

Abstract
To evaluate the vasoconstrictor component of PH in CHF by investigating the hemodynamic response to inhaled nitric oxide (iNO) and to determine whether this response was influenced by the phosphodiesterase 5 gene (PDE5) G(1142)T polymorphism. CHF patients underwent right heart catheterization at rest and after 20 ppm of iNO and plasma cGMP and PDE5 G(1142)T polymorphism determinations. Of the 72 included CHF patients (mean age, 53±1 years; mean left ventricular ejection fraction, 29±1%; and mean pulmonary artery pressure, 25.5±1.3 mmHg), 54% had ischemic heart disease. Proportions of patients with the TT, GT, and GG genotypes were 39%, 42% and 19% respectively. Baseline hemodynamic characteristics were not significantly different across PDE5 genotype groups, although pulmonary capillary wedge pressure (PCWP) tended to be lower in the TT group (P=0.09). Baseline plasma cGMP levels were significantly lower in the TT than in the GG and GT patients. With iNO, PVR diminished in TT (-33%) but not GG (-1.6%) or GT (0%) patients (P=0.002); and PCWP increased more in TT than in GT (P<0.05) or GG (P<0.003) patients. The PDE5 G(-1142) polymorphism is therefore a major contributor to the iNO-induced PVR decrease in CHF.
Keywords: heart failure, nitric oxide, phosphodiesterase
INTRODUCTION
Pulmonary hypertension (PH) is common and of adverse prognostic significance in patients with chronic heart failure (CHF).[1,2] PH in CHF patients is often associated with an increase in pulmonary vascular resistance (PVR), which may result from constriction and/or remodeling of the pulmonary vessels. Only those patients in whom the predominant mechanism is pulmonary vasoconstriction can be expected to benefit from vasodilator therapy after heart transplantation.[3–5]
A possible mechanism of pulmonary vasoconstriction in CHF is diminished relaxation of the arteriolar smooth muscle. Inhalation of nitric oxide (NO) is now considered useful for assessing the contribution of vasoconstriction to idiopathic PH or PH associated with an underlying disease. Inhaled NO is used to treat patients with PH associated with various conditions including congenital heart disease, valvular heart disease, respiratory failure after heart transplantation, and adult respiratory distress syndrome.[6] Exogenous NO diffuses rapidly through the tissues, increasing the NO exposure of smooth muscle cells in pulmonary resistance vessels and thereby producing a direct vasodilator effect. Similar to intravenous beta-1 agonist or phosphodiesterase 3 inhibitor administration, NO inhalation is often used to assess the PVR response to vasodilation in CHF patients considered for heart transplantation. In CHF patients, the PVR decrease produced by NO inhalation is ascribable to an increase in left ventricular (LV) filling pressure, as opposed to a decrease in pulmonary artery pressure.[7] The mechanisms of this LV filling pressure increase are unknown. However, studies have shown inter-individual variations in the inhaled-NO response that were independent from the cause of PH.[7–11]
NO signaling is mainly mediated by cyclic guanosine monophosphate (cGMP) accumulation within the cells as a consequence of increased guanylate cyclase activity. However, cGMP bioavailability is limited by the hydrolytic activity of phosphodiesterases (PDE), most notably PDE5.[12] A recently identified regulatory region of the PDE5 gene (GenBank accession n°AB001615) has two functionally relevant AP1 regulatory sequences,[13] of which one harbors a T/G polymorphism at position 1142.[14] We hypothesized that the PDE5 G(1142)T polymorphism affected inhaled-NO responses in CHF patients, indicating that it contributed to the inter-individual differences in basal pulmonary vasoconstriction across CHF patients.
We prospectively studied 72 consecutive CHF patients to evaluate potential associations linking the PDE5 G(1142)T polymorphism to the pulmonary hemodynamic characteristics and/or inhaled-NO response. We report here for the first time that the PDE5 G(-1142) polymorphism is associated with differences in cGMP levels in CHF patients and that the inhaled-NO response, used as a measure of pulmonary vasoconstriction, varies with the PDE5 genotype.
MATERIALS AND METHODS
Inclusion and exclusion criteria
Inclusion criteria were an LV ejection fraction (LVEF) less than 35% and a history of symptomatic CHF. Exclusion criteria were acute heart failure, severe tricuspid regurgitation, severe obstructive lung disease, pulmonary embolism, and valvular disease.
The patients were selected among participants in a genetic study of the causes of PH. It was supported by a publicly funded French research agency (DRRC: Délégation Régionale à la Recherche Clinique, www.drrc.aphp.fr, registration number, P020902) and approved by the ethics committee of the Henri Mondor Teaching Hospital. Written informed consent was obtained from all patients before inclusion.
Hemodynamic measurements
Maximal oxygen consumption (VO2) was determined and echocardiography (Vivid 7, General Electric) performed 24 hrs. before or after right-sided cardiac catheterization. During echocardiography, left and right ventricular functions were assessed as recommended by the American Society of Echocardiography[15] and tricuspid regurgitation was quantified using standard criteria.
All treatments were stopped 12 hrs. before right-sided cardiac catheterization. A Swan-Ganz catheter was introduced up to the pulmonary artery. Heart rate, right atrial pressure, mean pulmonary artery pressure (mPAP), cardiac output (Q) measured by thermodilution, and pulmonary capillary wedge pressure (PCWP) were measured while breathing air and after breathing 20 ppm of NO for 10 min. via a closed facemask. Pulmonary vascular resistance (PVR) was computed as follows: PVR = (mPAP – PCWP)/Q. After catheter removal, patients were monitored clinically for 4 hours.
Assay of cGMP
Venous blood was used for cGMP measurement in all patients. In 12 patients, cGMP was also measured after NO inhalation in blood collected at the distal pulmonary tip of the catheter. After immediate centrifugation at 4°C, the serum was separated and stored at –80°C for future use. A commercially available cGMP assay was used (Biovision, Calif., USA). The cGMP levels in the GG and GT groups were pooled and compared to those in the TT group.
Genetic analysis
Venous blood was collected during the initial evaluation. Genomic DNA was extracted from leukocytes using a DNA extraction kit (QIAamp DNA blood kit, QUIAGEN, Hilden, Germany). The published sequence of the PDE5A 5′-flanking region (AB001615) was analyzed and amplified by polymerase chain reaction (PCR, Ready-To-Go PCR kit, Amersham Pharmacia Biotech, Paris, France) in a total volume of 25 ml using 40 ng of DNA and 50 ng of each primer (upstream, 5’-TTGCTTTTCTTTGGTTGTGGCT-3’; and downstream, 5’GTGTCATCCTTGCTTTGTCTG-3’). Each of the 35 amplification cycles consisted of denaturation at 96°C for 50 s, annealing at 62°C for 50 sec, and elongation at 72°C for 1 min. (DNA Thermal Cycler, Perkin-Elmer/Cetus). An initial denaturation step was carried out at 97°C for 5 min. and a final elongation step at 72°C for 10 min. The PCR products were analyzed by gel electrophoresis using 2% Seakem LE agarose (BioWhittaker Molecular Applications, Rockland, Me., USA).
Statistical analysis
Normally distributed data were described as mean±SEM. Between-group comparisons of quantitative variables were done using ANOVA or the Student t test, as appropriate. Qualitative variables were compared using the χ2test. Differences between baseline and inhaled-NO values were compared using repeated measures ANOVA with a post hoc analysis using Fisher's probable least-significant difference test. P values smaller than 0.05 were considered significant. Statistical analyses were performed using Statview software (SAS Institute, Cary, NC, USA).
RESULTS
Study population, baseline characteristics
We studied 72 patients, whose baseline characteristics are listed in (Table 1). All patients were receiving optimal CHF medications. Hemodynamic parameter values on room air are listed in Table 2. Mean values of PCWP and PVR were elevated.
Table 1.
Baseline characteristics of the whole cohorte and divided by PDE5 G(1142)T polymorphism
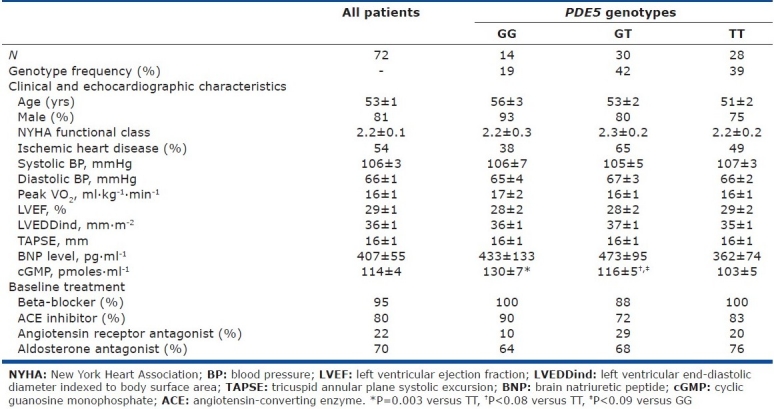
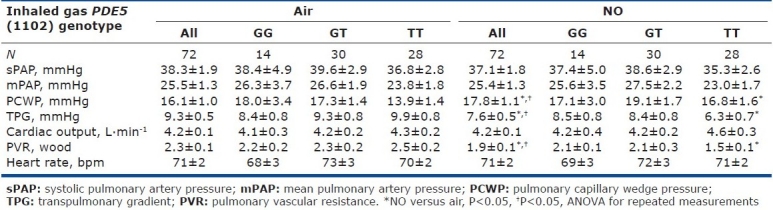
Table 2.
Hemodynamic characteristics at baseline and after NO inhalation depending on the PDE5 G(1142)T polymorphism
Genotype distribution and interaction with baseline characteristics
Of the 72 patients, 28 (38.9%) had the TT genotype, 30 (41.7%) the GT genotype, and 14 (19.4%) the GG genotype. The G(-1142) allele accounted for 40.3% of all alleles at position 1142.
Neither the clinical characteristics nor the plasma brain natriuretic peptide (BNP) levels differed significantly across the 3 genotype groups (Table 1). Plasma cGMP under basal conditions was significantly lower in the TT group than in the GG or GT group (Fig. 1), suggesting increased PDE5 activity in the TT group.
Figure 1.
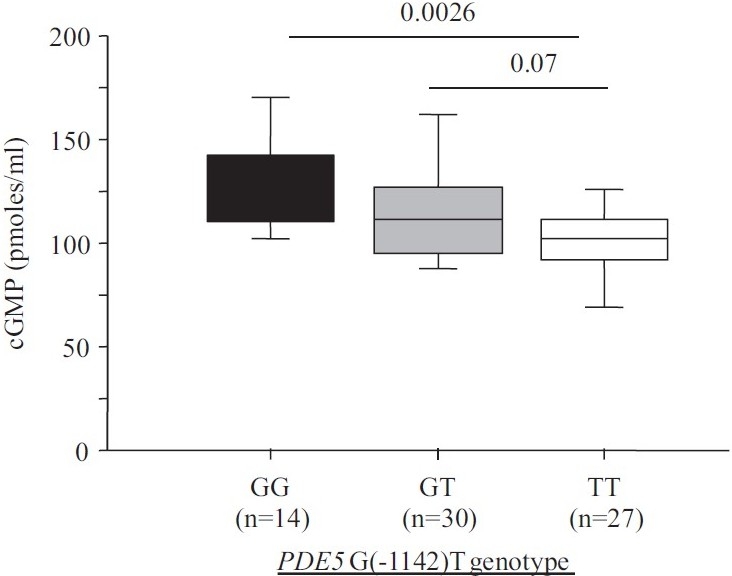
Plasma cGMP concentration depending on PDE5 G(1142)T polymorphism.
Baseline hemodynamic characteristics were not significantly different across the three genotype groups. A trend toward lower PCWP values was found in the TT group compared to the GT-GG group, despite the absence of significant differences in CHF severity or pulmonary artery pressure (Table 1). The transpulmonary gradient (TPG) normalized for mPAP was significantly higher in the TT group compared to the GG-GT group.
Effect of inhaled NO on hemodynamic parameters according to PDE5 genotype
NO was well tolerated, with no effects on heart rate, Q, or pulmonary artery pressure (Table 2). NO inhalation significantly decreased PVR (Table 2) by increasing PCWP and the TPG without decreasing mPAP.
The hemodynamic effect of NO inhalation was greatest in the TT group, where the PCWP increase was largest (Fig. 2). In TT, the PVR decrease was significant (Fig. 3) and was larger in the group with PH (Fig. 4). After NO inhalation, cGMP increased significantly in the TT and GT groups but not in GG (Fig. 5). Plasma cGMP elevation was not correlated with the PVR decrease.
Figure 2.
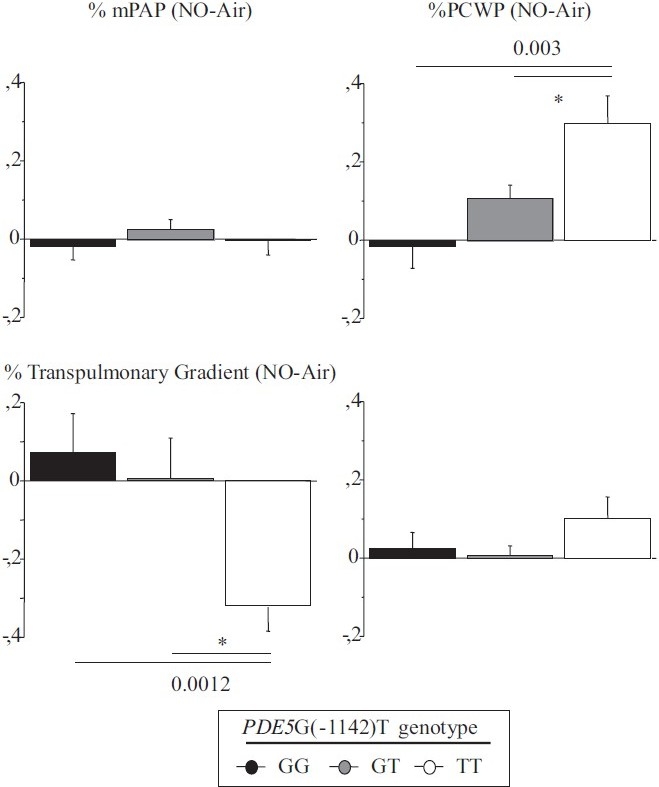
Percent of change between baseline and after inhaled NO in transpulmonary gradient, mean pulmonary artery pressure, pulmonary capillary wedge pressure and cardiac output.
Figure 3.
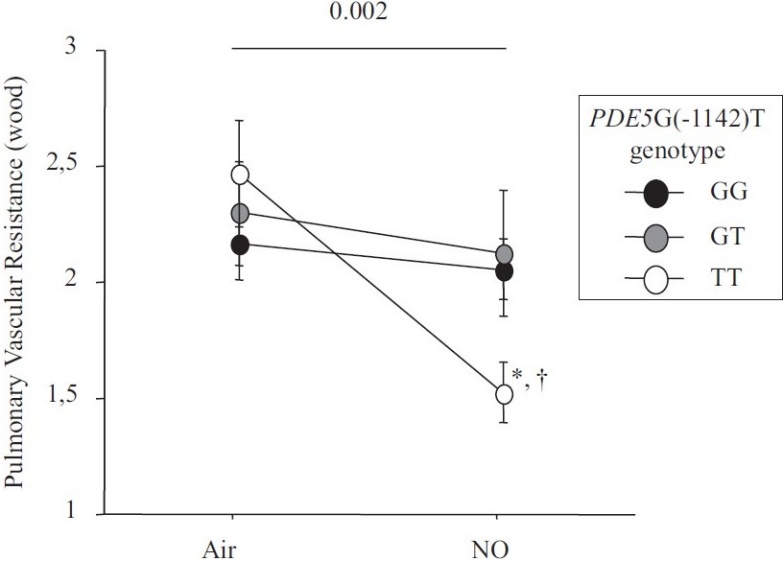
Change between baseline and after inhaled NO in pulmonary vascular resistance.
Figure 4.
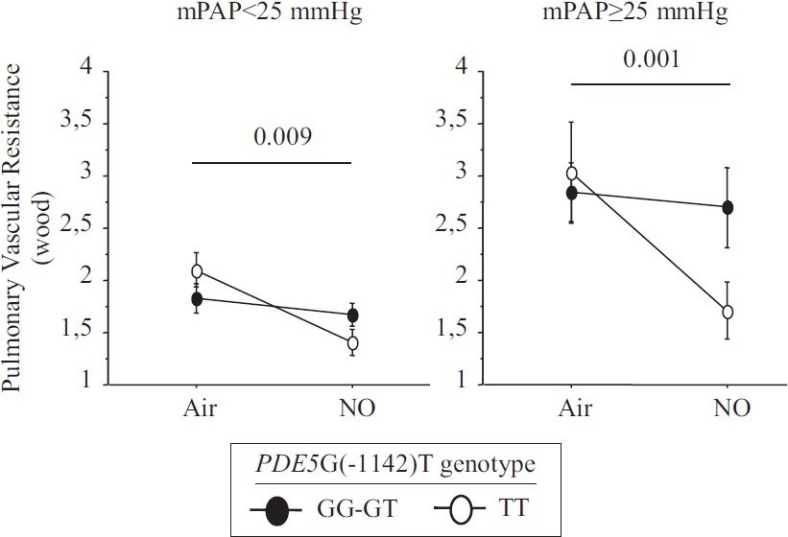
Change between baseline and after inhaled NO in pulmonary vascular resistance depending on mean pulmonary artery pressure subgroup.
Figure 5.
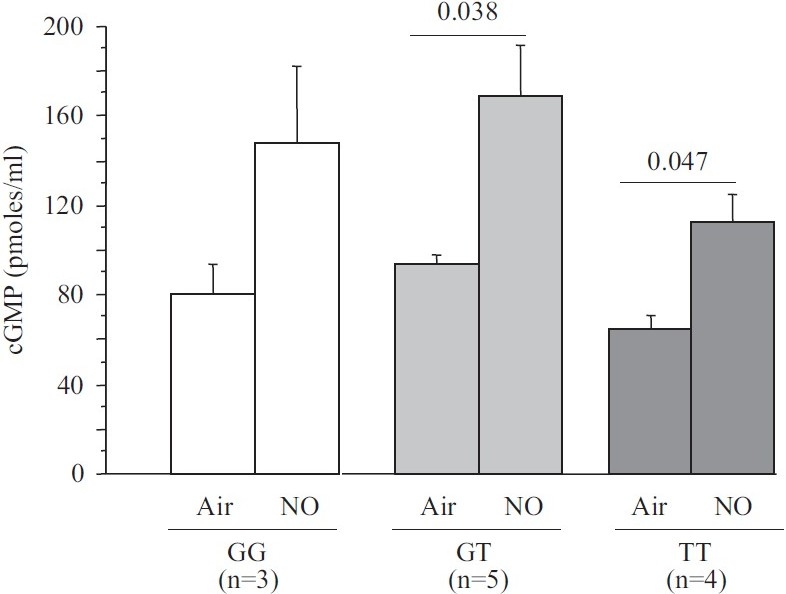
Plasma cGMP concentration depending on PDE5 G(1142)T polymorphism at baseline and after inhaled NO.
DISCUSSION
The main finding from this study is that the PDE5 G(- 1142) polymorphism influences plasma cGMP levels and hemodynamic parameter values under basal conditions and after NO inhalation in patients with CHF.
Baseline characteristics
The G(-1142) allele frequency and PDE5 genotype distribution were consistent with a previous report.[14] The only baseline feature that differed significantly across the three genotype groups (TT, GT, and GG) was plasma cGMP concentration, which was lower in the TT group (Fig. 1), suggesting a functional effect of the PDE5 polymorphism, with the TT genotype being associated with greater PDE5 activity and higher clearance of cGMP. In the TT group, a trend toward a lower filling pressure was noted (P=0.09). A recent in vitro study suggests that different PDE5 conformations involving the H-loop may affect the interaction of PDE5A1 with cGMP.[16] Furthermore, the H-loop variant may have a less critical interaction with PDE5 inhibitors,[16] However, plasma cGMP levels depend not only on cGMP clearance, but also on cGMP synthesis, which involves endogenous NO production (soluble guanylate cyclase) and atrial natriuretic peptides (particulate guanylate cyclase). However, in our study plasma BNP levels were not significantly different across genotype groups (Table 1), suggesting that the lower cGMP level in the TT group may have been due to greater PDE5 activity. PDE5 activity in pulmonary arteries, and therefore plasma cGMP levels, may affect pulmonary vascular tone. Conceivably, the higher cGMP levels in the GG and GT groups may explain the trend toward an increase in baseline PCWP in these genotype groups. Thus, the higher cGMP levels (due to diminished PDE5 activity) in the GG and GT groups may indicate greater pulmonary vasodilation with increased venous blood return to the left cardiac chambers and, therefore, increased basal PCWP in CHF patients. The TT group, in contrast, may have a higher basal pulmonary vessel tone associated with lower capillary pressures.
No significant differences were found across genotypes for baseline LV function (LVEF) or right ventricular function (TAPSE) (Table 1), suggesting that PDE5 activity did not affect ventricular function under basal conditions. Similarly, earlier experimental studies found no effect of PDE5 inhibition on basal contractility of human papillary muscle strips[17] or failing canine hearts.[18] Nevertheless, increased PDE5 expression has been found in myocardial specimens from patients with ventricular hypertrophy.[19]
Effect of inhaled NO on pulmonary artery
Our study agrees with a previous report that inhaled NO significantly diminished PVR and the TPG in patients with CHF, by increasing PCWP.[7,20] A possible explanation is that NO inhalation induced pulmonary dilatation with shifts of blood volume from the pulmonary arterial to the pulmonary venous compartment. LV failure causes an increase in pulmonary venous volume and, therefore, in LV filling pressure.[21,22] Here, we found that the PVR decrease in response to inhaled NO response was dependent on the PDE5 G(-1142) polymorphism. The TT genotype was associated with a greater PVR decrease due to larger increase in PCWP and decrease in TPG. NO inhalation increased plasma cGMP concentrations in all genotype groups. However, the effects of NO inhalation depend also on the severity of the hemodynamic abnormalities.[7] Loh et al. showed that the response to inhaled NO was greater in patients with decompensated CHF than in patients with compensated CHF. We included only patients with compensated CHF. Among them, those having mPAP values greater than 25 mmHg exhibited a stronger response to inhaled NO. However, in the TT-genotype patients with mPAP values lower than 25 mmHg, the PVR response remained significantly greater than in the other two genotype groups (Fig. 3).
Conceivably, changes in PDE5 activity related to the PDE5 G(1142) polymorphism may affect the regulation of pulmonary artery vasodilation at baseline and after NO inhalation. The TT genotype may be associated with greater PDE5 activity responsible for an increase in basal vascular tone and, therefore, for protection of the LV from excessive filling pressures.
We used a moderate NO dose (20 ppm). The use of higher dosages (40 or 80 ppm) would perhaps have eliminated the differences across genotypes. However, in earlier studies a moderate dose of inhaled-NO was as effective as a higher dose in decreasing PCWP in patients with congestive CHF.[23] Furthermore, most of the inhaled NO response was achieved with only 10 ppm in patients who had secondary or primary PH,[9] and inhaled NO was as effective as vasodilators selectively affecting the pulmonary vasculature.[24] Furthermore, in patients with stable severe CHF, high doses of NO have been reported to induce pulmonary edema.[25] In the 12 patients whose plasma cGMP levels were measured after NO inhalation, we found no correlation between the individual cGMP response to NO and the magnitude of the pulmonary vasorelaxation response, in keeping with a previous study.[8]
Limitations
This study has several limitations. First, the sample size is relatively small for a genetic study. However, our study sample is the largest included to date in an evaluation of the response to NO inhalation, and we studied the functional activity of a genetic polymorphism as opposed to its distribution frequency. Second, we cannot rule out an interaction between the medications taken by our patients and the NO response. In previous studies, the patients were not receiving beta-blockers. We discontinued beta-blocker therapy only 12 hrs. before right-sided cardiac catheterization to avoid undue risk to the patients. We found no significant effects of the PDE5 polymorphism on baseline hemodynamics, although there was a trend toward lower PCWP values in the TT group. Further studies are needed to evaluate the impact of the PDE5 polymorphism on the effect of PDE5 inhibitors. Recently, oral PDE5 inhibitors approved for the treatment of erectile dysfunction or primary PH were found to benefit patients with or CHF.[26,27] Some studies found that the hemodynamic response to PDE5 inhibitor administration was increased by concomitant NO inhalation.[20,28]
CONCLUSIONS
Our data suggest that the PDE5 G(-1142) polymorphism is functional and may affect pulmonary artery tone at baseline, thus influencing the response to inhaled NO in patients with CHF. Further studies are necessary to determine the clinical and therapeutic implications of this polymorphism in CHF patients.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Kjaergaard J, Akkan D, Iversen KK, Kjoller E, Kober L, Torp-Pedersen C, et al. Prognostic importance of pulmonary hypertension in patients with heart failure. Am J Cardiol. 2007;99:1146–50. doi: 10.1016/j.amjcard.2006.11.052. [DOI] [PubMed] [Google Scholar]
- 2.Damy T, Goode K, Kallvikbacka-Bennet A, Lewinter C, Hobkirk J, Nikitin N, et al. Determinants and prognostic value of pulmonary artery pressure in patients with chronic heart failure. Eur Heart J. 2010. [Last accessed on 2010 Aug 05]. Available from: http://eurheartj.oxfordjournals.org/content/early/2010/08/05/eurheartj.ehq245.full . [DOI] [PubMed]
- 3.Guglin M, Khan H. Pulmonary hypertension in Heart Failure. J Cardiac Fail. 2010;16:461–74. doi: 10.1016/j.cardfail.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 4.Simmoneau G, Robins I, Beghetti M, Channick R, Delcroix M, Denton CP, et al. Updated Clinical Classification of Pulmonary Hypertension. J Am Coll Cardiol. 2009;54:S43–54. doi: 10.1016/j.jacc.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 5.Badesch D, Champion HC, Gomez Sanchez M, Hoeper M, Loyd J, Manes A, et al. Diagnosis and Assessment of Pulmonary Artery Hypertension. J Am Coll Cardiol. 2009;54:S55–66. doi: 10.1016/j.jacc.2009.04.011. [DOI] [PubMed] [Google Scholar]
- 6.Germann P, Braschi A, Della Rocca G, Dinh-Xuan AT, Falke K, Frostell C, et al. Inhaled nitric oxide therapy in adults: European expert recommendations. Intensive Care Med. 2005;31:1029–41. doi: 10.1007/s00134-005-2675-4. [DOI] [PubMed] [Google Scholar]
- 7.Loh E, Stamler JS, Hare JM, Loscalzo J, Colucci WS. Cardiovascular effects of inhaled nitric oxide in patients with left ventricular dysfunction. Circulation. 1994;90:2780–5. doi: 10.1161/01.cir.90.6.2780. [DOI] [PubMed] [Google Scholar]
- 8.Ghofrani HA, Wiedemann R, Rose F, Weissmann N, Schermuly RT, Quanz K, et al. Lung cGMP release subsequent to NO inhalation in pulmonary hypertension: Responders versus nonresponders. Eur Respir J. 2002;19:664–71. doi: 10.1183/09031936.02.00982001. [DOI] [PubMed] [Google Scholar]
- 9.Krasuski RA, Warner JJ, Wang A, Harrison JK, Tapson VF, Bashore TM. Inhaled nitric oxide selectively dilates pulmonary vasculature in adult patients with pulmonary hypertension, irrespective of etiology. J Am Coll Cardiol. 2000;36:2204–11. doi: 10.1016/s0735-1097(00)00994-3. [DOI] [PubMed] [Google Scholar]
- 10.Lepore JJ, Dec GW, Zapol WM, Bloch KD, Semigran MJ. Combined administration of intravenous dipyridamole and inhaled nitric oxide to assess reversibility of pulmonary arterial hypertension in potential cardiac transplant recipients. J Heart Lung Transplant. 2005;24:1950–6. doi: 10.1016/j.healun.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 11.Semigran MJ, Cockrill BA, Kacmarek R, Thompson BT, Zapol WM, Dec GW, et al. Hemodynamic effects of inhaled nitric oxide in heart failure. J Am Coll Cardiol. 1994;24:982–8. doi: 10.1016/0735-1097(94)90859-1. [DOI] [PubMed] [Google Scholar]
- 12.Omori K, Kotera J. Overview of PDEs and their regulation. Circ Res. 2007;100:309–27. doi: 10.1161/01.RES.0000256354.95791.f1. [DOI] [PubMed] [Google Scholar]
- 13.Yanaka N, Kotera J, Ohtsuka A, Akatsuka H, Imai Y, Michibata H, et al. Expression, structure and chromosomal localization of the human cGMP-binding cGMP-specific phosphodiesterase PDE5 A gene. Eur J Biochem. 1998;255:391–9. doi: 10.1046/j.1432-1327.1998.2550391.x. [DOI] [PubMed] [Google Scholar]
- 14.Salvi F, Sarzani R, Giorgi R, Donatelli G, Pietrucci F, Micheli A, et al. Cardiovascular effects of sildenafil in hypertensive men with erectile dysfunction and different alleles of the type. 5 cGMP-specific phosphodiesterase (PDE5) Int J Impot Res. 2004;16:412–7. doi: 10.1038/sj.ijir.3901246. [DOI] [PubMed] [Google Scholar]
- 15.Gardin JM, Adams DB, Douglas PS, Feigenbaum H, Forst DH, Fraser AG, et al. Recommendations for a standardized report for adult transthoracic echocardiography: A report from the American Society of Echocardiography's Nomenclature and Standards Committee and Task Force for a Standardized Echocardiography Report. J Am Soc Echocardiogr. 2002;15:275–90. doi: 10.1067/mje.2002.121536. [DOI] [PubMed] [Google Scholar]
- 16.Wang H, Liu Y, Huai Q, Cai J, Zoraghi R, Francis SH, et al. Multiple conformations of phosphodiesterase-5: Implications for enzyme function and drug development. J Biol Chem. 2006;281:21469–79. doi: 10.1074/jbc.M512527200. [DOI] [PubMed] [Google Scholar]
- 17.Cremers B, Scheler M, Maack C, Wendler O, Schafers HJ, Sudkamp M, et al. Effects of sildenafil (viagra) on human myocardial contractility, in vitro arrhythmias, and tension of internal mammaria arteries and saphenous veins. J Cardiovasc Pharmacol. 2003;41:734–43. doi: 10.1097/00005344-200305000-00010. [DOI] [PubMed] [Google Scholar]
- 18.Senzaki H, Smith CJ, Juang GJ, Isoda T, Mayer SP, Ohler A, et al. Cardiac phosphodiesterase 5 (cGMP-specific) modulates beta-adrenergic signaling in vivo and is down-regulated in heart failure. FASEB J. 2001;15:1718–26. doi: 10.1096/fj.00-0538com. [DOI] [PubMed] [Google Scholar]
- 19.Nagendran J, Archer SL, Soliman D, Gurtu V, Moudgil R, Haromy A, et al. Phosphodiesterase type 5 is highly expressed in the hypertrophied human right ventricle, and acute inhibition of phosphodiesterase type 5 improves contractility. Circulation. 2007;116:238–48. doi: 10.1161/CIRCULATIONAHA.106.655266. [DOI] [PubMed] [Google Scholar]
- 20.Michelakis E, Tymchak W, Lien D, Webster L, Hashimoto K, Archer S. Oral sildenafil is an effective and specific pulmonary vasodilator in patients with pulmonary arterial hypertension: Comparison with inhaled nitric oxide. Circulation. 2002;105:2398–403. doi: 10.1161/01.cir.0000016641.12984.dc. [DOI] [PubMed] [Google Scholar]
- 21.Hare JM, Shernan SK, Body SC, Graydon E, Colucci WS, Couper GS. Influence of inhaled nitric oxide on systemic flow and ventricular filling pressure in patients receiving mechanical circulatory assistance. Circulation. 1997;95:2250–3. doi: 10.1161/01.cir.95.9.2250. [DOI] [PubMed] [Google Scholar]
- 22.Hare JM, Loh E, Creager MA, Colucci WS. Nitric oxide inhibits the positive inotropic response to beta-adrenergic stimulation in humans with left ventricular dysfunction. Circulation. 1995;92:2198–203. doi: 10.1161/01.cir.92.8.2198. [DOI] [PubMed] [Google Scholar]
- 23.Matsumoto A, Momomura S, Sugiura S, Fujita H, Aoyagi T, Sata M, et al. Effect of inhaled nitric oxide on gas exchange in patients with congestive heart failure.A randomized, controlled trial. Ann Intern Med. 1999;130:40–4. doi: 10.7326/0003-4819-130-1-199901050-00008. [DOI] [PubMed] [Google Scholar]
- 24.Pagano D, Townend JN, Horton R, Smith C, Clutton-Brock T, Bonser RS. A comparison of inhaled nitric oxide with intravenous vasodilators in the assessment of pulmonary haemodynamics prior to cardiac transplantation. Eur J Cardiothorac Surg. 1996;10:1120–6. doi: 10.1016/s1010-7940(96)80360-5. [DOI] [PubMed] [Google Scholar]
- 25.Bocchi EA, Bacal F, Auler JO, Jr, Carmone MJ, Bellotti G, Pileggi F. Inhaled nitric oxide leading to pulmonary edema in stable severe heart failure. Am J Cardiol. 1994;74:70–2. doi: 10.1016/0002-9149(94)90496-0. [DOI] [PubMed] [Google Scholar]
- 26.Lewis GD, Shah R, Shahzad K, Camuso JM, Pappagianopoulos PP, Hung J, et al. Sildenafil improves exercise capacity and quality of life in patients with systolic heart failure and secondary pulmonary hypertension. Circulation. 2007;116:1555–62. doi: 10.1161/CIRCULATIONAHA.107.716373. [DOI] [PubMed] [Google Scholar]
- 27.Lewis GD, Lachmann J, Camuso J, Lepore JJ, Shin J, Martinovic ME, et al. Sildenafil improves exercise hemodynamics and oxygen uptake in patients with systolic heart failure. Circulation. 2007;115:59–66. doi: 10.1161/CIRCULATIONAHA.106.626226. [DOI] [PubMed] [Google Scholar]
- 28.Lepore JJ, Maroo A, Bigatello LM, Dec GW, Zapol WM, Bloch KD, et al. Hemodynamic effects of sildenafil in patients with congestive heart failure and pulmonary hypertension: Combined administration with inhaled nitric oxide. Chest. 2005;127:1647–53. doi: 10.1378/chest.127.5.1647. [DOI] [PubMed] [Google Scholar]