

Pulmonary arterial hypertension (PAH) is a severe and fatal disease with a prevalence of 15 cases in a million[1] and characterized by increased pulmonary vascular resistance and right heart failure. PAH can be idiopathic (IPAH), familial (FPAH), or associated with other disorders. In 1984 the National Institute of Health (NIH) compiled the first large registry of PAH patients (all IPAH, FPAH, and anorexigen-associated) confirming poor survival and prognosis.[2] In an era of PAH-specific therapies, subsequent PAH registries (the Pulmonary Hypertension Connection [PHC] registry,[3] the French registry,[4] and the Registry to Evaluate Early and Long-term Pulmonary Arterial Hypertension Disease Management [REVEAL][5] ) have demonstrated improved prognosis (Table 1).
Table 1.
Incidence and survival of PAH based on different registries
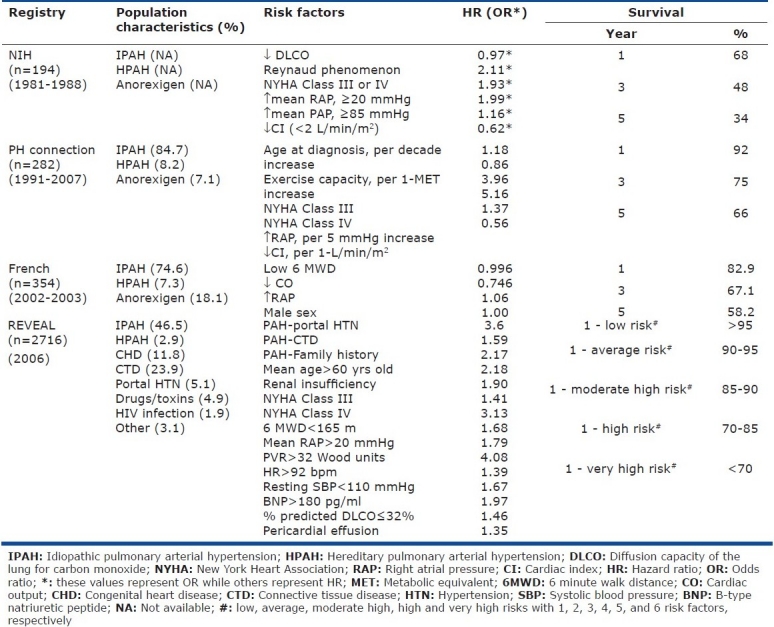
Each registry is defined by unique strengths and weak-nesses. The NIH registry is a landmark accomplishment, but may not be as applicable to the modern patient on PAH-specific therapy. The PHC validated many of the findings of the NIH registry, including various risk factors, and demonstrated improvements in survival. The REVEAL registry is the largest and, in contrast to other registries, includes associated forms of PAH. For the first time, this last distinction opens the potential applicability of the registry findings to nearly all patients with PAH beyond IPAH, HPAH, and anorexigen-associated PAH. Importantly, although both demonstrate consistency in improvement from the NIH registry, the REVEAL and the French registries have reported a significant difference in survival data at 1 year (95% and 83%, respectively). This discrepancy may be attributed to multiple factors including the consideration, especially in PAH clinical trials, of the proportion of incident versus prevalent patient populations during the estimation for prognosis. Specifically, given the association of poor survival with a subset of PAH patients with very high risk factors, each registry may have different contributions of survival bias based on the individual representation of these patients within each registry. Finally, although all registries establish risk factors associated with adverse outcomes, they fail to incorporate prognosis information after risk factor modification. Nonetheless, there is considerable overlap and validation of established risk factors by the various reports with new makers of prognosis based on functional status and other clinical markers that extend beyond the three hemodynamic-based parameters established by the NIH registry. The latter registries have also identified factors associated with protection from poor outcomes. Several risk scores (equations) have subsequently been developed based on the associated risk factors and are used to predict mortality in patients with PAH. The REVEAL registry, for example, provides the following equation to estimate survival in patient's with PAH:
P(t) = [H(t)] A(x,y,z),
where x, y, and z represent the values for individual risk factors. Similar scores are provided in the other registries.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.McLaughlin VV, Suissa S. Prognosis of pulmonary arterial hypertension: The power of clinical registries of rare diseases. Circulation. 2010;122:106–8. doi: 10.1161/CIRCULATIONAHA.110.963983. [DOI] [PubMed] [Google Scholar]
- 2.D’Alonzo GE, Barst RJ, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, et al. Survival in patients with primary pulmonary hypertension.Results from a national prospective registry. Ann Intern Med. 1991;115:343–9. doi: 10.7326/0003-4819-115-5-343. [DOI] [PubMed] [Google Scholar]
- 3.Thenappan T, Shah SJ, Rich S, Gomberg-Maitland M. A USA-based registry for pulmonary arterial hypertension: 1982-2006. Eur Respir J. 2007;30:1103–10. doi: 10.1183/09031936.00042107. [DOI] [PubMed] [Google Scholar]
- 4.Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation. 2010;122:156–63. doi: 10.1161/CIRCULATIONAHA.109.911818. [DOI] [PubMed] [Google Scholar]
- 5.Benza RL, Miller DP, Gomberg-Maitland M, Frantz RP, Foreman AJ, Coffey CS, et al. Predicting survival in pulmonary arterial hypertension: Insights from the registry to evaluate early and long-term pulmonary arterial hypertension disease management (REVEAL) Circulation. 2010;122:164–72. doi: 10.1161/CIRCULATIONAHA.109.898122. [DOI] [PubMed] [Google Scholar]