Because the mutations that drive HPAH show reduced penetrance, there can be multiple unaffected generations in a family pedigree, and thus family history can be apparently negative for pulmonary vascular disease. It is only with the advent of genetic testing for particular mutations that these patients have become apparent. In addition, genetic testing has allowed for the identification of what may be de novo mutations driving disease in patients previously classified as IPAH patients.[17]
The overall incidence of PAH is difficult to determine with accuracy, as very few studies have actually reported incidence and prevalence data, and clinical under-recognition of the disease has been a challenge until recent years. One of the most recent studies to assess incidence was a large multicenter French study looking at 17 university hospitals across France during a 1-year period from October 2002 to October 2003.[18] In this study, 18% (121 of 674) of the cases were new diagnoses during the period of the study. The low end of the range of estimated incidence was 2.4 cases per million adult population per year. Estimated prevalence was 15 cases per million, with an estimated 5.9 cases of IPAH per million adults. Within the cohort, 39.2% were classified as IPAH, and 3.9% were classified as familial. In the initial report of their study, the investigators described a less severe clinical presentation for the familial PAH patients compared to IPAH. None of the familial patients presented with NYHA class IV heart failure, and as a group the familial patients had a better 6-minute walk distance than the IPAH patients. However, hemodynamics were similar in both groups. Subsequently, these same investigators compared BMPR2 mutation carriers (28 familial and 40 idiopathic PAH patients) to non-carriers (155 IPAH patients) and found that mutation carriers are diagnosed and die approximately 10 years earlier and with worse hemodynamics (e.g., higher mean pulmonary artery pressure, lower cardiac index, and lower mixed venous oxygen saturation) compared to non-carriers. Mutation carriers also have shorter times from diagnosis to death or lung transplantation, but the overall survival is similar between mutation carriers and non-carriers.[19] Following this, these investigators have examined the clinical presentation in HPAH caused by mutations in the activin A receptor Type II-like kinase-1 (ACVRL1 or ALK1), a receptor in the TGF-β receptor family. Mutations in ALK1 are associated with PAH and with hereditary hemorrhagic telangiectasia (HHT). Comparing a small group (32 patients) of ALK1 mutation carriers to both BMPR2 mutation carriers and IPAH non-carriers, the ALK1 mutation carriers presented at a younger age than even the BMPR2 mutation carriers. Despite better hemodynamics at the time of diagnosis and despite similar PAH therapies, the ALK1 mutation carriers had shorter survival times compared to BMPR2 carriers and to non-carriers.[20] Overall, then, the data from the French Registry and others suggest that while histopathologically identical, there may be subtle differences between HPAH and IPAH that impact the clinical presentation and progression of disease in the two groups.[21]
The most recent and largest observational cohort study of PAH to date has been compiled by the Actelion-sponsored Registry to Evaluate Early and Long-term PAH Disease Management (REVEAL) database. The registry was designed to enroll 3,000 prevalent and/or incident patients from 54 centers in the United States with WHO Group 1 PAH and to study their baseline characteristics as well as to examine their clinical progression and responses to therapy in a prospective way.[22] The baseline characteristics on the first 2,525 adult patients enrolled between March 2006 and September 2007 have been reported.[23] The proportions of PAH defined as IPAH and familial PAH were 46.2% and 2.7%, respectively, in line with what was described for the French Registry initially. Of note, subgroups of WHO Group 1 PAH were determined by the clinician enrolling the patient in the registry, based upon his/her assessment of what was the most likely etiology of the patient's PAH. Thus, genetic testing was not a necessary criterion, and the FPAH patients defined as such are not identical to the HPAH patients that have been described in the later analyses from the French Registry. Some of the salient differences between REVEAL and the French Registry as well as the 1987 NIH Registry have recently been summarized.[24] No studies from the REVEAL Registry comparing FPAH to IPAH have been forthcoming.
One of the most interesting findings to come out of the REVEAL Registry to date that may have a major impact on the future understanding of PAH disease and disease-modifying genetics has been the finding that 79.5% of the adult PAH patients in the registry are female. A female predominance has been noted in previous studies both in the U.S. and in the French Registry, with the female predominance being even more pronounced among blacks in these studies. The female-to-male ratio of 4.1:1 is much higher than that reported in the 1987 NIH registry (1.7:1)[4] or in the French Registry (1.9:1), but is in keeping with what was observed in the Surveillance of Pulmonary Hypertension in America Registry (4.3:1).[25] The prevalent hypothesis is that estrogen or its metabolites have an influence on development of disease and/or survival, and indeed recent studies have supported this notion (see below: Modifiers of BMPR2-mediated PAH, subsection Steroid hormones).
Heritable PAH and BMPR2
Dresdale first recognized a familial or heritable component to PAH in 1954, soon after the initial description in the literature of the disease itself.[12] In the 30 years that followed, a total of 13 families were described in the English literature with features of a heritable disease fitting the description of PAH. In 1984, these 13 families were reviewed and the pedigrees expanded by the PAH group at Vanderbilt, and a 14th family was added with 6 PAH deaths (all in women) already identified at that time.[26] We have continued to follow this family (Fig. 1), which now has 36 members diagnosed with HPAH (29 females and 7 males) and at least another 48 members who are unaffected but who are obligate carriers of what has been identified as a mutation in the ligand-binding domain of BMPR2.
Figure 1.
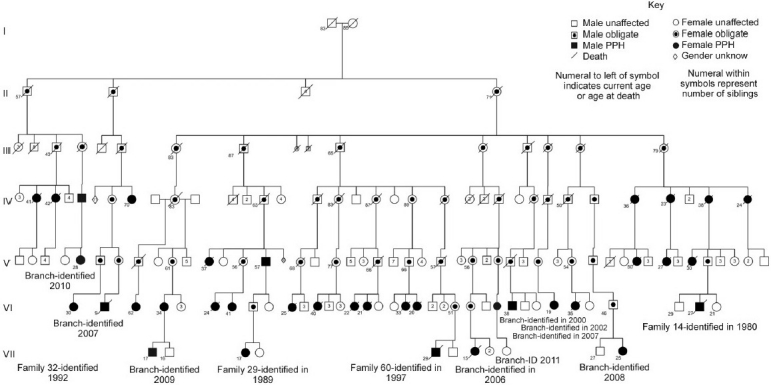
Updated pedigree of the 14th family reported in the literature with HPAH. Symbols are standard for pedigree analysis and are explained in the figure caption. This family carries a mutation in the ligand-binding domain of BMPR2.
Prior to the identification of BMPR2 as the major heritable risk factor for HPAH, there were a number of things known about the genetic behavior of HPAH, not all of which have been explained since the discovery of the culprit gene. The inheritance pattern in HPAH is best described as autosomal dominant with reduced penetrance.[26,27] The penetrance is highly variable, from 20%-80%. This strongly suggests the presence of modifying factors, genetic or environmental, that confer increased or decreased risk. Age of onset or diagnosis of the disease is highly variable, with HPAH being diagnosed any time from infancy out to 70 years of age. As mentioned above, there is a female predominance in PAH that is seen in all forms of WHO Group 1 PAH including HPAH. Interestingly, there have been several reports of HPAH occurring at earlier and earlier ages in subsequent generations, a phenomenon known as genetic anticipation.[28]
There are two major genetic mechanisms by which anticipation is known to occur. The first is trinucleotide repeat expansion, first described for Fragile X syndrome and now recognized to be causative or contributory in 40 or more neurologic diseases, most of which exhibit genetic anticipation.[29] The second mechanism is progressive telomere shortening, as has been described to explain the genetic anticipation observed with dyskeratosis congenita.[30] This is a disease characterized by mutations in telomerase reverse transcriptase, which is one of the enzymes responsible for maintaining telomere length in humans. Both of these mechanisms have been investigated in HPAH, and to date neither has proven to be the explanation for the apparent genetic anticipation in families with PAH.[31] There is a single report in the literature from Uziel et al. describing apparent genetic anticipation of neurologic disease associated with the mitochondrial mutation T8993G in 6 pedigrees. In this relatively small study, anticipation of symptoms correlated with increasing degree of heteroplasmy for the mitochondrial mutation.[32] This potential mechanism has not been investigated in HPAH, and to date no mutations in the mitochondrial genome itself have been associated with HPAH. An important alternative explanation to consider is statistical artifact or bias, particularly ascertainment bias. With progressive improvements in genetic and clinical screening in families with PAH, it could simply be that family members are diagnosed earlier or with a greater frequency at younger ages than they would have been in prior generations. Or, sufficient time has not passed to allow for additional “older age” diagnoses to occur in more recent generations. However, given the particularly poor survival in HPAH, the temporal distance between diagnosis and death is short enough that profound differences in age at diagnosis according to ascertainment bias are less likely. Regardless, until a biological mechanism can be demonstrated to explain the anticipation phenomenon observed in HPAH, the possibility of statistical artifact will remain.
With this background information, two teams working independently first began to localize the gene responsible for the majority of HPAH in the mid-1990s. Using linkage analysis referenced to short tandem repeats and to other microsatellite markers, both groups identified chromosome 2q31-33 as the region of interest and published their findings in 1997.[33,34] Again working independently, both groups subsequently found that the mutated gene responsible was BMPR2. Deng et al. used a modified candidate gene approach, examining 3 genes by denaturing high performance liquid chromatography (DHPLC) and identifying 5 nonsense and 2 missense mutations in BMPR2, which were not present in 196 control samples.[35] Lane et al. used positional cloning of patient genomic samples and a candidate gene approach to examine 17 possible genes in the region of 2q33 and identified 2 frame-shift, 2 nonsense, and 3 missense mutations in BMPR2.[36] Subsequent to these reports in 2000, there have been numerous studies that have identified nearly 300 different mutations in BMPR2 using methods as diverse as direct sequencing, melting curve analysis, DHPLC, Southern blotting, and multiplex ligation-dependent probe amplification.[17] Mutations in BMPR2 account for 75%-80% of the cases of HPAH, with a small percentage of cases attributed to known mutations in other TGF-β family members (e.g., ACVRL1/ALK1 or endoglin, ENG). The remaining cases of HPAH that are negative for known mutations may well have as yet unidentified alterations in genes in the TGF-β pathway, as exemplified by a case report describing a Smad 8 gene mutation in a patient with sporadic PAH.[37]
At the time of its identification as the gene responsible for the majority of cases of HPAH, BMPR2 possessed biological plausibility. It was already known that mutations in ACVRL1/ALK1 or in ENG were causative in HHT and were associated with pulmonary arterial hypertension.[38] ACVRL1/ALK1 is a Type I TGF-β receptor, and endoglin is its co-receptor. Moreover, altered TGF-β signaling had already been described in remodeling pulmonary arteries in PAH.[39] Finally, TGF-β signaling was known to influence cell proliferation and survival, tissue growth and repair, and inflammation,[40] and all of these processes seemed to be dis-regulated in PAH. Surprisingly, despite identification of BMPR2 and implication of the TGF-β pathway, and despite the subsequent development of powerful investigative tools, a clear mechanistic connection between altered BMPR2 and/or TGF-β signaling and PAH has remained elusive. More unfortunately, knowing the genetic underpinning of HPAH has not yet allowed the development of novel therapeutics that target the underlying molecular pathogenesis of the disease.
A number of important questions were immediately logical follow-ups to the identification of the gene responsible for HPAH. These were questions that were raised and addressed with very little knowledge of the function of the gene product or its molecular pathogenesis. The first of these was to ask whether BMPR2 mutations were responsible for sporadic PAH in addition to HPAH, perhaps even serving as a common cause of all forms of PAH. Very soon after the report of BMPR2 as the causative gene in familial PAH, Thomson et al. reported the detection of germline BMPR2 mutations in 11 out of a sample of 50 unrelated patients with IPAH (sporadic IPAH without family history or known genetic association to explain PAH).[41] These mutations encompassed the same spectrum of mutation types (frame shift, missense, and nonsense) as that described for familial PAH. Other groups have subsequently reported BMPR2 mutations in cases previously identified as sporadic PAH.[42,43] The prevalence of BMPR2 mutations among sporadic PAH patients is estimated to be between 11% and 40%, with the most recent report from the French Registry at 14.8% (49 of 332 patients) as of April 2009.[20] BMPR2 mutations have also been examined in other forms of PAH. In a series of 106 children and adults with congenital heart defects of various types, BMPR2 mutations were detected in 6 patients.[44] Most of these were atrioventricular canal or septation defects, and this was in keeping with prior studies in animal models showing that BMP signaling is important for normal cardiac septation and outflow tract formation in a BMPR2 hypomorph mouse model.[45] In small studies of less than 25 patients each, BMPR2 mutations were not found in patients with PAH associated with scleroderma or in HIV-infected patients with PAH.[46,47] In a larger series of 103 patients with chronic thromboembolic PAH, perhaps not surprisingly, no BMPR2 mutations were detected.[48] Taken together, the data suggest that BMPR2 mutations are responsible for the majority of HPAH and for a significant subset of sporadic PAH patients initially identified as IPAH.
Molecular mechanisms underlying BMPR2 mutations
The next step, following the identification of a culprit gene for a complex disease, would be to ask what types of mutations are present and how those mutations affect the gene product—e.g., haploinsufficiency, loss of function, gain of function, dominant negative, etc. From the initial investigations, frame shift, missense, and nonsense mutations were identified in the BMPR2 coding region.[35,36] On further investigation of intron/exon boundaries, splice site single nucleotide mutations have been identified.[49] In addition, larger disruptions of the BMPR2 gene, including both small and large rearrangements (exon or partial gene deletions or insertions and duplications) have been described.[50] It has subsequently been demonstrated that at least some of the nonsense mutations identified result in haploinsufficiency through the process of nonsense-mediated decay (NMD).[50,51] This is a feature of cellular quality control that exists between transcription and translation whereby nonsense mutations that result in significantly truncated transcripts are identified and degraded before they ever undergo translation.[52] In a study of 45 families with HPAH caused by BMPR2 mutations, 24 of the families were found to have a mutation that results in NMD. Other studies have estimated that the rate of NMD-causing BMPR2 mutations in HPAH may be as high as 70%. Other BMPR2 mutations have been shown to act in what seems to be a dominant negative fashion, and these mutations tend to have a more severe clinical phenotype. The major mechanism for dominant negative mutations is thought to be due to failed trafficking of the mutant receptor to the cell surface and the formation of nonfunctional intracellular heteromers composed of mutant and wild-type receptors, which effectively traps the wild-type receptor in the cytoplasm.[53] The BMPR2 gene is comprised of 13 exons that encode for 4 major functional domains of the receptor—the extracellular ligand binding domain (exons 2-3), the transmembrane domain (exon 4), the serine/threonine kinase domain (exons 5-11), and a long cytoplasmic tail (exons 12- 13) that is unique amongst the TGF-β receptor family members for its length. Mutations have been identified in all of these functional domains. Perhaps not surprisingly, the extracellular domain, kinase domain, and cytoplasmic tail domain are the sites of the vast majority of disease-causing BMPR2 mutations, representing 187 out of 210 distinct mutations. Of these 187 mutations, 105 were found to affect the kinase domain.[49]
Before engaging in a detailed biochemical investigation of how mutations affect BMPR2 signaling and how this leads to PAH, a final question to ask from the standpoint of HPAH genetics is whether BMPR2 mutation has any detectable impact on the clinical presentation or behavior of disease. As mentioned above, the French Registry examined BMPR2 mutation carriers and non-carriers and compared their clinical disease course. The mutation carriers were younger at diagnosis, had worse hemodynamic parameters at diagnosis, progressed faster to death or lung transplantation, and died at younger ages, but their overall survival was the same as for non-carriers. BMPR2 mutations have also been associated with decreased or absent vasoreactivity in PAH. Elliott et al. examined 67 unrelated patients with PAH (52 idiopathic, 15 familial) for BMPR2 mutations. Non-synonymous BMPR2 mutations were found in 16 of the idiopathic patients and 11 of the familial patients. Vasoreactivity at right heart catheterization was demonstrated for only 1 of the patients with non-synonymous BMPR2 variations, compared to 14 of the 40 patients who did not have non-synonymous BMPR2 changes (P=0.003).[54] Rosenzweig et al. looked at vasoreactivity in a larger cohort of 147 patients, comprised of 69 adults and 78 children, with 114 IPAH patients and 33 familial PAH patients. Of the 147 patients, 23 were positive for a BMPR2 mutation. The mutation carriers in this study were found to be much less likely to exhibit acute vasoreactivity at right heart catheterization (4% vs. 33% for non-carriers, P<0.003). Patients with BMPR2 mutations also had lower mixed venous oxygen saturations and lower cardiac index, similar to what was described in the French Registry.[55] Taken together, these findings suggest that BMPR2 mutation carriers are less likely to exhibit acute vasoreactivity and are more likely to have worse hemodynamics at right heart catheterization. This suggests more severe disease at the time of diagnosis. Thus far, however, this has not seemed to translate into excess mortality in the studies that have been reported to date.
Identification of BMPR2 as the gene responsible for HPAH has allowed for a number of subsequent informative inquiries into the nature of BMPR2 mutations and how these affect the clinical presentation of disease. It has also allowed for widespread targeted genetic testing for HPAH. However, the central point to investigate following the identification of the culprit gene(s) for any complex disease is the central point of molecular pathogenesis. By elucidating how a mutant gene mechanistically leads to disease, the hope is that novel and specific therapeutic targets can be identified and pharmacologically engaged with the eventual goal of developing more effective treatments, and perhaps even curative or preventative therapies.
TGF-β/BMP signaling
Much was known about the TGF-β superfamily at the time of the discovery of BMPR2 as the gene responsible for HPAH. The cytokine ligands in the TGF-β family are encoded by 42 open reading frames in humans. These ligands function as dimers and interact with heterotetrameric complexes consisting of 2 Type I receptors (7 subtypes identified) and 2 Type II receptors (5 subtypes identified, including BMPR2). The Type II receptor then phosphorylates the Type I receptor's kinase domain, activating it. The Type I receptor's kinase domain phosphorylates and activates 1 or more of the R-Smad proteins (Smads 1, 2, 3, 5, and 8). The phosphorylated R-Smads then interact with the Co-Smad, Smad 4. This complex then translocates to the nucleus to affect the transcription of target genes (Fig. 2). Regulation of TGF-β signaling is very complex, with negative regulators present at the level of ligand binding (e.g., ligand binding traps, decoy receptors), of intracellular receptor-binding proteins, and of Smad complexes (e.g., the I-Smads, which are Smads 6 and 7). TGF-β signaling has multiple roles in cellular differentiation during development, determination and maintenance of cell fate, apoptosis, cell proliferation, and regulation of inflammation.[17,40] In general, TGF-β signaling has a negative effect on cell growth in the post-development period, and loss of TGF-β signaling has been linked to tumorigenesis.[56]
Figure 2.
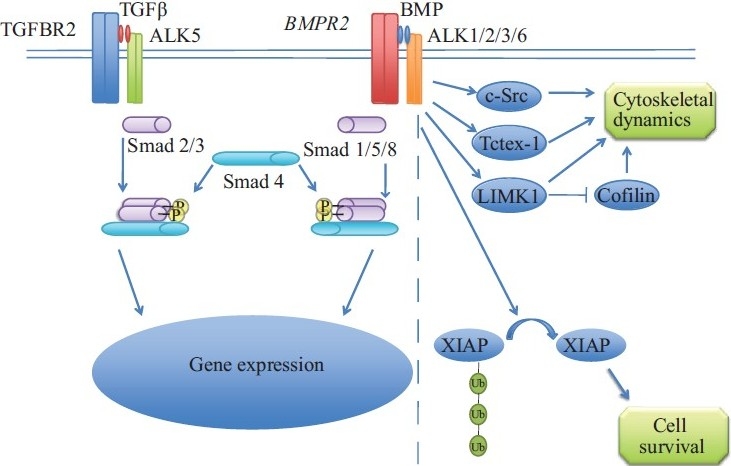
Simplified canonical TGF-β/BMP signaling and non-canonical signaling unique to BMPR2. The dashed line delineates canonical TGF-β signaling (on the left) from some of the pathways interacting with BMPR2via its long cytoplasmic tail.
Central to the pathology of PAH are the findings of muscularization of the smallest pulmonary arteries, occlusion of small pulmonary arteries by aggregations of cells of unclear lineage, and loss of the smallest branches of the pulmonary vascular tree (so-called “pruning”). Given the neointimal and smooth muscle proliferation and the vaso-occlusive plexiform lesions seen in histologic sections of PAH lungs, as well as findings of altered cellular proliferation and apoptosis in cells isolated from PAH patients, the TGF-β pathway seems optimally positioned to mediate these effects. BMPR2 is expressed in smooth muscle cells and endothelial cells in the human pulmonary vasculature, though its effects appear to be opposing in these two cell types.[57–59] In pulmonary artery endothelial cells, loss of BMP responsiveness confers increased sensitivity to apoptosis with abnormal repopulation/repair responses. In pulmonary artery smooth muscle cells, decreases in BMP signaling result in a loss of growth restriction and abnormal smooth muscle proliferation.[60] These phenotypes are likely mediated not so much by widespread and severe loss of BMP signaling but rather by more subtle changes in receptor multimer formation and makeup that shift the overall phenotype in the directions described.
BMPR2 participates in TGF-β signaling at the level of a Type II receptor. The most obvious hypothesis to link BMPR2 mutation and PAH would be through altered Smad signaling. While there has been some evidence linking defective Smad signaling to abnormalities in pulmonary vascular homeostasis that are similar to what is seen in PAH,[59,61–63] the molecular pathogenesis is more complicated than this. Mutations in the cytoplasmic tail domain of BMPR2 that have been identified in HPAH patients have been shown to have essentially intact Smad signaling when expressed in vitro.[64,65] The unusually long cytoplasmic tail of BMPR2 is thought to participate in Smad-independent signaling and has been shown to interact with a number of other proteins, including Tctex-1 (a dynein light chain), LIMK-1 (involved in regulation of actin cytoskeletal dynamics), XIAP (a negative regulator of apoptosis), p38 MAPK, c-Src, and RACK-1 (Fig. 2).[65–72]
TheBMPR2 transgenic mouse model of PAH
One of the best tools available for studying the molecular pathogenesis of any complex disease is a robust animal model. For laboratory research purposes, this often implies a rodent model, as rodents possess the necessary organ system complexity to more accurately reproduce human disease while still being easy to work with and maintain as well as being genetically tractable. This is worth mentioning in light of the fact that there are strains of fowl and of cattle that develop pulmonary hypertension.[73,74] Prior to the identification of BMPR2 in HPAH, several rodent models of PAH existed, including monocrotaline treatment, chronic hypoxia with or without treatment with the VEGF receptor antagonist Sugen 5416, extrarenal overexpression of mouse renin in the Ren-2 rat, and endothelial nitric oxide synthase (eNOS) knockout plus hypoxia. The major animal models of PAH in current experimental use have been recently reviewed elsewhere.[75] The fundamental problem with all of these models is that they required a condition, either an environmental exposure or a genetic condition, which was not present in patients with PAH. While this is a problem common to many animal models of complex diseases, and while subsequent studies have described altered BMPR2 expression and signaling in at least some of these models, the discovery of the culprit gene in HPAH allowed for the development of much more robust transgenic animal models. This, however, was not as straightforward as it initially appeared.
Because TGF/BMP signaling plays a large role in development, terminal differentiation, and determination of cell fate, animals with severe germline loss of BMPR2, such as BMPR2 hypomorphs or homozygous deletion of BMPR2, exhibit an embryonic lethal phenotype.[76] Homozygous deletion leads to growth arrest at the early gastrulation/egg cylinder stage, and BMPR2 hypomorphs have failure of septation of the cardiac outflow tract, as mentioned above.[45] BMPR2+/- heterozygotes show mildly elevated right ventricular systolic pressure (RVSP) and modest muscularization of small pulmonary arterioles in some studies, but not in others.[77,78] To attempt to achieve a more dramatic and consistent phenotype, a transgenic mouse was made that expresses a dominant negative BMPR2 allele only in smooth muscle, under the control of the Sm22 promoter. These animals showed more consistent elevation of RVSP and some muscularization of the small pulmonary arterioles, but many of the features of human PAH were not present.[79] Hong et al. subsequently demonstrated that conditional knockout of BMPR2 in pulmonary endothelial cells resulted in elevated RVSP with associated RV hypertrophy, muscularization of the distal pulmonary arteries, and perhaps some evidence of early “onion skin” lesions.[80] Though still not a true mimic of the human disease, these studies are important for demonstrating that neither the pulmonary endothelial cell nor the vascular smooth muscle cell is clearly the dominant cell type in PAH, as both seem to drive disease in these animal models.
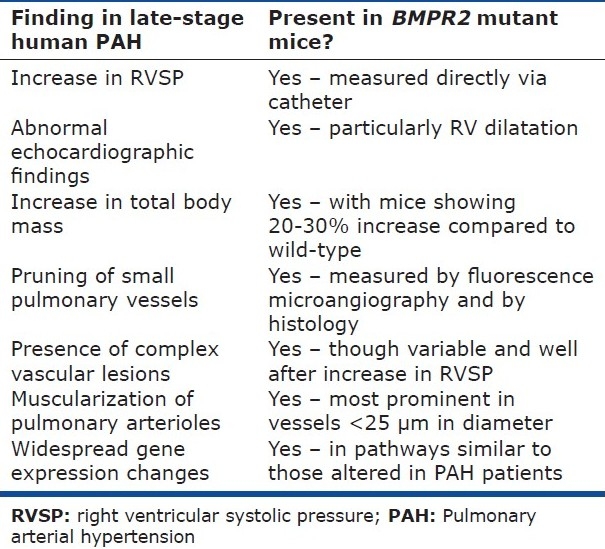
Further progress in the creation of a robust animal model of HPAH was realized when West et al. created a double transgenic mouse that expresses rtTA under the control of the Sm22 promoter and expresses a BMPR2 receptor with a dominant negative truncation mutation in the cytoplasmic tail (R899X) under the control of the TetO(7) promoter. These mice thus express the mutant BMPR2 only in smooth muscle cells and only when exposed to doxycycline. Not only does this circumvent the problems associated with a BMPR2 mutation expressed from conception, but these animals also exhibit elevated RVSP, muscularization of distal pulmonary arteries, and larger pulmonary vascular structural changes.[81] Importantly, the particular mutation expressed in these animals is the most common BMPR2 mutation in the cohort of HPAH patients followed at Vanderbilt University. A further refinement to the model has been the creation of a transgenic mouse that expresses the same BMPR2 mutation only in the presence of doxycycline, but expresses the mutant gene in every cell in the body.[82] This is actually a better representation of what occurs in the human disease, as BMPR2 and its mutations exhibit widespread tissue expression. Moreover, it avoids the inherent bias of restricting expression of the mutant gene to one particular cell type. Finally, these animals recapitulate nearly every aspect of the human disease (Table 1). In addition to elevated RVSP and muscularization of the distal pulmonary arteries, these animals show plexiform lesions, influx of perivascular inflammatory cells, increase in oxidative stress, and alterations in actin cytoskeletal dynamics. Finally, and perhaps most interestingly, these animals show reduced penetrance of the disease phenotype. While perhaps a frustrating aspect of the animal model in one sense, this suggests that this transgenic animal model is the most robust available for the study of the molecular pathogenesis of HPAH.
Table 1.
Comparison of changes observed in PAH patients and in mice expressing mutant BMPR2
Modifiers of BMPR2 -mediated PAH
Steroid hormones
As mentioned above, the reduced penetrance of BMPR2 mutations in HPAH strongly suggests the presence of modifiers that increase and/or decrease disease risk. It is likely that these modifiers represent both genetic and environmental factors, as evidenced by the fact that the most robust transgenic mouse models are incompletely penetrant and should have very similar or identical genetic backgrounds from one individual to the next. Nonetheless, the evidence is strongest for genetic modifiers. The earliest identified modifier, noted in all of the epidemiologic studies of PAH discussed above, is female gender, which increases the incidence of the development of disease up to 4-fold. This would seem to be due either to a detrimental effect of estrogen or its metabolites or to a protective effect of testosterone or its metabolites, or perhaps a combination of these. It is also possible that more complex changes at the chromosomal level, such as aberrant X-inactivation, may play a role, though there is only scant data to support this idea.[61] West et al. used expression arrays to examine EBV-immortalized B lymphocytes from HPAH patients with BMPR2 mutations, from family members who were mutation carriers but had not developed disease, and from non-carrier family members, in an attempt to discover modifier genes not previously identified. Overall, the study concluded that pathway analysis was more informative than single gene changes. However, changes in the expression level of CYP1B1, an estrogen metabolizing enzyme, were highly correlated with disease penetrance in female but not in male PAH patients in the study. CYP1B1 showed 10-fold lower expression levels in female patients compared to controls.[83] This enzyme metabolizes estrogens to 2-hydroxy and 4-hydroxy metabolites.[84] This reaction is in direct competition with other cytochrome P450 enzymes that metabolize estrogens to 16-alpha-hydroxy metabolites, which have been shown to possess significant mitogenic activity and to be protumorigenic.[85,86]
Austin et al. followed up the expression array findings by examining CYP1B1 polymorphisms in 140 BMPR2 mutation carriers. Of the 140 subjects, 92 had HPAH (62 of whom were female) and 48 were unaffected mutation carriers (24 of whom were female). Genotyping for a CYP1B1 polymorphism (N435S mutation, previously associated with breast, endometrial, and prostate cancers) was done, and from the female subjects, a nested case-control study examining the urinary metabolites of estrogen (2-OHE and 16α-OHE1 metabolites) in 5 female HPAH patients and 6 female unaffected mutation carriers. Among female mutation carriers, there was a 4-fold higher penetrance of disease for those homozygous for the N/N CYP1B1 allele compared to those who were heterozygous (N/S) or homozygous (S/S) for the polymorphism (P=0.005 for Chi-squared analysis). In the nested case-control portion of the study, the 2-OHE/16α-OHE1 ratio was 2.3-fold lower (0.65 vs. 1.48 ng/mg creatinine/mL, P=0.006) in the 5 female HPAH patients compared to the unaffected mutation carriers.[87] Taken together, these data strongly establish CYP1B1 as a potentially important modifier of BMPR2-mediated PAH, at least in female patients.
The influence of estrogen and estrogen metabolites is likely a bit more complex, as estrogen has a number of metabolic fates in vivo. For example, estradiol is converted to 2-OHE by CYP1A1/CYP1B1, which is converted to 2-methoxyestradiol (2-ME) by catechol-O-methyltransferase (COMT). 2-ME has been demonstrated in at least some animal models of PAH to have potent antimitogenic and overall beneficial therapeutic effects, and it is possible that the overall balance of estrogen and its metabolites is the biologically relevant variable.[88,89] However, it is likely that not all of the enzymes involved in estrogen synthesis and metabolism are equally important. The critical control point may still be CYP1B1, as there are no published reports linking PAH and COMT polymorphisms or COMT inhibitors.
Whether diagnosis of PAH and propensity to survive after diagnosis reflect distinct processes is unclear. Reports from the two large epidemiologic registries of PAH in France and in the US found reduced survival among males, although it was unclear if this was due to effects on the pulmonary vasculature, effects on the ability of the right ventricle to respond to stress, or both.[90,91] Furthermore, any survival disadvantage for males may be specific to men over the age of 60 years. This was the case for the REVEAL study in terms of men over age 60 compared with younger men and compared with women at any age. Detailed prospective studies with validation will be necessary to fully appreciate the degree to which gender participates in survival, if at all.
A logical extension of the sex hormone discussion is to androgens. To date, though studies have indicated a potential protective role for testosterone via its actions as a pulmonary vasodilator,[92,93] no investigations have revealed an association between polymorphisms in any of the androgen synthesis, signaling, or metabolic machinery and increased or decreased penetrance of HPAH in males or females. However, a study conducted by Roberts et al. examining genetic risk factors (independent of BMPR2 or the serotonin transporter, SERT) for the development of portopulmonary hypertension in males and females did find that 2 polymorphisms in the gene for aromatase, which is the rate-limiting step in the conversion of androgens to estradiol, conferred an increased risk for developing portopulmonary hypertension. The increased risk, correlated with increasing levels of estradiol in a dose-dependent fashion, controlled for gender.[94] One possible explanation for this finding is a change in the balance of estradiol and its metabolites, but another possibility is a decreased protective effect of androgens due to increased conversion to estradiol. This study did not measure androgens or estrogen metabolites, so the question remains an open one.
Variation in the TGFβ/BMP/BMPR2/Smad axis
Interestingly, one modifier of disease expression has been described involving the BMPR2 gene itself. It has been estimated that as many as 70% of the total mutations in BMPR2 would be predicted to result in truncated transcripts that would likely be subject to nonsense-mediated decay.[49] Indeed, NMD has clearly been demonstrated for some BMPR2 mutations, which is predicted to result in functional haploinsufficiency. However, even amongst subjects harboring NMD+ mutations, disease penetrance is not uniform, and only 20% of mutation carriers will go on to develop HPAH. Hamid et al. hypothesized that variations in the expression of the wild-type BMPR2 allele in these individuals may be an important disease-modifying factor by contributing to variable degrees of haploinsufficiency. To test this, they examined EBV-immortalized B lymphocytes from members of 4 families, each family with a different NMD+ BMPR2 mutation. From each family, immortalized lymphocytes from both HPAH patients and related unaffected mutation carriers were analyzed for expression levels of wild-type BMPR2. Compared to unaffected family members, HPAH patients had significantly lower expression levels of the wild-type BMPR2 allele (P<0.005), a finding that was independent of the specific NMD+ mutation in the other allele.[95]
Just as variations in the BMPR2 receptor cause or contribute to disease, variations in the gene for at least one of the ligands in the TGF-β/BMP pathway, TGFβ1, have been shown to influence disease penetrance. TGFβ1 SNPs were separated into least active, intermediate active, or most active groups and examined in 81 HPAH patients and 39 unaffected BMPR2 mutation carriers. In the context of NMD-resistant BMPR2 mutations, the relative activity of TGFβ1 mutations present influenced both age of disease onset and penetrance. More active TGFβ1 SNPs correlated with earlier age at disease onset; and the least, intermediate, and most active SNP groups showed penetrances of 33%, 72%, and 80%, respectively (P<0.003). There also appeared to be a dose effect, as those with 0-1, 2, or 3-4 active SNPs had penetrances of 33%, 72%, and 75%, respectively (P<0.005). Phosphorylated Smad-2 was shown to be increased on immunohistochemical analysis of lung sections from HPAH patients, though this was not clearly a difference in expression but more likely a difference in activity of the TGF-β pathway.[96] Interestingly, there appear to be only two reports in the literature of genetic alteration of any Smad gene in a patient with PAH. Both are case reports of alterations in the Smad-8 gene. One patient had a germline BMPR2 mutation and a somatic deletion on chromosome 13 that involved loss of the gene for Smad-8.[61] The other had a nonsense mutation reported in the Smad-8 gene as mentioned above.[37]
Other modifiers and gene arrays
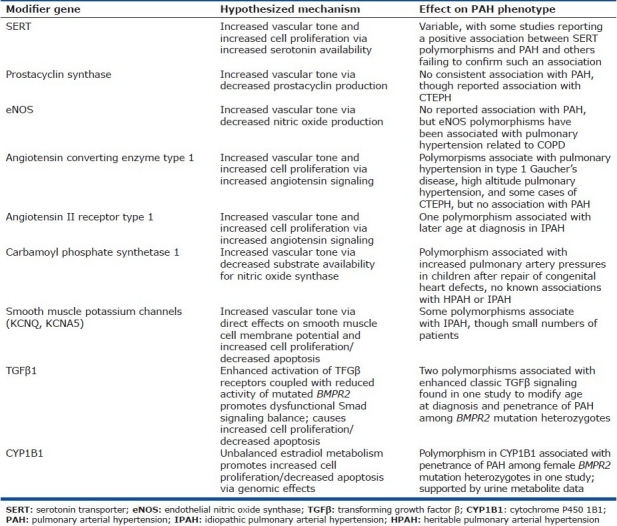
Alterations in a number of genes outside the BMPR2/TGF-β pathway have been associated with HPAH or IPAH. Most of these have been examined on the basis of perceived biological plausibility, as they primarily are in pathways that contribute to the regulation of vascular tone (including the renin/angiotensin pathway, the prostacyclin pathway, potassium channels, endothelial nitric oxide synthase, carbamyl phosphate synthetase 1 through its influence on nitric oxide production, and vasoactive intestinal peptide through vasodilator action that is at least partially dependent upon nitric oxide) and/or to cell proliferation (e.g., the serotonin transport pathway).[97–107] Several issues should be considered when interpreting the findings from these various studies. Many of these are relatively small studies, making it more difficult to detect weaker associations and increasing the likelihood of statistical artifact. For those candidate risk factors that have been examined in multiple studies, such as polymorphisms in the serotonin transporter (SERT or 5-HTT), the data are conflicting, with some studies reporting an association with SERT polymorphisms and PAH while others have not shown any relationship.[108–110] A number of these potential risk factors have been shown to associate with varieties of PAH in the pediatric population (such as ACE polymorphisms and persistent pulmonary hypertension of the newborn or CPS-1 polymorphisms and pulmonary hypertension following surgical correction of congenital cardiac defects) but not in the adult PAH population. Polymorphisms in several of these candidate genes have been shown to modify the expression of adult pulmonary hypertension in very specific circumstances, such as pulmonary hypertension associated with COPD, chronic thromboembolic pulmonary hypertension, exercise-induced pulmonary hypertension, and portopulmonary hypertension. The best available data suggest that the pathologies involved in these various settings are quite different from one another and from HPAH or IPAH, so the ability to generalize these findings is in question. For the most part, each of these has been examined as an independent risk factor or contributor, as opposed to being a modifier for the expression of disease mediated by other known heritable risk factors such as BMPR2 mutations. One larger study by Machado et al. did examine SERT polymorphisms in PAH patients, a subgroup of whom had known BMPR2 mutations, and was unable to detect any modifying influence of SERT polymorphisms on the expression of disease (e.g., severity, age of onset, clinical course, etc.).[109] These and other proposed modifier genes are summarized in (Table 2).
Table 2.
Proposed modifier genes, their hypothesized mechanisms of action, and the observed effect on human PAH phenotype
While the examination of polymorphisms in biologically plausible modifiers for HPAH and IPAH has proven to be only minimally informative, the increasing availability of gene array studies has allowed for the identification of potential modifier genes or pathways previously unsuspected. In particular, changes in pathways involved in actin cytoskeleton regulation, stress response, cellular metabolism, and inflammation have all been identified as potential contributors to IPAH and/or HPAH.[111] These studies have been performed in mouse models of disease, in human lung samples, and in circulating cells from human patients and controls. Mouse models have the disadvantage of not representing the actual human disease but only an approximation, and have the additional possibility of representing pathways that are only relevant in rodents. Studies of human patients are inherently biased because PAH has already affected the person to a degree severe enough to permit clinical diagnosis, and it can therefore be difficult to know what is contributory and what is response. The caveats and limitations germane to these studies, as well as some of the common important findings, have been recently reviewed.[112] Despite the known limitations, gene array studies have been and will continue to be very informative for ongoing inquiry into the pathogenesis of PAH.
Current and future therapies for PAH
The current therapies for PAH largely reflect the older notion of the disease as primarily a perturbation of pulmonary vasoreactivity. Current FDA approved therapies include prostacyclin analogs, endothelin-1 receptor antagonists, and phosphodiesterase Type V inhibitors (which potentiate nitric oxide's actions by inhibiting the intracellular breakdown of cyclic GMP). In addition, calcium channel antagonists can be highly effective pulmonary vasodilators in a small subset of patients who have a very robust acute pulmonary vasodilator response, but these drugs do not have FDA approval for this use. These therapies were developed prior to the confirmation of the importance of BMPR2 and the TGF-β pathway in PAH. Unfortunately, of the hundreds of interventional clinical trials currently registered with clinicaltrials.gov that have “pulmonary arterial hypertension” as a key search term, only a very small number are specifically designed to investigate possible interventions in adult PAH that do not directly impact upon the pathways targeted by existing therapies. There are registered trials of administration of endothelial progenitor cells; trials examining the tyrosine kinase inhibitors imatinib and nilotinib; a small phase IV trial examining the effect of pioglitazone in PAH patients with insulin resistance; a trial of ranolazine to improve angina from RV ischemia related to PAH; a trial of dichloroacetate sodium in IPAH, HPAH, or anorexigen-associated PAH; and a trial of administration of coenzyme-Q10, an important intermediate in the mitochondrial electron transport chain that also has intrinsic antioxidant properties. Other compounds have shown promise in pre-clinical studies, including fasudil (a Rho-kinase inhibitor), vasoactive intestinal peptide, and angiotensin converting enzyme Type 2.[113–117] However, none of these has been tested in large clinical trials in PAH patients. Clearly, there remains a significant gap between genetic investigations of PAH to date and an understanding of the pathophysiology of the disease that is detailed enough to permit identification and investigation of novel therapeutic targets in humans.
Genetic testing for PAH
Genetic testing for known mutations in BMPR2, ALK1, and ENG are currently available from several laboratories in North America and Europe. Unless there is a known family history of HHT or a strong clinical suspicion for the disease, clinical genetic testing specific to PAH should focus on testing for BMPR2 mutations. There is no current indication to incorporate testing for common genetic variants into the clinical testing approach. Likewise, there are no other pulmonary hypertension-specific genetic variants for other forms of the disease that support routine clinical genetic testing. However, testing for underlying causative diseases or syndromes is appropriate when clinically indicated.
Before undertaking clinical genetic testing for BMPR2 mutations, patients should receive professional genetic counseling from trained individuals prior to testing. This ensures that all involved understand the possible results of the testing and what these results might imply for both the patient and his or her family members. A brief discussion of the possible outcomes of genetic testing is in order. For the asymptomatic offspring of a BMPR2 mutation carrier, there is an overall pre-test probability of developing disease of approximately 1 in 10, or 10%, because there is an approximately 50% chance that the mutation was inherited, modified by the approximately 20% chance that the mutation would actually cause disease (because of reduced penetrance). If clinical genetic testing is undertaken in this asymptomatic person and found to be negative for BMPR2 mutations, the predicted risk of developing PAH drops to the level observed in the general population, which is a risk of less than 1 in 500,000 and which represents a 50,000-fold risk reduction. If this asymptomatic person is found to have inherited the mutant BMPR2 allele, the risk of developing PAH increases to approximately 20%, the known incidence of PAH in BMPR2 mutation positive individuals. This represents only a modest, approximately 2-fold increase in the risk of developing disease. This logic is depicted schematically in (Fig. 3). Thus, for these individuals, a negative genetic screen is extremely reassuring, and a positive test only modestly increases the risk of developing HPAH.
Figure 3.
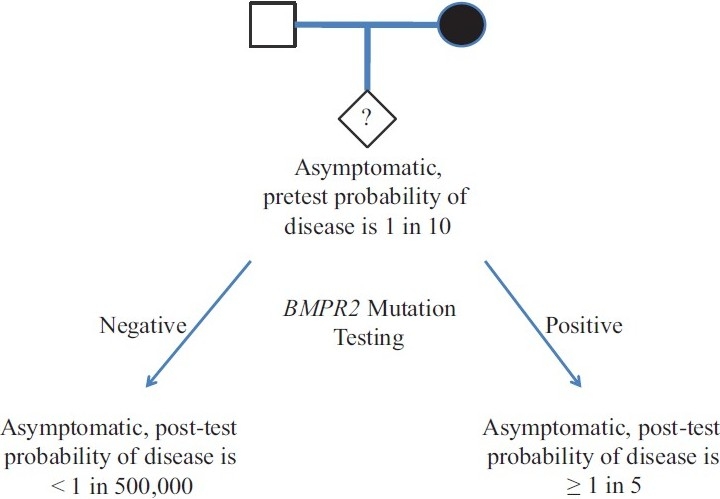
Diagram of pre- and post-test probabilities for developing HPAH in an unaffected family member, and the mathematical impact of testing for BMPR2mutation.
The question of whether to test a patient with IPAH for mutations in BMPR2 is worth consideration as well. Approximately 20% of incident IPAH is thought to be driven by mutations in BMPR2, suggesting that a significant portion of IPAH may in fact have a heritable component. Though detection of a BMPR2 mutation does not guarantee that the mutation is a germline mutation, detection of the mutant allele in cells/tissues distant from the site of disease (e.g., peripheral blood mononuclear cells or a buccal wash/swab) strongly suggests such. Detection of a BMPR2 mutation is often surprising, disappointing, and anxiety-provoking for a patient who previously thought that he or she had a “sporadic” disease, particularly if the individual has children. This is often the first time that increased risk in the patient's family members is perceived. There can be significant emotional stress both for the patient, who can experience what has been termed the “guilt of heritability,” as well as for other family members (if informed of the result), who can have significant difficulty with the inherent uncertainty of HPAH and its genetic underpinnings.[118] These issues thus highlight the importance of genetic counseling prior to testing.[119]
The decision of whether or not to test a subject for BMPR2 mutations or other single gene variants related to PAH is a complicated one, and particular caution should be used with children. Clinical genetic testing should only be considered at this time for children with diagnosed PAH, or for healthy children within a family affected by HPAH or IPAH. Several factors further complicate this issue in particular for children, including potentially profound psychological effects (e.g., seeing oneself as “sick” or “diseased” at a period of vulnerability during psychological development); concerns about future insurability and employer discrimination; and the uncertainty caused by the reduced penetrance and variable disease expressivity noted above, which can be especially difficult for both children and parents.
The question then arises of how to manage individuals who have tested positive for a mutation but who do not yet have any symptoms or evidence of clinical disease. To date there have been no studies specifically designed to investigate the best strategy for screening and early detection of clinically significant disease. The most current recommendation for these asymptomatic, mutation positive individuals is to have clinical and non-invasive echocardiographic screening every 3-5 years.[120] Those members of HPAH families who do undergo genetic testing and are found to be negative do not require future screening. The further question of the optimal time to start therapies that can be expensive, complicated, disruptive to normal routines, and associated with sometimes significant side effects, has not been adequately addressed.
With specific regard to PAH in families, given the vast number of potential mutations in the large BMPR2 gene, screening for mutations best starts with the patient, so that if present the specific mutation in the family can be identified.[17] The current cost in U.S. dollars of genetic testing ranges between $1,000 and $3,000 to screen the entire BMPR2 gene, with mutation-specific testing then costing approximately $300 to $500 once a mutation is known. Genetic testing should only be provided in concert with professional genetic counseling by experienced counselors for the reasons noted above.[119,120]
Summary and future directions
There are fundamental questions that stem from the current knowledge of PAH genetics that remain to be answered. It is not clear how or why a disease caused by a mutation in a gene expressed widely in many tissues has its primary manifestations only in the pulmonary vasculature (and perhaps the right ventricle). It is known that the pulmonary vasculature differs from systemic vascular beds in many ways, but whether any of these differences alone or in combination explains the phenotype of PAH is not known. There is mounting evidence that there are detectable abnormalities outside of the heart and lungs in PAH. It is possible that these are changes secondary to the known pathophysiology of PAH. However, it is also entirely possible that PAH is truly a systemic disease, with systemic manifestations that are fundamental to the development of elevated pulmonary vascular resistance and right heart failure, and that “pulmonary arterial hypertension” is a misnomer derived from the most easily observed and life-limiting manifestations of the disease. A systemic disease might actually make more intuitive sense given the expression of BMPR2 in many tissues outside the heart and lungs. As discussed above, the reduced penetrance of BMPR2 mutation continues to represent a large gap in our understanding of the pathophysiology of PAH. More broadly stated, the fundamental understanding of how a mutation in this gene leads to PAH is far from complete or what would even be described as robust. The canonical signaling pathways downstream from the receptor do not explain the development of PAH, and indeed, in many animal and human studies do not even seem to be directly related to the disease. Investigation of genetic modifiers has begun to yield insights into how penetrance is modified in HPAH, and thus how reduced penetrance in other “single gene” diseases might be investigated. Given the similarities between HPAH and IPAH in particular, a better understanding of the genetic and molecular pathogenesis of HPAH should further inform IPAH and all types of PAH, and thus make the study of HPAH critical to the community of PAH patients, families, and researchers.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Tuder RM, Abman SH, Braun T, Capron F, Stevens T, Thistlethwaite PA, et al. Development and pathology of pulmonary hypertension. J Am Coll Cardiol. 2009;54:S3–9. doi: 10.1016/j.jacc.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 2.Runo JR, Loyd JE. Primary pulmonary hypertension. Lancet. 2003;361:1533–44. doi: 10.1016/S0140-6736(03)13167-4. [DOI] [PubMed] [Google Scholar]
- 3.Simonneau G, Robbins IM, Beghetti M, Channick RN, Delcroix M, Denton CP, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2009;54:S43–54. doi: 10.1016/j.jacc.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 4.Rich S, Dantzker DR, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, et al. Primary pulmonary hypertension. A national prospective study. Ann Intern Med. 1987;107:216–23. doi: 10.7326/0003-4819-107-2-216. [DOI] [PubMed] [Google Scholar]
- 5.Blasko E, Haskell CA, Leung S, Gualtieri G, Halks-Miller M, Mahmoudi M, et al. Beneficial role of the GPR30 agonist G-1 in an animal model of multiple sclerosis. J Neuroimmunol. 2009;214:67–77. doi: 10.1016/j.jneuroim.2009.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation. 2010;122:156–63. doi: 10.1161/CIRCULATIONAHA.109.911818. [DOI] [PubMed] [Google Scholar]
- 7.Humbert M, Sitbon O, Yaici A, Montani D, O’Callaghan DS, Jais X, et al. Survival in incident and prevalent cohorts of patients with pulmonary arterial hypertension. Eur Respir J. 2010;36:549–55. doi: 10.1183/09031936.00057010. [DOI] [PubMed] [Google Scholar]
- 8.McLaughlin VV, Shillington A, Rich S. Survival in primary pulmonary hypertension: the impact of epoprostenol therapy. Circulation. 2002;106:1477–82. doi: 10.1161/01.cir.0000029100.82385.58. [DOI] [PubMed] [Google Scholar]
- 9.Zhang R, Dai LZ, Xie WP, Yu ZX, Wu BX, Pan L, et al. Survival of Chinese Patients with Pulmonary Arterial Hypertension in the Modern Management Era. Chest. 2011;140:301–9. doi: 10.1378/chest.10-2327. [DOI] [PubMed] [Google Scholar]
- 10.Rich S, Pogoriler J, Husain AN, Toth PT, Gomberg-Maitland M, Archer SL. Long-term effects of epoprostenol on the pulmonary vasculature in idiopathic pulmonary arterial hypertension. Chest. 2010;138:1234–9. doi: 10.1378/chest.09-2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dresdale DT, Schultz M, Michtom RJ. Primary pulmonary hypertension. I. Clinical and hemodynamic study. Am J Med. 1951;11:686–705. doi: 10.1016/0002-9343(51)90020-4. [DOI] [PubMed] [Google Scholar]
- 12.Dresdale DT, Michtom RJ, Schultz M. Recent studies in primary pulmonary hypertension, including pharmacodynamic observations on pulmonary vascular resistance. Bull N Y Acad Med. 1954;30:195–207. [PMC free article] [PubMed] [Google Scholar]
- 13.Deng Z, Haghighi F, Helleby L, Vanterpool K, Horn EM, Barst RJ, et al. Fine mapping of PPH1, a gene for familial primary pulmonary hypertension, to a 3-cM region on chromosome 2q33. Am J Respir Crit Care Med. 2000;161:1055–9. doi: 10.1164/ajrccm.161.3.9906051. [DOI] [PubMed] [Google Scholar]
- 14.Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA, 3rd, Loyd JE, et al. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. The International PPH Consortium. Nat Genet. 2000;26:81–4. doi: 10.1038/79226. [DOI] [PubMed] [Google Scholar]
- 15.Badesch DB, Champion HC, Sanchez MA, Hoeper MM, Loyd JE, Manes A, et al. Diagnosis and assessment of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54:S55–66. doi: 10.1016/j.jacc.2009.04.011. [DOI] [PubMed] [Google Scholar]
- 16.McLaughlin VV, Archer SL, Badesch DB, Barst RJ, Farber HW, Lindner JR, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association developed in collaboration with the American College of Chest Physicians; American Thoracic Society, Inc.; and the Pulmonary Hypertension Association. J Am Coll Cardiol. 2009;53:1573–619. doi: 10.1016/j.jacc.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 17.Machado RD, Eickelberg O, Elliott CG, Geraci MW, Hanaoka M, Loyd JE, et al. Genetics and genomics of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54:S32–42. doi: 10.1016/j.jacc.2009.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, et al. Pulmonary arterial hypertension in France: Results from a national registry. Am J Respir Crit Care Med. 2006;173:1023–30. doi: 10.1164/rccm.200510-1668OC. [DOI] [PubMed] [Google Scholar]
- 19.Sztrymf B, Coulet F, Girerd B, Yaici A, Jais X, Sitbon O, et al. Clinical outcomes of pulmonary arterial hypertension in carriers of BMPR2 mutation. Am J Respir Crit Care Med. 2008;177:1377–83. doi: 10.1164/rccm.200712-1807OC. [DOI] [PubMed] [Google Scholar]
- 20.Girerd B, Montani D, Coulet F, Sztrymf B, Yaici A, Jais X, et al. Clinical outcomes of pulmonary arterial hypertension in patients carrying an ACVRL1 (ALK1) mutation. Am J Respir Crit Care Med. 2010;181:851–61. doi: 10.1164/rccm.200908-1284OC. [DOI] [PubMed] [Google Scholar]
- 21.Sztrymf B, Yaici A, Jais X, Sitbon O, Simonneau G, Humbert M. Idiopathic pulmonary hypertension: What did we learn from genes? Sarcoidosis Vasc Diffuse Lung Dis. 2005;22(Suppl 1):S91–100. [PubMed] [Google Scholar]
- 22.McGoon MD, Krichman A, Farber HW, Barst RJ, Raskob GE, Liou TG, et al. Design of the REVEAL registry for US patients with pulmonary arterial hypertension. Mayo Clin Proc. 2008;83:923–31. doi: 10.4065/83.8.923. [DOI] [PubMed] [Google Scholar]
- 23.Badesch DB, Raskob GE, Elliott CG, Krichman AM, Farber HW, Frost AE, et al. Pulmonary arterial hypertension: Baseline characteristics from the REVEAL Registry. Chest. 2010;137:376–87. doi: 10.1378/chest.09-1140. [DOI] [PubMed] [Google Scholar]
- 24.Frost AE, Badesch DB, Barst RJ, Benza RL, Elliott CG, Farber HW, et al. The changing picture of patients with pulmonary arterial hypertension in the United States: How REVEAL differs from historic and non-US Contemporary Registries. Chest. 2011;139:128–37. doi: 10.1378/chest.10-0075. [DOI] [PubMed] [Google Scholar]
- 25.Walker AM, Langleben D, Korelitz JJ, Rich S, Rubin LJ, Strom BL, et al. Temporal trends and drug exposures in pulmonary hypertension: an American experience. Am Heart J. 2006;152:521–6. doi: 10.1016/j.ahj.2006.02.020. [DOI] [PubMed] [Google Scholar]
- 26.Loyd JE, Primm RK, Newman JH. Familial primary pulmonary hypertension: Clinical patterns. Am Rev Respir Dis. 1984;129:194–7. doi: 10.1164/arrd.1984.129.1.194. [DOI] [PubMed] [Google Scholar]
- 27.Loyd JE, Slovis B, Phillips JA, 3rd, Butler MG, Foroud TM, Conneally PM, et al. The presence of genetic anticipation suggests that the molecular basis of familial primary pulmonary hypertension may be trinucleotide repeat expansion. Chest. 1997;111:82S–3S. doi: 10.1378/chest.111.6_supplement.82s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Loyd JE, Butler MG, Foroud TM, Conneally PM, Phillips JA, 3rd, Newman JH. Genetic anticipation and abnormal gender ratio at birth in familial primary pulmonary hypertension. Am J Respir Crit Care Med. 1995;152:93–7. doi: 10.1164/ajrccm.152.1.7599869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pearson CE, Nichol Edamura K, Cleary JD. Repeat instability: Mechanisms of dynamic mutations. Nat Rev Genet. 2005;6:729–42. doi: 10.1038/nrg1689. [DOI] [PubMed] [Google Scholar]
- 30.Armanios M, Chen JL, Chang YP, Brodsky RA, Hawkins A, Griffin CA, et al. Haploinsufficiency of telomerase reverse transcriptase leads to anticipation in autosomal dominant dyskeratosis congenita. Proc Natl Acad Sci U S A. 2005;102:15960–4. doi: 10.1073/pnas.0508124102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Austin ED, Loyd JE, Phillips JA., 3rd Genetics of pulmonary arterial hypertension. Semin Respir Crit Care Med. 2009;30:386–98. doi: 10.1055/s-0029-1233308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Uziel G, Moroni I, Lamantea E, Fratta GM, Ciceri E, Carrara F, et al. Mitochondrial disease associated with the T8993G mutation of the mitochondrial ATPase 6 gene: A clinical, biochemical, and molecular study in six families. J Neurol Neurosurg Psychiatry. 1997;63:16–22. doi: 10.1136/jnnp.63.1.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nichols WC, Koller DL, Slovis B, Foroud T, Terry VH, Arnold ND, et al. Localization of the gene for familial primary pulmonary hypertension to chromosome 2q31-32. Nat Genet. 1997;15:277–80. doi: 10.1038/ng0397-277. [DOI] [PubMed] [Google Scholar]
- 34.Morse JH, Jones AC, Barst RJ, Hodge SE, Wilhelmsen KC, Nygaard TG. Mapping of familial primary pulmonary hypertension locus (PPH1) to chromosome 2q31-q32. Circulation. 1997;95:2603–6. doi: 10.1161/01.cir.95.12.2603. [DOI] [PubMed] [Google Scholar]
- 35.Deng Z, Morse JH, Slager SL, Cuervo N, Moore KJ, Venetos G, et al. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet. 2000;67:737–44. doi: 10.1086/303059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA, 3rd, Loyd JE, et al. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat Genet. 2000;26:81–4. doi: 10.1038/79226. [DOI] [PubMed] [Google Scholar]
- 37.Shintani M, Yagi H, Nakayama T, Saji T, Matsuoka R. A new nonsense mutation of SMAD8 associated with pulmonary arterial hypertension. J Med Genet. 2009;46:331–7. doi: 10.1136/jmg.2008.062703. [DOI] [PubMed] [Google Scholar]
- 38.Marchuk DA. Genetic abnormalities in hereditary hemorrhagic telangiectasia. Curr Opin Hematol. 1998;5:332–8. doi: 10.1097/00062752-199809000-00005. [DOI] [PubMed] [Google Scholar]
- 39.Botney MD, Bahadori L, Gold LI. Vascular remodeling in primary pulmonary hypertension.Potential role for transforming growth factor-beta. Am J Pathol. 1994;144:286–95. [PMC free article] [PubMed] [Google Scholar]
- 40.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 41.Thomson J, Machado R, Pauciulo M, Morgan N, Yacoub M, Corris P, et al. Familial and sporadic primary pulmonary hypertension is caused by BMPR2 gene mutations resulting in haploinsufficiency of the bone morphogenetic protein tuype II receptor. J Heart Lung Transplant. 2001;20:149. doi: 10.1016/s1053-2498(01)00259-5. [DOI] [PubMed] [Google Scholar]
- 42.Aldred MA, Vijayakrishnan J, James V, Soubrier F, Gomez-Sanchez MA, Martensson G, et al. BMPR2 gene rearrangements account for a significant proportion of mutations in familial and idiopathic pulmonary arterial hypertension. Hum Mutat. 2006;27:212–3. doi: 10.1002/humu.9398. [DOI] [PubMed] [Google Scholar]
- 43.Eickelberg O, Morty RE. Transforming growth factor beta/bone morphogenic protein signaling in pulmonary arterial hypertension: Remodeling revisited. Trends Cardiovasc Med. 2007;17:263–9. doi: 10.1016/j.tcm.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 44.Roberts KE, McElroy JJ, Wong WP, Yen E, Widlitz A, Barst RJ, et al. BMPR2 mutations in pulmonary arterial hypertension with congenital heart disease. Eur Respir J. 2004;24:371–4. doi: 10.1183/09031936.04.00018604. [DOI] [PubMed] [Google Scholar]
- 45.Delot EC, Bahamonde ME, Zhao M, Lyons KM. BMP signaling is required for septation of the outflow tract of the mammalian heart. Development. 2003;130:209–20. doi: 10.1242/dev.00181. [DOI] [PubMed] [Google Scholar]
- 46.Morse J, Barst R, Horn E, Cuervo N, Deng Z, Knowles J. Pulmonary hypertension in scleroderma spectrum of disease: Lack of bone morphogenetic protein receptor 2 mutations. J Rheumatol. 2002;29:2379–81. [PubMed] [Google Scholar]
- 47.Nunes H, Humbert M, Sitbon O, Morse JH, Deng Z, Knowles JA, et al. Prognostic factors for survival in human immunodeficiency virus-associated pulmonary arterial hypertension. Am J Respir Crit Care Med. 2003;167:1433–9. doi: 10.1164/rccm.200204-330OC. [DOI] [PubMed] [Google Scholar]
- 48.Suntharalingam J, Machado RD, Sharples LD, Toshner MR, Sheares KK, Hughes RJ, et al. Demographic features, BMPR2 status and outcomes in distal chronic thromboembolic pulmonary hypertension. Thorax. 2007;62:617–22. doi: 10.1136/thx.2006.070680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Machado RD, Aldred MA, James V, Harrison RE, Patel B, Schwalbe EC, et al. Mutations of the TGF-beta type II receptor BMPR2 in pulmonary arterial hypertension. Hum Mutat. 2006;27:121–32. doi: 10.1002/humu.20285. [DOI] [PubMed] [Google Scholar]
- 50.Cogan JD, Pauciulo MW, Batchman AP, Prince MA, Robbins IM, Hedges LK, et al. High frequency of BMPR2 exonic deletions/duplications in familial pulmonary arterial hypertension. Am J Respir Crit Care Med. 2006;174:590–8. doi: 10.1164/rccm.200602-165OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cogan JD, Vnencak-Jones CL, Phillips JA, 3rd, Lane KB, Wheeler LA, Robbins IM, et al. Gross BMPR2 gene rearrangements constitute a new cause for primary pulmonary hypertension. Genet Med. 2005;7:169–74. doi: 10.1097/01.gim.0000156525.09595.e9. [DOI] [PubMed] [Google Scholar]
- 52.Khajavi M, Inoue K, Lupski JR. Nonsense-mediated mRNA decay modulates clinical outcome of genetic disease. Eur J Hum Genet. 2006;14:1074–81. doi: 10.1038/sj.ejhg.5201649. [DOI] [PubMed] [Google Scholar]
- 53.Li W, Dunmore BJ, Morrell NW. Bone morphogenetic protein type II receptor mutations causing protein misfolding in heritable pulmonary arterial hypertension. Proc Am Thorac Soc. 2010;7:395–8. doi: 10.1513/pats.201002-024AW. [DOI] [PubMed] [Google Scholar]
- 54.Elliott CG, Glissmeyer EW, Havlena GT, Carlquist J, McKinney JT, Rich S, et al. Relationship of BMPR2 mutations to vasoreactivity in pulmonary arterial hypertension. Circulation. 2006;113:2509–15. doi: 10.1161/CIRCULATIONAHA.105.601930. [DOI] [PubMed] [Google Scholar]
- 55.Rosenzweig EB, Morse JH, Knowles JA, Chada KK, Khan AM, Roberts KE, et al. Clinical implications of determining BMPR2 mutation status in a large cohort of children and adults with pulmonary arterial hypertension. J Heart Lung Transplant. 2008;27:668–74. doi: 10.1016/j.healun.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 56.Bierie B, Moses HL. Tumour microenvironment: TGFbeta: The molecular Jekyll and Hyde of cancer. Nat Rev Cancer. 2006;6:506–20. doi: 10.1038/nrc1926. [DOI] [PubMed] [Google Scholar]
- 57.Atkinson C, Stewart S, Upton PD, Machado R, Thomson JR, Trembath RC, et al. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation. 2002;105:1672–8. doi: 10.1161/01.cir.0000012754.72951.3d. [DOI] [PubMed] [Google Scholar]
- 58.Richter A, Yeager ME, Zaiman A, Cool CD, Voelkel NF, Tuder RM. Impaired transforming growth factor-beta signaling in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2004;170:1340–8. doi: 10.1164/rccm.200311-1602OC. [DOI] [PubMed] [Google Scholar]
- 59.Frank DB, Abtahi A, Yamaguchi DJ, Manning S, Shyr Y, Pozzi A, et al. Bone morphogenetic protein 4 promotes pulmonary vascular remodeling in hypoxic pulmonary hypertension. Circ Res. 2005;97:496–504. doi: 10.1161/01.RES.0000181152.65534.07. [DOI] [PubMed] [Google Scholar]
- 60.Jurasz P, Courtman D, Babaie S, Stewart DJ. Role of apoptosis in pulmonary hypertension: from experimental models to clinical trials. Pharmacol Ther. 2010;126:1–8. doi: 10.1016/j.pharmthera.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 61.Aldred MA, Comhair SA, Varella-Garcia M, Asosingh K, Xu W, Noon GP, et al. Somatic chromosome abnormalities in the lungs of patients with pulmonary arterial hypertension. Am J Respir Crit Care Med. 2010;182:1153–60. doi: 10.1164/rccm.201003-0491OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gallione CJ, Richards JA, Letteboer TG, Rushlow D, Prigoda NL, Leedom TP, et al. SMAD4 mutations found in unselected HHT patients. J Med Genet. 2006;43:793–7. doi: 10.1136/jmg.2006.041517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huang Z, Wang D, Ihida-Stansbury K, Jones PL, Martin JF. Defective pulmonary vascular remodeling in Smad8 mutant mice. Hum Mol Genet. 2009;18:2791–801. doi: 10.1093/hmg/ddp214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nishihara A, Watabe T, Imamura T, Miyazono K. Functional heterogeneity of bone morphogenetic protein receptor-II mutants found in patients with primary pulmonary hypertension. Mol Biol Cell. 2002;13:3055–63. doi: 10.1091/mbc.E02-02-0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rudarakanchana N, Flanagan JA, Chen H, Upton PD, Machado R, Patel D, et al. Functional analysis of bone morphogenetic protein type II receptor mutations underlying primary pulmonary hypertension. Hum Mol Genet. 2002;11:1517–25. doi: 10.1093/hmg/11.13.1517. [DOI] [PubMed] [Google Scholar]
- 66.Foletta VC, Lim MA, Soosairajah J, Kelly AP, Stanley EG, Shannon M, He W, et al. Direct signaling by the BMP type II receptor via the cytoskeletal regulator LIMK1. J Cell Biol. 2003;162:1089–98. doi: 10.1083/jcb.200212060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Machado RD, Rudarakanchana N, Atkinson C, Flanagan JA, Harrison R, Morrell NW, et al. Functional interaction between BMPR-II and Tctex-1, a light chain of Dynein, is isoform-specific and disrupted by mutations underlying primary pulmonary hypertension. Hum Mol Genet. 2003;12:3277–86. doi: 10.1093/hmg/ddg365. [DOI] [PubMed] [Google Scholar]
- 68.Zakrzewicz A, Hecker M, Marsh LM, Kwapiszewska G, Nejman B, Long L, et al. Receptor for activated C-kinase 1, a novel interaction partner of type II bone morphogenetic protein receptor, regulates smooth muscle cell proliferation in pulmonary arterial hypertension. Circulation. 2007;115:2957–68. doi: 10.1161/CIRCULATIONAHA.106.670026. [DOI] [PubMed] [Google Scholar]
- 69.Adachi-Yamada T, Nakamura M, Irie K, Tomoyasu Y, Sano Y, Mori E, et al. p38 mitogen-activated protein kinase can be involved in transforming growth factor beta superfamily signal transduction in Drosophila wing morphogenesis. Mol Cell Biol. 1999;19:2322–9. doi: 10.1128/mcb.19.3.2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Morty RE, Nejman B, Kwapiszewska G, Hecker M, Zakrzewicz A, Kouri FM, et al. Dysregulated bone morphogenetic protein signaling in monocrotaline-induced pulmonary arterial hypertension. Arterioscler Thromb Vasc Biol. 2007;27:1072–8. doi: 10.1161/ATVBAHA.107.141200. [DOI] [PubMed] [Google Scholar]
- 71.Liu Z, Shen J, Pu K, Katus HA, Ploger F, Tiefenbacher CP, et al. GDF5 and BMP2 inhibit apoptosis via activation of BMPR2 and subsequent stabilization of XIAP. Biochim Biophys Acta. 2009;1793:1819–27. doi: 10.1016/j.bbamcr.2009.09.012. [DOI] [PubMed] [Google Scholar]
- 72.Wong WK, Knowles JA, Morse JH. Bone morphogenetic protein receptor type II C-terminus interacts with c-Src: Implication for a role in pulmonary arterial hypertension. Am J Respir Cell Mol Biol. 2005;33:438–46. doi: 10.1165/rcmb.2005-0103OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wideman RF, Jr, Eanes ML, Hamal KR, Anthony NB. Pulmonary vascular pressure profiles in broilers selected for susceptibility to pulmonary hypertension syndrome: Age and sex comparisons. Poult Sci. 2010;89:1815–24. doi: 10.3382/ps.2010-00754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rhodes J. Comparative physiology of hypoxic pulmonary hypertension: historical clues from brisket disease. J Appl Physiol. 2005;98:1092–100. doi: 10.1152/japplphysiol.01017.2004. [DOI] [PubMed] [Google Scholar]
- 75.Stenmark KR, Meyrick B, Galie N, Mooi WJ, McMurtry IF. Animal models of pulmonary arterial hypertension: the hope for etiological discovery and pharmacological cure. Am J Physiol Lung Cell Mol Physiol. 2009;297:L1013–32. doi: 10.1152/ajplung.00217.2009. [DOI] [PubMed] [Google Scholar]
- 76.Beppu H, Kawabata M, Hamamoto T, Chytil A, Minowa O, Noda T, et al. BMP type II receptor is required for gastrulation and early development of mouse embryos. Dev Biol. 2000;221:249–58. doi: 10.1006/dbio.2000.9670. [DOI] [PubMed] [Google Scholar]
- 77.Beppu H, Ichinose F, Kawai N, Jones RC, Yu PB, Zapol WM, et al. BMPR-II heterozygous mice have mild pulmonary hypertension and an impaired pulmonary vascular remodeling response to prolonged hypoxia. Am J Physiol Lung Cell Mol Physiol. 2004;287:L1241–7. doi: 10.1152/ajplung.00239.2004. [DOI] [PubMed] [Google Scholar]
- 78.Long L, MacLean MR, Jeffery TK, Morecroft I, Yang X, Rudarakanchana N, et al. Serotonin increases susceptibility to pulmonary hypertension in BMPR2-deficient mice. Circ Res. 2006;98:818–27. doi: 10.1161/01.RES.0000215809.47923.fd. [DOI] [PubMed] [Google Scholar]
- 79.West J, Fagan K, Steudel W, Fouty B, Lane K, Harral J, et al. Pulmonary hypertension in transgenic mice expressing a dominant-negative BMPRII gene in smooth muscle. Circ Res. 2004;94:1109–14. doi: 10.1161/01.RES.0000126047.82846.20. [DOI] [PubMed] [Google Scholar]
- 80.Hong KH, Lee YJ, Lee E, Park SO, Han C, Beppu H, et al. Genetic ablation of the BMPR2 gene in pulmonary endothelium is sufficient to predispose to pulmonary arterial hypertension. Circulation. 2008;118:722–30. doi: 10.1161/CIRCULATIONAHA.107.736801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.West J, Harral J, Lane K, Deng Y, Ickes B, Crona D, et al. Mice expressing BMPR2R899X transgene in smooth muscle develop pulmonary vascular lesions. Am J Physiol Lung Cell Mol Physiol. 2008;295:L744–55. doi: 10.1152/ajplung.90255.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Talati M, West J, Blackwell TR, Loyd JE, Meyrick B. BMPR2 mutation alters the lung macrophage endothelin-1 cascade in a mouse model and patients with heritable pulmonary artery hypertension. Am J Physiol Lung Cell Mol Physiol. 2010;299:L363–73. doi: 10.1152/ajplung.00295.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.West J, Cogan J, Geraci M, Robinson L, Newman J, Phillips JA, et al. Gene expression in BMPR2 mutation carriers with and without evidence of Pulmonary Arterial Hypertension suggests pathways relevant to disease penetrance. BMC Med Genomics. 2008;1:45. doi: 10.1186/1755-8794-1-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tsuchiya Y, Nakajima M, Yokoi T. Cytochrome P450-mediated metabolism of estrogens and its regulation in human. Cancer Lett. 2005;227:115–24. doi: 10.1016/j.canlet.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 85.Belous AR, Hachey DL, Dawling S, Roodi N, Parl FF. Cytochrome P450 1B1-mediated estrogen metabolism results in estrogen-deoxyribonucleoside adduct formation. Cancer Res. 2007;67:812–7. doi: 10.1158/0008-5472.CAN-06-2133. [DOI] [PubMed] [Google Scholar]
- 86.Missmer SA, Eliassen AH, Barbieri RL, Hankinson SE. Endogenous estrogen, androgen, and progesterone concentrations and breast cancer risk among postmenopausal women. J Natl Cancer Inst. 2004;96:1856–65. doi: 10.1093/jnci/djh336. [DOI] [PubMed] [Google Scholar]
- 87.Austin ED, Cogan JD, West JD, Hedges LK, Hamid R, Dawson EP, et al. Alterations in oestrogen metabolism: implications for higher penetrance of familial pulmonary arterial hypertension in females. Eur Respir J. 2009;34:1093–9. doi: 10.1183/09031936.00010409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tofovic SP. Estrogens and development of pulmonary hypertension: Interaction of estradiol metabolism and pulmonary vascular disease. J Cardiovasc Pharmacol. 2010;56:696–708. doi: 10.1097/FJC.0b013e3181f9ea8d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tofovic SP, Jones TJ, Bilan VP, Jackson EK, Petrusevska G. Synergistic therapeutic effects of 2-methoxyestradiol with either sildenafil or bosentan on amelioration of monocrotaline-induced pulmonary hypertension and vascular remodeling. J Cardiovasc Pharmacol. 2010;56:475–83. doi: 10.1097/FJC.0b013e3181f215e7. [DOI] [PubMed] [Google Scholar]
- 90.Benza RL, Miller DP, Gomberg-Maitland M, Frantz RP, Foreman AJ, Coffey CS, et al. Predicting survival in pulmonary arterial hypertension: insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL) Circulation. 2010;122:164–72. doi: 10.1161/CIRCULATIONAHA.109.898122. [DOI] [PubMed] [Google Scholar]
- 91.Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation. 2010;122:156–63. doi: 10.1161/CIRCULATIONAHA.109.911818. [DOI] [PubMed] [Google Scholar]
- 92.Smith AM, Jones RD, Channer KS. The influence of sex hormones on pulmonary vascular reactivity: Possible vasodilator therapies for the treatment of pulmonary hypertension. Curr Vasc Pharmacol. 2006;4:9–15. doi: 10.2174/157016106775203090. [DOI] [PubMed] [Google Scholar]
- 93.Smith AM, Bennett RT, Jones TH, Cowen ME, Channer KS, Jones RD. Characterization of the vasodilatory action of testosterone in the human pulmonary circulation. Vasc Health Risk Manag. 2008;4:1459–66. doi: 10.2147/vhrm.s3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Roberts KE, Fallon MB, Krowka MJ, Brown RS, Trotter JF, Peter I, et al. Genetic risk factors for portopulmonary hypertension in patients with advanced liver disease. Am J Respir Crit Care Med. 2009;179:835–42. doi: 10.1164/rccm.200809-1472OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hamid R, Cogan JD, Hedges LK, Austin E, Phillips JA, 3rd, Newman JH, et al. Penetrance of pulmonary arterial hypertension is modulated by the expression of normal BMPR2 allele. Hum Mutat. 2009;30:649–54. doi: 10.1002/humu.20922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Phillips JA, 3rd, Poling JS, Phillips CA, Stanton KC, Austin ED, Cogan JD, et al. Synergistic heterozygosity for TGFbeta1 SNPs and BMPR2 mutations modulates the age at diagnosis and penetrance of familial pulmonary arterial hypertension. Genet Med. 2008;10:359–65. doi: 10.1097/GIM.0b013e318172dcdf. [DOI] [PubMed] [Google Scholar]
- 97.Abraham WT, Raynolds MV, Badesch DB, Wynne KM, Groves BM, Roden RL, et al. Angiotensin-converting enzyme DD genotype in patients with primary pulmonary hypertension: Increased frequency and association with preserved haemodynamics. J Renin Angiotensin Aldosterone Syst. 2003;4:27–30. doi: 10.3317/jraas.2003.003. [DOI] [PubMed] [Google Scholar]
- 98.Chung WK, Deng L, Carroll JS, Mallory N, Diamond B, Rosenzweig EB, et al. Polymorphism in the angiotensin II type 1 receptor (AGTR1) is associated with age at diagnosis in pulmonary arterial hypertension. J Heart Lung Transplant. 2009;28:373–9. doi: 10.1016/j.healun.2009.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tanabe N, Amano S, Tatsumi K, Kominami S, Igarashi N, Shimura R, et al. Angiotensin-converting enzyme gene polymorphisms and prognosis in chronic thromboembolic pulmonary hypertension. Circ J. 2006;70:1174–9. doi: 10.1253/circj.70.1174. [DOI] [PubMed] [Google Scholar]
- 100.Nana-Sinkam P, Oyer RJ, Stearman RS, Sotto-Santiago S, Moore MD, Bull TM, et al. Prostacyclin synthase promoter regulation and familial pulmonary arterial hypertension. Chest. 2005;128:612S. doi: 10.1378/chest.128.6_suppl.612S. [DOI] [PubMed] [Google Scholar]
- 101.Tuder RM, Cool CD, Geraci MW, Wang J, Abman SH, Wright L, et al. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med. 1999;159:1925–32. doi: 10.1164/ajrccm.159.6.9804054. [DOI] [PubMed] [Google Scholar]
- 102.Wipff J, Dieude P, Guedj M, Ruiz B, Riemekasten G, Cracowski JL, et al. Association of a KCNA5 gene polymorphism with systemic sclerosis-associated pulmonary arterial hypertension in the European Caucasian population. Arthritis Rheum. 2010;62:3093–100. doi: 10.1002/art.27607. [DOI] [PubMed] [Google Scholar]
- 103.Remillard CV, Tigno DD, Platoshyn O, Burg ED, Brevnova EE, Conger D, et al. Function of Kv1.5 channels and genetic variations of KCNA5 in patients with idiopathic pulmonary arterial hypertension. Am J Physiol Cell Physiol. 2007;292:C1837–53. doi: 10.1152/ajpcell.00405.2006. [DOI] [PubMed] [Google Scholar]
- 104.Loukanov T, Hoss K, Tonchev P, Klimpel H, Arnold R, Sebening C, et al. Endothelial nitric oxide synthase gene polymorphism (Glu298Asp) and acute pulmonary hypertension post cardiopulmonary bypass in children with congenital cardiac diseases. Cardiol Young. 2011;21:161–9. doi: 10.1017/S1047951110001630. [DOI] [PubMed] [Google Scholar]
- 105.Yildiz P, Oflaz H, Cine N, Erginel-Unaltuna N, Erzengin F, Yilmaz V. Gene polymorphisms of endothelial nitric oxide synthase enzyme associated with pulmonary hypertension in patients with COPD. Respir Med. 2003;97:1282–8. doi: 10.1016/j.rmed.2003.06.001. [DOI] [PubMed] [Google Scholar]
- 106.Canter JA, Summar ML, Smith HB, Rice GD, Hall LD, Ritchie MD, et al. Genetic variation in the mitochondrial enzyme carbamyl-phosphate synthetase I predisposes children to increased pulmonary artery pressure following surgical repair of congenital heart defects: A validated genetic association study. Mitochondrion. 2007;7:204–10. doi: 10.1016/j.mito.2006.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Anaid S, Petkov V, Baykuscheva-Gentscheva T, Hoeger H, Painsipp E, Holzer P, et al. Involvement of endothelial NO in the dilator effect of VIP on rat isolated pulmonary artery. Regul Pept. 2007;139:102–8. doi: 10.1016/j.regpep.2006.10.012. [DOI] [PubMed] [Google Scholar]
- 108.Willers ED, Newman JH, Loyd JE, Robbins IM, Wheeler LA, Prince MA, et al. Serotonin transporter polymorphisms in familial and idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2006;173:798–802. doi: 10.1164/rccm.200509-1361OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Machado RD, Koehler R, Glissmeyer E, Veal C, Suntharalingam J, Kim M, et al. Genetic association of the serotonin transporter in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2006;173:793–7. doi: 10.1164/rccm.200509-1365OC. [DOI] [PubMed] [Google Scholar]
- 110.Roberts KE, Fallon MB, Krowka MJ, Benza RL, Knowles JA, Badesch DB, et al. Serotonin transporter polymorphisms in patients with portopulmonary hypertension. Chest. 2009;135:1470–5. doi: 10.1378/chest.08-1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.West J, Cogan J, Geraci M, Robinson L, Newman J, Phillips JA, et al. Gene expression in BMPR2 mutation carriers with and without evidence of pulmonary arterial hypertension suggests pathways relevant to disease penetrance. BMC Med Genomics. 2008;1:45. doi: 10.1186/1755-8794-1-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Menon S, Fessel J, West J. Microarray studies in pulmonary arterial hypertension. Int J Clin Pract Suppl. 2011:19–28. doi: 10.1111/j.1742-1241.2010.02604.x. [DOI] [PubMed] [Google Scholar]
- 113.Mouchaers KT, Schalij I, de Boer MA, Postmus PE, van Hinsbergh VW, van Nieuw Amerongen GP, Vonk et al. Fasudil reduces monocrotaline-induced pulmonary arterial hypertension: comparison with bosentan and sildenafil. Eur Respir J. 2010;36:800–7. doi: 10.1183/09031936.00130209. [DOI] [PubMed] [Google Scholar]
- 114.Nagaoka T, Gebb SA, Karoor V, Homma N, Morris KG, McMurtry IF, et al. Involvement of RhoA/Rho kinase signaling in pulmonary hypertension of the fawn-hooded rat. J Appl Physiol. 2006;100:996–1002. doi: 10.1152/japplphysiol.01028.2005. [DOI] [PubMed] [Google Scholar]
- 115.Petkov V, Mosgoeller W, Ziesche R, Raderer M, Stiebellehner L, Vonbank K, et al. Vasoactive intestinal peptide as a new drug for treatment of primary pulmonary hypertension. J Clin Invest. 2003;111:1339–46. doi: 10.1172/JCI17500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Shenoy V, Ferreira AJ, Qi Y, Fraga-Silva RA, Diez-Freire C, Dooies A, et al. The angiotensin-converting enzyme 2/angiogenesis-(1-7)/Mas axis confers cardiopulmonary protection against lung fibrosis and pulmonary hypertension. Am J Respir Crit Care Med. 2010;182:1065–72. doi: 10.1164/rccm.200912-1840OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ferreira AJ, Shenoy V, Yamazato Y, Sriramula S, Francis J, Yuan L, et al. Evidence for angiotensin-converting enzyme 2 as a therapeutic target for the prevention of pulmonary hypertension. Am J Respir Crit Care Med. 2009;179:1048–54. doi: 10.1164/rccm.200811-1678OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Jones DL, Sandberg JC, Rosenthal MJ, Saunders RC, Hannig VL, Clayton EW. What patients and their relatives think about testing for BMPR2. J Genet Couns. 2008;17:452–8. doi: 10.1007/s10897-008-9172-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Newman JH, Trembath RC, Morse JA, Grunig E, Loyd JE, Adnot S, et al. Genetic basis of pulmonary arterial hypertension: Current understanding and future directions. J Am Coll Cardiol. 2004;43:33S–9S. doi: 10.1016/j.jacc.2004.02.028. [DOI] [PubMed] [Google Scholar]
- 120.McGoon M, Gutterman D, Steen V, Barst R, McCrory DC, Fortin TA, et al. Screening, early detection, and diagnosis of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest. 2004;126:14S–34S. doi: 10.1378/chest.126.1_suppl.14S. [DOI] [PubMed] [Google Scholar]