

Abstract
Background
Clonorchis sinensis is a zoonotic parasite causing clonorchiasis-associated human disease such as biliary calculi, cholecystitis, liver cirrhosis, and it is currently classified as carcinogenic to humans for cholangiocarcinoma. MicroRNAs (miRNAs) are non-coding, regulating small RNA molecules which are essential for the complex life cycles of parasites and are involved in parasitic infections. To identify and characterize miRNAs expressed in adult C. sinensis residing chronically in the biliary tract, we developed an integrative approach combining deep sequencing and bioinformatic predictions with stem-loop real-time PCR analysis.
Results
Here we report the use of this approach to identify and clone 6 new and 62,512 conserved C. sinensis miRNAs which belonged to 284 families. There was strong bias on families, family members and sequence nucleotides in C. sinensis. Uracil was the dominant nucleotide, particularly at positions 1, 14 and 22, which were located approximately at the beginning, middle and end of conserved miRNAs. There was no significant "seed region" at the first and ninth positions which were commonly found in human, animals and plants. Categorization of conserved miRNAs indicated that miRNAs of C. sinensis were still innovated and concentrated along three branches of the phylogenetic tree leading to bilaterians, insects and coelomates. There were two miRNA strategies in C. sinensis for its parasitic life: keeping a large category of miRNA families of different animals and keeping stringent conserved seed regions with high active innovation in other places of miRNAs mainly in the middle and the end, which were perfect for the parasite to perform its complex life style and for host changes.
Conclusions
The present study represented the first large scale characterization of C. sinensis miRNAs, which have implications for understanding the complex biology of this zoonotic parasite, as well as miRNA studies of other related species such as Opisthorchis viverrini and Opisthorchis felineus of human and animal health significance.
Background
Fish-borne clonorchiasis, caused by the oriental liver fluke Clonorchis sinensis, is endemic in many Asian countries and over 35 million people globally are infected with C. sinensis, including an estimated 15 million in China [1]. The parasite has major socioeconomic impacts in other parts of Asia as well. In Korea, infection in humans is one of the most prevalent [2]. While in Vietnam, the prevalence reaches 79% in the Haiphong and Hanoi area [3]. This infection is also becoming increasingly common in non-endemic regions and in developed countries due to growing international markets, improved transportation systems, and demographic changes such as population movements [2,3]. Epidemiological data suggested that clonorchiasis has an increasing human-health impact resulted from the greater consumption of raw, frozen, dried, or pickled freshwater fish imported from endemic areas [1,4].
Adult C. sinensis flukes reside chronically in the biliary tract and cause periductal inflammation, fibrosis, pyogenic cholangitis, biliary calculi, cholecystitis, liver cirrhosis and pancreatitis [2,5]. Like Opistorchis viverrini, C. sinensis is classified as carcinogenic to humans by the International Agency for Research on Cancer for cholangiocarcinoma in 2009 [6]. The disease arises from metaplastic changes of biliary epithelial cells and usually occurs in the secondary intrahepatic bile duct, where the fluke is preferentially situated. However, the exact mechanisms of the carcinogenesis are not clearly elucidated [3,7].
The prevention and control strategies for this parasite include fecal examination and treatment of individual patients with praziquantel. The World Health Organization has also recommended mass chemotherapy in humans in endemic areas as the most practical and immediately effective control strategy [2]. Other efforts to control the parasite include interrupting transmission at the intermediate host level. However, there has been little effect on the impact of snail populations (the first intermediate host of the parasite) or on the practice of eating raw fish [1].
MicroRNAs (miRNAs) are 18-22 nucleotide, non-coding, small RNA molecules found in diverse organisms from viruses [8], plants [9], flies [10] to mammals [11,12], which regulate gene expression at the post-transcriptional level. They are essential for the complex life cycle of pathogenic parasites for their ability to respond to environmental and developmental signals and are now considered as a key mechanism of gene regulation [13]. The discovery of miRNA function sheds new light on the control of these parasites. However, there was no miRNAs being identified experimentally in C. sinensis.
In light of the probability that miRNA species are involved in gene regulation in C. sinensis, here we investigated the expression profile of miRNAs and detected potential novel miRNAs in C. sinensis adults. Due to the similarity in morphology, life cycle and modes of transmission among members of the Opisthorchiidae [3], miRNA profile research in C. sinensis will shed light on the miRNA studies of other species such as O. felineus and O. viverrini.
Results
Profile characteristics of short RNAs from C. sinensis
Deep sequencing yielded 14.8 million reads (Additional file 1 for flowchart), and the raw sequencing data was deposited in GEO of NCBI http://www.ncbi.nlm.nih.gov/geo/ with accession number GSE22244. After 5' and 3' adaptors, contamination formed by adaptor-adaptor ligation and low quality tags were removed, a total of 12.14 million reads with high quality were obtained. Length distribution analysis showed that most reads were distributed among 20-23 nt. The highest percentage was 18.73% with reads of 21 nt long, followed by 18.68% of 22 nt reads (Figure 1). In the next analysis step, reads smaller than 18 nt (6.05%) were removed, and a total of 11.19 million clean reads remained with 2.74 million (24.44%) unique sequences. Among the 11.19 million clean reads, a total of 2.18 million (19.46%) were perfectly mapped to the Schistosoma japonicum genome, including 30,558 (1.12%) unique sequences; and 16.13% of these have only one location on the genome.
Figure 1.
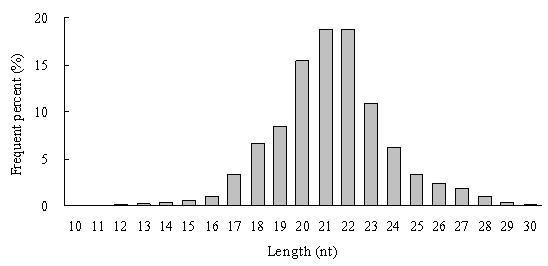
Length distribution of small RNAs from Clonorchis sinensis identified and analyzed by deep sequencing. Analysis of 12,138,350 high quality reads after filtering low quality tags, 5' and 3' adaptor and contamination formed by adaptor-adaptor ligation.
Among the 11.19 million reads, 1,222,173 (10.92%) were ncRNAs, including rRNA, tRNA, snRNA and snoRNA. Repeat-associated small RNAs (1,909; 0.02%) coming from high-repeat regions of genome or transposon-regions were found to belong to the two types of repeat: LINE/RTE:0 and LINE/RTE:1. The percentage of known miRNAs was 18.71% with 2,093,879 reads which includes 62,512 unique sequences (Additional file 2). Except for the miRNA, rRNA and repeats mentioned above, 7,875,451 (70.36%) sequences (2,610,289 unique reads) had no match and were marked as un-annotated (Table 1).
Table 1.
Summary of reads that match various RNAs
| Locus class | Unique reads | Total reads |
|---|---|---|
| rRNAetc | 61946 (2.26%) | 1222173 (10.92%) |
| Repeat | 732 (0.03%) | 1909 (0.02%) |
| Known miRNAs | 62512 (2.29%) | 2093879 (18.71%) |
| other small RNAs | 2610289 (95.42%) | 7875451 (70.36%) |
| Total | 2735479 (100%) | 11193412 (100%) |
Identification of miRNA* and 6 novel miRNAs
A total of 17,535 un-annotated unique reads (out of 30,558 unique ones) that can match elements of the S. japonicum genome were marked as potential novel miRNA candidates. The secondary structure of the inverted repeat predicted by Mfold and evaluated by MirCheck showed that 6 conserved reads were found (Additional files 3, 4). Many miRNAs were found in multiple locations of the S. japonicum genome, for example, a novel miRNA named cis-miR-001, corresponds to 14 different locations on different chains of the genome (Additional file 5). Although we found the homologs of known miRNA* of other organisms, we did not obtain any miRNA* sequences of this 6 novel miRNAs in the C. sinensis data set. The sequences and locations of the 6 predicted novel reads are shown in Table 2 and the predicted stem-loop structures for their miRNA precursors are shown in Figure 2.
Table 2.
Sequences of the six novel miRNAs identified in Clonorchis sinensis and their location within the published Schistosoma japonicum genome
| miRNA | Sequence (5'-3') | Size | Locia | Countb | ΔGc | Express leveld |
|---|---|---|---|---|---|---|
| cis-mir-001 | UGGAAAAGAGAUACGGCUGCU | 21 | 14 | 12 | -23.1 | 0.30 ± 0.07 |
| cis-mir-002 | CUGGUCAUCAUCAUCAUCAUA | 21 | 1 | 5 | -23.0 | 0.10 ± 0.02 |
| cis-mir-006 | UAUCACAGCCGUGCUUAAGGGC | 22 | 1 | 108 | -28.4 | 0.001 ± 0.00 |
| cis-mir-010 | UAUUAUGCAACGUUUCACUCU | 21 | 1 | 8 | -37.9 | 0.02 ± 0.01 |
| cis-mir-018 | GAGAGAUUUGUGGAUACCUU | 20 | 2 | 13 | -21.9 | 0.0005 ± 0.00 |
| cis-mir-019 | UAGAGGAAUUGACGGAAGGGCA | 22 | 1 | 5 | -19.9 | 67.32 ± 12.87 |
a Location number of the miRNA sequence with the published genome sequence of S. japonicum of LSBI, Shanghai.
b The number of each miRNA appeared in the clean reads.
c ΔG means the energy of pre-miRNA hairpin, kcal/mol.
d The expression levels of the six novel miRNA relative to actin gene, and the data represent the means and standard deviation (SD) for triplicate reactions independently.
Figure 2.
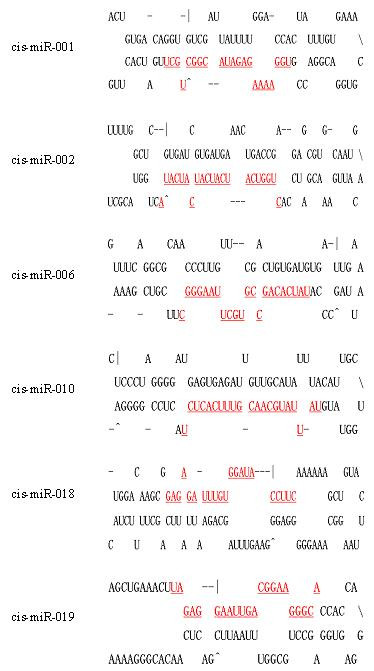
The predicted stem-loop structures for the six novel miRNA precursors in Clonorchis sinensis. The mature miRNA sequences are shown in red and underlined. The actual size of each putative precursor was not identified experimentally and may be slightly shorter or longer than represented. There are two possible stem-loop structures for cis-miR-006 and three predicted stem-loop structures for cis-miR-019, the structures of these two precursors shown in this figure are the ones recommended by the software of Mfold.
Most miRNA* homologs had only one copy in the C. sinensis dataset and the most abundant unique reads was homologs of gga-miR-1627* with only 238 counts (Additional file 6). Although there were abundant miRNA* sequences in Caenorhabditis elegans and other organisms in Sanger miRBase [14] (Release 13), all these miRNA* homologs belonged to different kinds of organisms including vertebrates, insects, virus and some coelomates, such as Amphimedon queenslandica, Schmidtea mediterranea, Locusta migratoria, Xenopus tropicalis, Mus musculus and Gallus gallus, with the exception of parasites and nematodes including Schistosoma mansoni, S. japonicum, C. elegans, C. briggsae and all kinds of mosquitoes including Anopheles gambiae, Aedes aegypti and Culex quinquefasciatus, which were deposited in the database (Additional file 6). It was reported that though the miRNA: miRNA* duplex were complementary, their base-pairing was imperfect and miRNA* was less stable than the mature miRNA [15]. The phenomenon above might indicate a fast degradation mature mechanism of miRNA* in parasites, at least in C. sinensis.
The phylogenetic evolution of miRNAs
Some families included many members and showed distribution bias in the C. sinensis dataset. Totally, 284 conserved miRNAs families were found in C. sinensis out of 2,093,879 reads (with 62,512 unique sequences). These conserved families were presented in large category of vertebrates, insects and nematodes, and can be sorted into 6 groups based on their phylogenetic distribution. Fifty conserved miRNA families are showed in Figure 3a as examples. Four families were found to present among vertebrates, insects and nematodes, including let-7, miR-1, miR-34 and miR-124; Some families were present in vertebrates and insects, but no nematodes; Some were restricted respectively to vertebrates, insects, and invertebrates (insects and nematodes); while some were nematode-specific. This phenomenon showed that C. sinensis miRNAs were distributed widely, some of which specifically belonged to other animals and there might be a redundant miRNA expression in C. siensis. It was reported that there were 50 conserved families and 185 potential locust-specific miRNA families in locust, and these families can only be divided into 4 groups [15]. It was also reported that 16 miRNAs belonged to 13 miRNA families in S. japonicum [16], but no detailed information about miRNA families' distribution was reported. Considering the complex parasitic life of C. sinensis and the key regulation function of miRNAs, possessing miRNAs of different kinds of animals would be a perfect strategy for its parasitic life, which could help them to adapt and modify the host and parasitic environments quickly and conveniently. On the other hand, for a large number and kinds of miRNA expressed, nucleotides of miRNA* need to be reused quickly, and this might be the reason why there were so little miRNA* sequences found in this kind of parasite. It is known that C. sinensis is parasitic in animal hosts such as snails, cats, dogs, fishes, mice and human beings. However, due to the widely distribution of its miRNA families, we believe that this parasite can easily enlarge its host range when the parasitic situation is convenient.
Figure 3.
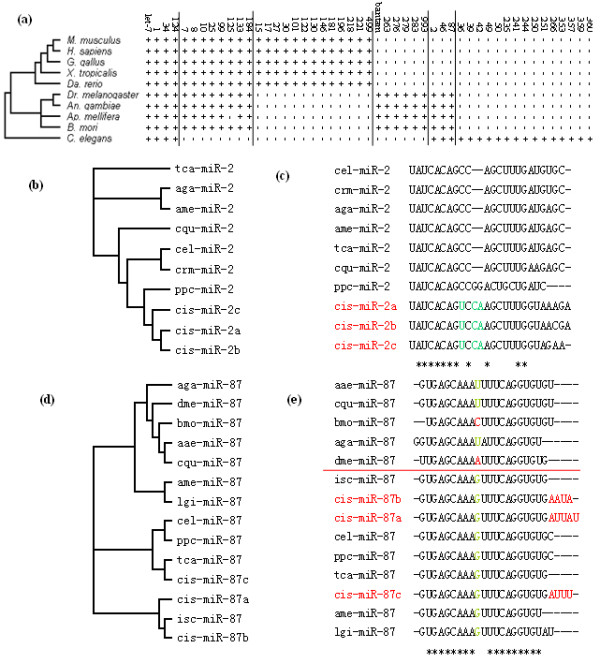
Phylogenetic evolution of 50 conserved miRNA families in Clonorchis sinensis. (a) Phylogenetic distribution of 50 conserved miRNA families of C. sinensis. A plus (+) symbol indicates this miRNA family exists in the species named on the left. (b and c) Phylogenetic analysis of miR-2 family shows middle evolution approach of miRNA in C. sinensis. (d and e) Phylogenetic analysis of miR-87 family shows another evolution approach of miRNA in C. sinensis which is conserved in middle and changed in the tail.
Categorization of conserved miRNAs indicated that miRNAs of C. sinensis were still in the process of evolution. Some of the miRNAs belonged to ancient families, such as miR-1 and miR-34; while some others appeared to be much younger, such as C. sinensis specific miRNAs. The innovation was concentrated along three branches of the phylogenetic tree leading to bilaterians, insects and coelomates. Age differences indicated that there was an ongoing process of miRNA evolution and the birth and death of a kind of miRNA family was a common phenomenon [15].
There might be two innovation approaches of miRNA in C. sinensis, including evolution in the middle and tail. We randomly elected 4 conserved families, miR-2, miR-7, miR-133 and miR-87, and analyzed all of the members of each in miRBase. miR-7 and miR-133 belonged to vertebrates, insects and nematodes, while miR-2 and miR-87 were present in invertebrates only (Figure 3a). If a miRNA family of C. sinensis had more than one of its members, we chose the most abundant 2 or 3 ones, for they were the main part of the family. Phylogenetic analysis of miR-2 family showed that the cis-miR-2 can be easily found for its substitutions and insertions in the middle of miRNA, just behind the "seed region" (Figure 3c); In contrast, the cis-miR-87 was conserved in the middle, and mosquitoes (above red line) and some other insects including B. mori and D. melanogaster innovated at the position. However, it can be found that cis-miR-87 evolved from the tail (Figure 3e). Different members of the cis-miR family showed different innovation rate indicated by phylogenetic trees (Figure 3b, d). The same phenomenon can also be found in families of miR-7 and miR-133 (Additional file 7). It was found that the seed regions of all the species were conserved, while one nucleotide right behind the seed region was changed from U to G and another one was deleted in cis-miR-7 of miR-7 family. In miR-133, one nucleotide was changed from U to C in the middle which was the same as sja-miR-133 (S. japonicum), and another one was changed from U to A which was the same as bma-miR-133 (B. malayi). We supposed that there might be a kind of site-directed evolution and mutagenesis for parasitic life in parasites, however, more miRNA information of other kinds of parasites are needed, which are absent in public databases at present.
Strong family member bias of miRNAs in C. sinensis
Almost one third of the reads belonged to miR-71 family with a total percentage of 33.24%, which included miR-71 (26.79%), miR-71a (0.01%), miR-71b (0.03%) and miR-71c (6.41%). It was followed by the family miR-2 with most of the reads focusing on miR-2b. This bias phenomenon has also been found in other families, such as miR-277 and miR-1. Except for miR-1 which was common in different kinds of animals, miRNAs including miR-71, miR-2 and miR-277 all belonged to invertebrates (insects and nematodes).
Some families included more members than others. let-7 was the second miRNA found in nematode C. elegans in 2000 [17] and it was found that the let-7 family was the largest one among the conserved miRNAs of C. sinensis, including 15 members from let-7a to let-7j and star sequences such as let-7b* and let-7g*. The miR-1422 family was the second largest family represented in the current dataset, which included 13 members. The reads in these families showed strong bias. For example, 95.12% of the reads focused on let-7 in the let-7 family.
Expression predominance of some miRNAs in C. sinensis
Some kinds of miRNAs were expressed with high predominance. miR-71 had the most abundant reads accounting for 26.79% (597,871) of the total reads. It was followed by miR-277b with a percentage of 7.97% (98,845 reads). The third most abundant miRNA is miR-71c with a percentage of 6.41% (Additional file 8). The miRNA named lin-4, which was firstly found in the nematode C. elegans in 1993 [18], was also found in C. sinensis (with only 5 reads). Some other miRNAs were not found in C. sinensis, including miR-40, miR-46, miR-79, and miR-103.
There were 756 kinds of miRNA with copy numbers (reads) fewer than 1,000, and 45 kinds of them with only 1 copy. Seventy-eight kinds of miRNAs had reads of between 1,000 and 10, 000, while there were only 30 kinds of miRNA with copy numbers between 10,000 and 100,000. When the copy numbers were higher than 100,000, only 4 kinds of miRNA were represented (miR-71, miR-277b, miR-71c and miR-215). This expression predominance phenomenon was also found in the 6 novel miRNAs. For example, there were 108 copies of cis-miR-006, while only 5 copies of cis-miR-002 and cis-miR-019 were found, respectively (Table 2).
Nucleotides bias of miRNAs in C. sinensis
The first nucleotide bias analysis of the 2,093,879 reads revealed that nucleotide uracil (U) was the most frequently used first nucleotide in miRNAs of C. sinensis (70.07% incidence), while G or C was seldom used as the first nucleotide with only 10.31% and 3.32%, respectively (Figure 4). (A+U) was found most abundantly with a percentage of 86.37% on average, reaching a percentage of 95.75% and 95.23% at the 22 and 23 nt positions, respectively (Figure 4).
Figure 4.
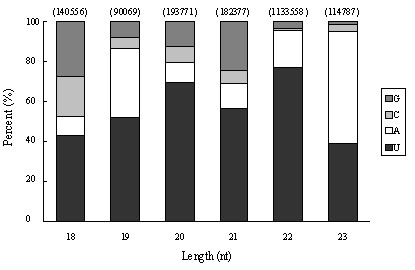
Analysis of first nucleotide bias of miRNAs in Clonorchis sinensis.
Nucleotide bias analysis at each position showed that A and U mainly appeared at the beginnings and the ends of reads, while C and G occupied a very high percentage at the second position and the positions of 6-9th, which belonged to the "seed region" in other species [19,20]. Particularly at the beginning of seed region, the G+C content reached as high as 80.06% (Additional file 9). For a broad range of miRNA families which were presented in C. sinensis (Figure 3a), the seed regions of them might tend to be conserved. This speculation can be proven by the phylogenetic analysis of the two families of miR-2 and miR-87 (Figure 3). Although there were innovations to the families of miR-2 and miR-87 in C. sinensis, nucleotide change only happened in the middle and the end. Considering the complex life history of the parasite, keeping steady seed region and flexible in other places of miRNA might be another strategy for the parasite to adapt to different parasitic situations in different tissues of different hosts.
The analysis showed that U had a high frequency in the 1st, 14th and 22nd positions with percentages of 68.01%, 61.38% and 66.43% respectively, while it seldom appeared at the 2nd and 24th positions with percentages of 3.80% and 2.44%. (A+U) was distributed mainly in the front of reads, at the first and 3-5th nucleotides (86.76%, 80.67%, 69.92%, and 79.84% respectively) but less at the second position (19.94%). C had the lowest percentage (1.73%) at 15th position. G showed high percentage incidence at the positions of almost every 3 nucleotides, such as 6th, 12th and 18th with 69.54%, 50.77% and 58.61%, respectively, and it seldom appeared at the end positions, such as the 22nd position (0.93%). The highest percentage of (G+C) was located at the second position with 80.06%, and it also predominated at the positions of 6-9th and 16-18th. The phenomenon that most A and U were distributed in the front of reads with G and C being focused on the 6-18 positions might be concerned with the mechanisms of miRNA action, such as binding with the targets for gene regulation.
Quantification of C. sinensis miRNA expression
Using the modified stem-loop RT-PCR, the relative expression levels of the six novel miRNAs relative to the actin gene were calculated (Additional file 10, Table 2, expression level). Among them, cis-miR-019 showed a high expression level while that of cis-miR-018 and cis-miR-006 were very low.
Discussion
miRNAs are now considered as key regulators of gene expression at the post-transcriptional level and perform a variety of significant functions within cells such as regulation of growth, metabolism, development and cell differentiation [21-23]. Due to the complex life cycle of parasites with several developmental stages in vertebrate and invertebrate hosts, it is particularly important to elucidate the roles of miRNAs in the growth and development of parasites and their abilities to regulate infection of mammalian hosts. miRNAs are involved not only in the normal functioning of eukaryotic cells, but also associated with dysregulation during disease. miRNAs can be used as potential new tools for disease diagnostics and gene therapy [24] and a manually curated database (miR2Disease) is publicly available aiming at documenting known relationships between miRNA dysregulation and human disease [25].
miRNAs in C. sinensis of zoonotic significance were identified and characterized by deep sequencing in the present study. We found that the percentage of reads matching the S. japonicum genome was very low (19.46%) and only 30,558 unique reads out of 2,735,479 were perfectly matched, whereas this figure can be as high as 70.5% or higher in some other species [26]. The most likely reason for this phenomenon might be that the reference genome used for matching analysis was the S. japonicum genome, rather than C. sinensis, for the C. sinensis genome (or other species of Opisthorchiidae) was not available at present. Although C. sinensis and S. japonicum belong to a common phylogenetic group (Trematoda), there are some significant differences between them, and some miRNAs appeared to be species-specific [27].
The distinguishing characteristic of miRNAs from other endogenous small RNAs is that miRNA precursors have the inverted repeat sequence that can form hairpin structures [28]. Six novel miRNAs were predicted from 17,535 un-annotated genome-matched unique reads. We cannot exclude the possibility that there might be more miRNA types in C. sinensis adults.
Amongst the conserved miRNAs of C. sinensis, bias was found within both kinds of families and family members. The miR-71 and its families possessed the highest proportion of total reads, and the miR-71 family was conserved in S. mansoni, S. japonicum, Ixodes scapularis, and Anopheles gambiae being indexed by Sanger miRBase, which indicated that miR-71 is essential for the life of this flatworm. Like miR-71, some miRNAs had particularly high copies in C. sinensis, including miR-277b, miR-71c and miR-215. Considering the living environment of the adult parasites in the biliary tract of the hosts, as well as the normal function of miRNAs, we inferred that these families might be mainly involved in metabolism of the worm.
Nucleotides A and U were distributed mainly in the front of reads including the first and the 3-5th nucleotides with the exception of the second position (80.06% of G+C). U was the dominant nucleotide in mature miRNAs, especially in the first nucleotide position, and it showed a high frequency in the 1st, 14th and 22nd position, almost at the beginning, the middle, and the end of reads. It was reported by Zhang et al. (2009) that the 1st, 9th and the terminal positions were enriched with U and the 1st and 9th positions were the limits of the "seed region" of a miRNA, which was responsible for targeting mRNAs for gene regulation [20]. We showed a similar result in C. sinensis miRNAs at the first and the end positions, but there was only 20.13% on average at the ninth position, while it was 50.66% and 61.38% respectively at the 11th and the 14th positions. At the positions of 6-9th, G or C occupied a high percentage. Because the previously analyzed miRNAs were mainly derived from vertebrates including human, rat, mouse and pig, and only a few parasites were involved, we cannot exclude the possibility of species- or even order-specific differences and there might be a different or shifted "seed region" for miRNAs in C. sinensis compared with other species [20].
Conclusions
The present study represented the first large scale characterization of C. sinensis miRNAs, which will help us understand the complex life cycle of this zoonotic parasite, which in turn may have implications for the development of novel approaches for the effective control of this parasite. These results will also assist the miRNA studies of other related species such as Opisthorchis felineus and O. viverrini of human and animal health significance.
Methods
Parasites
Adults of C. sinensis were collected from the bile ducts of 8 wild cats obtained in the suburbs of Guangzhou, China, with 20 worms in average from each cat. The worms were immediately transferred to sterile physiological saline (37°C) in a sterile beaker, followed by washing five times with saline on a rotary shaker to remove contamination from the hosts. Thereafter, the flukes were transferred to Dulbecco's modification of Eagle's medium (DMEM) and incubated at 37°C with 10% CO2 for 3 h, allowing the flukes to regurgitate the gut contents until the flukes appeared pale and uniform. The flukes were then transferred to RNase-free 1.5 ml screw-top cryotube containing RNAlater (Sigma) and kept at 4°C for overnight, followed by storage at -70°C.
The maintenance and care of animals used in this study were handled in strict accordance with good animal practice as defined by the relevant national and/or local animal welfare bodies, and all animal work was approved by the appropriate committee.
RNA preparation
Total RNA from flukes was prepared using Trizol reagent (Invitrogen) according to the manufacturer's protocol with some modifications. Briefly, at the step of precipitation, 100% and 50% isopropanol were used to gain a clear mix solution, followed by incubation for 1 h at -70°C instead of 5 min at room temperature to enhance the precipitation of low-molecular-weight (LMW) RNAs. The purity and integrity of total RNA were examined by standard agarose gel electrophoresis, and the concentration was determined using a BioPhotometer (Eppendorf). The purified total RNA was stored at -70°C until use.
Small RNA isolation and high-throughput sequencing
The small RNA isolation was performed as described previously [29]. RNA fragments of 20-30 bases long were isolated and purified from 10 μg total RNA using a Novex 15% TBE-Urea gel. The 5' and 3' adaptors (Illumina) were added to the ends of fragments. Reverse transcription PCR (RT-PCR) was performed using a RT-PCR kit (Invitrogen). The fragments were purified using a 6% TBE PAGE gel and used for high-throughput sequencing with a Solexa sequencer at Huada Genomics Institute Co. Ltd, China. All the gels and kit for small RNA purification and amplification were bought from Invitrogen Co. Ltd.
Computational analysis
After masking of adaptor sequences and removal of redundancy and reads smaller than 18 nt as well, the cleans reads were screened against GenBank and Rfam database (version 9.0) http://www.sanger.ac.uk/software/Rfam/mirna to remove non-coding RNA, such as rRNA, tRNA, snRNA, snoRNA, and other ncRNA. The sequences of candidate precursors were analyzed using RepeatMasker http://www.repeatmasker.org to eliminate the repetitive sequences and then the left reads were searched against the Sanger miRBase (version 13.0) to identify the conserved miRNAs. Based on the nomenclature of miRNAs, reads showing high similarity to known miRNAs of other organisms (mismatches ≤ 2) were classified into the same miRNA family [15,30]. The family distribution of conserved miRNA and the nucleotide bias were gathered statistically to analysis the expression and coding characters of miRNAs. Reads that cannot match any database above were marked as unannotation. Cleans reads were then mapped onto the genome of Schistosoma japonicum http://lifecenter.sgst.cn/schistosoma/cn/genomeProject.do using the program of Short Oligo nucleotide Analysis Package (SOAP) [31]. The perfectly matched unannotation reads were predicted with Mfold http://www.bioinfo.rpi.edu/applications/mfold and then evaluated by MirCheck. Thereafter, the precursors (hairpin) of miRNAs were inspected manually in order to remove false predictions. Predicted precursors that miRNAs and miRNA* can be found in its both arms were deemed as high probability and stem-loop hairpins were considered typical when the mature miRNAs present in one arm instead of loop of hairpin precursors and with free energy hybridization lower than -18 kcal/mol.
Analysis of novel miRNA expression
Stem-loop real-time reverse transcription polymerase chain reaction (RT-PCR) with SYBR Green was used for the analysis of novel miRNA expression in C. sinensis adults. The stem-loop primers were used to quantify the miRNA expression because it can provide more specificity and sensitivity than linear primers [32]. All of the primers were synthesized by Shenggong Co, Ltd., China.
Real-time quantitative PCR was performed using an ABI PRISM® 7300 Sequence Detection System and SYBR Green PCR Master Mix (TOYOBO) in a 20 μl reaction. All reactions were carried out in triplicate. The PCR mix included 5 μl cDNA for each miRNA (in 1:20 dilution), 5 μM forward and reverse primers, respectively, 10 μl 2× SYBR Green PCR Master Mix. The C. sinensis actin gene (EU109284) was used as the endogenous control. The primer pairs were as follows: forward 5'-ATGGGTGATGAGGACGTTGCAGCT-3' and reverse 5'-CATGATCGAGTTGTA CG TCGTCTC-3' [33]. The cycle conditions were as follows: 95°C 10 min, followed by 40 cycles of 95°C for 15 s, 65°C for 30 s, and 72°C for 30 s. The threshold cycle (Ct) was defined as the cycle number at which the fluorescence intensity passed a predetermined threshold. The quantification of each miRNA relative to actin gene was calculated using the equation: N = 2-ΔCt, ΔCt = CtmiRNA-Ctacin [34,35].
Authors' contributions
XQZ and MJX conceived and designed the experiments. MJX, XQC, CY and XHH performed the experiments. MJX, QL, AJN, RQL and ZGY analyzed the data. HQS and XHH contributed reagents/materials. MJX, QL, AJN and XQZ wrote the manuscript. All authors read and approved the final manuscript.
Authors' information
1Department of Parasitology, College of Veterinary Medicine, South China Agricultural University, Guangzhou, Guangdong Province 510642, PR China. 2State Key Laboratory of Veterinary Etiological Biology, Key Laboratory of Veterinary Parasitology of Gansu Province, Lanzhou Veterinary Research Institute, CAAS, Lanzhou, Gansu Province 730046, PR China. 3Laboratory of Parasitology, Veterinary Institute, AMMS, 1068 Qinglong Road, Changchun 130062, PR China. 4Parasitology Division, Moredun Research Institute, Pentlands Science Park, Midlothian EH26 0PZ, Scotland. 5Zhongshan Entry-Exit Inspection and Quarantine Bureau, Zhongshan, Guangdong Province 528403, PR China. 6College of Animal Science and Technology, Yunnan Agricultural University, Kunming, Yunnan Province 650201, PR China.
Supplementary Material
Analyzing flowchart of Clonorchis sinensis miRNAs.
Known miRNAs of Clonorchis sinensis.
Six predicted novel miRNAs in Clonorchis sinensis.
Detailed information of the six novel miRNAs in Clonorchis sinensis.
Precusors of cis-miR-001 in Clonorchis sinensis.
Known miRNA star homologs in Clonorchis sinensis.
Phylogenetic analysis of the miR-7 and miR-133 families in Clonorchis sinensis.
The miRNA distribution in Clonorchis sinensis.
The nucleotide bias percentage at each position in miRNAs of Clonorchis sinensis.
The amplification and melting curves of the six novel miRNAs of Clonorchis sinensis by Real-Time quantitative PCR.
Contributor Information
Min-Jun Xu, Email: mjxu@yahoo.cn.
Quan Liu, Email: liuquan1973@hotmail.com.
Alasdair J Nisbet, Email: Alasdair.Nisbet@moredun.ac.uk.
Xian-Quan Cai, Email: lin0127@126.com.
Chao Yan, Email: yanchao6957@163.com.
Rui-Qing Lin, Email: rqlin@scau.edu.cn.
Zi-Guo Yuan, Email: zxx_yzg@yahoo.com.cn.
Hui-Qun Song, Email: songhuiqun@scau.edu.cn.
Xian-Hui He, Email: nswssda@163.com.
Xing-Quan Zhu, Email: xingquanzh@scau.edu.cn.
Acknowledgements
This work was supported by the Program for Changjiang Scholars and Innovative Research Team in University (Grant No. IRT0723), the Yunnan Provincial Program for Introducing High-level Scientists (Grant No. 2009CI125) and the State Key Laboratory of Veterinary Etiological Biology, Lanzhou Veterinary Research Institute, CAAS to XQZ, and China Postdoctoral Science Foundation (Grant No. 20090460064) to MJX.
References
- Lun ZR, Gasser RB, Lai DH, Li AX, Zhu XQ, Yu XB, Fang YY. Clonorchiasis: a key foodborne zoonoses in China. Lancet Infect Dis. 2005;5:31–41. doi: 10.1016/S1473-3099(04)01252-6. [DOI] [PubMed] [Google Scholar]
- Lee M, Chung YB, Lee SK, Chung BS, Li S, Choi MH, Hong ST. Identification of a Clonorchis sinensis gene encoding an antigenic egg protein. Parasitol Res. 2005;95:224–226. doi: 10.1007/s00436-004-1277-3. [DOI] [PubMed] [Google Scholar]
- Chai JY, Darwin Murrell K, Lymbery AJ. Fish-borne parasitic zoonoses: status and issues. Int J Parasitol. 2005;35:1233–1254. doi: 10.1016/j.ijpara.2005.07.013. [DOI] [PubMed] [Google Scholar]
- Fan PC. Viability of metacercariae of Clonorchis sinensis in frozen or salted freshwater fish. Int J Parasitol. 1998;28:603–605. doi: 10.1016/S0020-7519(97)00215-4. [DOI] [PubMed] [Google Scholar]
- Sripa B. Pathobiology of opisthorchiasis: an update. Acta Trop. 2003;88:209–220. doi: 10.1016/j.actatropica.2003.08.002. [DOI] [PubMed] [Google Scholar]
- Shin HR, Oh JK, Masuyer E, Curado MP, Bouvard V, Fang YY, Wiangnon S, Sripa B, Hong ST. Epidemiology of cholangiocarcinoma: an update focusing on risk factors. Cancer Sci. 2010;101(3):579–85. doi: 10.1111/j.1349-7006.2009.01458.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MG, Lu Y, Hua X, Mott KE. Progress in assessment of morbidity due to Clonorchis sinensis infection: a review of recent literature. Trop Dis Bull. 1994;91:R7–R65. [Google Scholar]
- Hussain M, Taft RJ, Asgari S. An insect virus-encoded microRNA regulates viral replication. J Virol. 2008;82:9164–9170. doi: 10.1128/JVI.01109-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhoades MW, Reinhart BJ, Lim LP, Burge CB, Bartel B, Bartel DP. Prediction of plant microRNA targets. Cell. 2002;110:513–520. doi: 10.1016/S0092-8674(02)00863-2. [DOI] [PubMed] [Google Scholar]
- Aravin AA, Lagos-Quintana M, Yalcin A, Zavolan M, Marks D, Snyder B, Gaasterland T, Meyer J, Tuschl T. The small RNA profile during Drosophila melanogaster development. Dev Cell. 2003;5:337–350. doi: 10.1016/S1534-5807(03)00228-4. [DOI] [PubMed] [Google Scholar]
- Bentwich I, Avniel A, Karov Y, Aharonov R, Gilad S, Barad O, Barzilai A, Einat P, Einav U, Meiri E, Sharon E, Spector Y, Bentwich Z. Identification of hundreds of conserved and nonconserved human microRNAs. Nat Genet. 2005;37:766–770. doi: 10.1038/ng1590. [DOI] [PubMed] [Google Scholar]
- Zhang B, Wang Q, Pan X. MicroRNAs and their regulatory roles in animals and plants. J Cell Physiol. 2007;210:279–289. doi: 10.1002/jcp.20869. [DOI] [PubMed] [Google Scholar]
- Lin WC, Li SC, Lin WC, Shin JW, Hu SN, Yu XM, Huang TY, Chen SC, Chen HC, Chen SJ, Huang PJ, Gan RR, Chiu CH, Tang P. Identification of microRNA in the protist Trichomonas vaginalis. Genomics. 2009;93:487–493. doi: 10.1016/j.ygeno.2009.01.004. [DOI] [PubMed] [Google Scholar]
- Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools formicroRNA genomics. Nucleic acids research. 2008. pp. D154–158. [DOI] [PMC free article] [PubMed]
- Wei Y, Chen S, Yang P, Ma Z, Kang L. Characterization and comparative profiling of the small RNA transcriptomes in two phases of locust. Genome Biol. 2009;10(1):R6. doi: 10.1186/gb-2009-10-1-r6. Epub 2009 Jan 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao L, Cai P, Jiang N, Wang H, Chen Q. Identification and characterization of microRNAs and endogenous siRNAs in Schistosoma japonicum. BMC Genomics. 2010;11:55. doi: 10.1186/1471-2164-11-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, Rougvie AE, Horvitz HR, Ruvkun G. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature. 2000;403:901–906. doi: 10.1038/35002607. [DOI] [PubMed] [Google Scholar]
- Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–854. doi: 10.1016/0092-8674(93)90529-Y. [DOI] [PubMed] [Google Scholar]
- Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- Zhang B, Stellwag EJ, Pan X. Large-scale genome analysis reveals unique features of microRNAs. Gene. 2009;443:100–109. doi: 10.1016/j.gene.2009.04.027. [DOI] [PubMed] [Google Scholar]
- Lim LP, Glasner ME, Yekta S, Burge CB, Bartel DP. Vertebrate microRNA genes. Science. 2003;299:1540. doi: 10.1126/science.1080372. [DOI] [PubMed] [Google Scholar]
- Wienholds E, Plasterk RH. MicroRNA function in animal development. FEBS Lett. 2005;579:5911–5922. doi: 10.1016/j.febslet.2005.07.070. [DOI] [PubMed] [Google Scholar]
- Du T, Zamore PD. Beginning to understand microRNA function. Cell Res. 2007;17:661–663. doi: 10.1038/cr.2007.67. [DOI] [PubMed] [Google Scholar]
- Zhang B, Farwell MA. microRNAs: a new emerging class of players for disease diagnostics and gene therapy. J Cell Mol Med. 2008;12:3–21. doi: 10.1111/j.1582-4934.2007.00196.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q, Wang Y, Hao Y, Juan L, Teng M, Zhang X, Li M, Wang G, Liu Y. miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 2009;37:D98–104. doi: 10.1093/nar/gkn714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue LJ, Zhang JJ, Xue HW. Characterization and expression profiles of miRNAs in rice seeds. Nucleic Acids Res. 2009;37:916–930. doi: 10.1093/nar/gkn998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit E, Linsen SE, Cuppen E, Berezikov E. Repertoire and evolution of miRNA genes in four divergent nematode species. Genome Res. 2009;19:2064–2074. doi: 10.1101/gr.093781.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Lau NC, Lim LP, Weinstein EG, Bartel DP. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science. 2001;294:858–862. doi: 10.1126/science.1065062. [DOI] [PubMed] [Google Scholar]
- Chen X, Li Q, Wang J, Guo X, Jiang X, Ren Z, Weng C, Sun G, Wang X, Liu Y, Ma L, Chen JY, Wang J, Zen K, Zhang J, Zhang CY. Identification and characterization of novel amphioxus microRNAs by Solexa sequencing. Genome Biol. 2009;10(7):R78. doi: 10.1186/gb-2009-10-7-r78. Epub 2009 Jul 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Li Y, Kristiansen K, Wang J. SOAP: short oligonucleotide alignment program. Bioinformatics (Oxford, England) 2008;24(5):713–714. doi: 10.1093/bioinformatics/btn025. [DOI] [PubMed] [Google Scholar]
- Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee DH, Nguyen JT, Barbisin M, Xu NL, Mahuvakar VR, Andersen MR, Lao KQ, Livak KJ, Guegler KJ. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005;33:e179. doi: 10.1093/nar/gni178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Na BK, Kang JM, Sohn WM. CsCF-6, a novel cathepsin F-like cysteine protease for nutrient uptake of Clonorchis sinensis. Int J Parasitol. 2008;38:493–502. doi: 10.1016/j.ijpara.2007.09.001. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Xue X, Sun J, Zhang Q, Wang Z, Huang Y, Pan W. Identification and characterization of novel microRNAs from Schistosoma japonicum. PLoS One. 2008;3:e4034. doi: 10.1371/journal.pone.0004034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Analyzing flowchart of Clonorchis sinensis miRNAs.
Known miRNAs of Clonorchis sinensis.
Six predicted novel miRNAs in Clonorchis sinensis.
Detailed information of the six novel miRNAs in Clonorchis sinensis.
Precusors of cis-miR-001 in Clonorchis sinensis.
Known miRNA star homologs in Clonorchis sinensis.
Phylogenetic analysis of the miR-7 and miR-133 families in Clonorchis sinensis.
The miRNA distribution in Clonorchis sinensis.
The nucleotide bias percentage at each position in miRNAs of Clonorchis sinensis.
The amplification and melting curves of the six novel miRNAs of Clonorchis sinensis by Real-Time quantitative PCR.