

Abstract
Trapping membrane proteins in the confines of a crystal lattice obscures dynamic modes essential for interconversion between multiple conformations in the functional cycle. Moreover, lattice forces could conspire with detergent solubilization to stabilize a minor conformer in an ensemble thus confounding mechanistic interpretation. Spin labeling in conjunction with electron paramagnetic resonance (EPR) spectroscopy offers an exquisite window into membrane protein dynamics in the native–like environment of a lipid bilayer. Systematic application of spin labeling and EPR identifies sequence-specific secondary structures, defines their topology and their packing in the tertiary fold. Long range distance measurements (60-80Å) between pairs of spin labels enable quantitative analysis of equilibrium dynamics and triggered conformational changes. This review highlights the contribution of spin labeling to bridging structure and mechanism. Efforts to develop methods for determining structures from EPR restraints and to increase sensitivity and throughput promise to expand spin labeling applications in membrane protein structural biology.
Spin Labeling in the Structural Biology Paradigm: the fourth dimension of protein structure
Membrane proteins are key control points in cell communication, movement of molecules across membrane barriers, flow and use of energy, as well as in triggering the initiation of numerous signaling pathways. After decades of slow progress, completion of genome sequencing projects, advances in protein expression and purification, and technological innovations overcame long-standing barriers and bottlenecks spurring a spectacular acceleration in the pace of membrane protein structure determination. The structures of these high value drug targets are elucidating the architectural principles that define classes of membrane proteins, exposing motifs that determine their stability and enable them to inhabit the lipid bilayer (Bowie, 2001), and unlocking secrets of ion channel selectivity, transporter specificity (Gouaux and Mackinnon, 2005), receptor/ligand interactions (Kobilka and Schertler, 2008) and catalysis in the membrane (Ha, 2007).
Transition from structure to mechanism is the next frontier. While time-averaged crystallographic snapshots frame biochemical and functional data in a structural context, achieving a mechanistic description of biological function requires an understanding of dynamics, the fourth dimension of protein structure. The function of channels, transporters, and receptors is intimately associated with their ability to execute movements that enable opening of a gate, alternate access of a substrate binding pocket to different sides of the membrane, or exposure of signaling sequences. Excursions between these conformers can be thermally activated; a view in stark contrast to the static picture communicated by crystal structures. In some cases, models of conformational changes can be inferred from a “patchwork of different homologs fortuitously crystallized in different states” (Krishnamurthy et al., 2009), but the caveat is that the observed distribution of structures may reflect “the idiosyncrasies of the different homologs” (Rees et al., 2009) rather than different intermediates in the functional cycle. Protein dynamics and conformational sampling can be altered by the crystallization process. Crystal contacts can act as a conformational selectivity filter distorting highly flexible but functionally critical segments and/or stabilizing conformations that may be sparsely populated in solution. Moreover, membrane proteins’ natural milieu is the lipid bilayer, which differs in its physico-chemical properties from detergent micelles, the preferred crystallography solvent. Accentuating this concern, detergent selection criteria often emphasize crystal and diffraction qualities at the expense of functional considerations thus dictating the use of harsh detergents. Together these factors may conspire to cloud the mechanistic interpretation of crystal structures (Cross et al., 2011). A detailed understanding of membrane protein functional cycles requires a description of the nature, amplitude and time scale of conformational equilibria and/or triggered conformational changes in a native-like environment.
Dynamics is the realm of spectroscopy by excellence. Liberated from the confines of the crystal lattice, proteins sample equilibrium dynamic modes or undergo triggered conformational changes. These movements can be probed on a multitude of time scales to determine their amplitude and extent. Although nuclear magnetic resonance allows direct detection of protein dynamics (Mittermaier and Kay, 2009), its promise has been hindered by mediocre sensitivity, the need for isotopic labeling and molecular mass limitations that exclude the vast majority of membrane proteins. In contrast, sensitivity and size are not limiting for probe-based spectroscopic approaches such as fluorescence (Wahl and Weber, 1967) and spin labeling EPR (Hubbell et al., 1996; Ogawa and McConnell, 1967), where proteins can be studied in an environment more closely resembling the native membranes. Both methodologies interpret spectral properties of site-specifically incorporated probes to deduce local structural features. The advent of single molecule fluorescence presents opportunities for the complementary use of the two methods drawing on their unique sensitivities to structure and dynamics. This review focuses on the contribution of spin labeling and EPR to the emerging field of membrane protein dynamics, describing the methodological tool kit and highlighting its application to key systems.
The DEER age of EPR spectroscopy
In parallel to advances in membrane protein structural biology, the EPR tool kit was revolutionized by the development of pulsed EPR methods to measure long range distances between spin labels. As originally conceived by McConnell (Ogawa and McConnell, 1967), spin labeling EPR analysis relied on the dependence of spin label dynamics, or its mobility relative to the protein, on local conformation. Capitalizing on advances in site-directed mutagenesis, Hubbell and colleagues introduced the concept of nitroxide scanning wherein spin labels are sequentially introduced along a stretch of residues (Altenbach et al., 1990; Mchaourab et al., 1996). The parameter set was expanded to include quantitative measurements of spin label solvent accessibility to membrane and water-soluble reagents (Altenbach et al., 2005; Hubbell et al., 1996). Systematic application of this approach, referred to as Site-Directed Spin Labeling (SDSL) enables the identification of secondary structures and their orientations in the membrane (Hubbell et al., 1996). Rabenstein and Shin (1995) complemented accessibility and mobility with a spectroscopic ruler to measure distances between spin label pairs in the 8-20Å range using the continuous wave (CW) EPR spectrum. Together, the EPR-derived constraints, in the form of sequence-specific secondary structure, topology and tertiary contacts, have been applied successfully to elucidate aspects of membrane protein structure and to detect conformational changes (Reviewed in Hubbell et al., 2000; Hubbell et al., 1996). However, the local nature of mobility and accessibility and the short range of distance constraints limit their utility.
Pulsed EPR methods extend the measurable distance between two electron spins (Figure 1A) up to 60 Å, and in favorable cases to 80 Å, by separating the dipolar term in the spin Hamiltonian for exclusive detection (Borbat et al., 2002; Jeschke and Polyhach, 2007). Although appropriate pulse sequences (e.g. Figure 1B) have long been developed (Milov et al., 1984; Pannier et al., 2000), the widespread application of dipolar EPR spectroscopy was spurred by commercialization of high sensitivity pulsed EPR spectrometers and the model-free analysis of dipolar interactions (Figure 1C) to calculate the distance distribution between two electron spins (Figure 1D) (Chiang et al., 2005; Jeschke et al., 2004). Freed and coworkers (Borbat et al., 2002) developed pulse sequences to detect double quantum coherence between spins which promises an order of magnitude in increased sensitivity. However, double electron-electron resonance (DEER), or pulsed electron double resonance (PELDOR), is the most commonly used method for distance measurements between spin labels.
Figure 1. From raw DEER signals to the distance distribution.

(A) A pair of spin labels (A and B) is depicted on the surface of a membrane protein embedded in a liposome. The spin echo decay has contributions from dipolar coupling between spins on the same protein molecule (rintra; blue arrow) and from intermolecular dipolar coupling between spins on neighboring molecules (rinter; red arrow).
(B) Typical four-pulse DEER sequence. An inversion pulse is applied to spin B at time t while observing the echo of spin A.
(C) Spin echo intensity decays as time t is increased. The observed signal (black) is the result of modulation of echo intensities from intramolecular coupling (blue) and intermolecular coupling, or background decay (red). The decays are based on simulations using the DEER2010 package (Jeschke et al., 2006).
(D) The distribution of distances between spins A and B is derived from the spin echo decay in (C).
(E) Reconstitution in Nanodiscs lowers the effective concentration of proteins by allowing proteins to occupy three dimensions while retaining the lipid bilayer environment.
(F) Increasing the intermolecular distance between spins by using Nanodiscs reduces the contribution of the background decay, relative to that of proteoliposomes, to the spin echo decay.
DEER Analysis of Structure and Conformational Changes
DEER distance distribution
DEER instrumentation and analysis have been extensively reviewed (Jeschke and Polyhach, 2007) and will not be discussed here. Briefly, spin echo decay of spin label A is modulated by intramolecular dipolar interaction with spin label B on the same protein molecule and by intermolecular dipolar interaction with spins A or B on a separate molecule (Figure 1A). The former leads to an oscillating echo decay the period of which directly reflects the average distance (Figure 1C). In contrast, the contribution of intermolecular dipolar interactions, referred to as the background, is an exponential decay that dampens the oscillation. The echo decay is analyzed to remove the background contribution and calculate the probability of a distance between the two spins yielding a distance distribution characterized by the weighted average distance, rav, and a standard deviation σ (Figure 1D). DEER experiments have to be carried out in the solid state, typically in the 50-80K temperature range. The spin label phase memory time, which defines the time scale of the echo decay (x-axis in Figure 1C) and therefore the upper distance range, is too short at room temperature.
Because the distance between spins in neighboring molecules is dependent on protein concentrations, the background decay introduces a tradeoff between the measurable distance range and sensitivity. It is particularly problematic for membrane proteins where high effective concentrations in the two-dimensional environment of a proteoliposome (Figure 1A) accentuate the background contribution (Figure 1, B &E) thereby imposing severe limits on sensitivity, distance range and experimental throughput. To overcome these limitations, Zou and Mchaourab (2010) reported a general methodology which relies on reconstitution of spin labeled membrane proteins into Nanodiscs (also referred to as nanoscale bilayers). These bilayers are a class of soluble nanoscale assemblies of lipids surrounded by a belt of amphipathic protein derived from apolipoprotein A1 (Bayburt and Sligar, 2010). By careful manipulation of the molar ratios between the three components, it is possible to reconstitute a single membrane protein per bilayer disk resetting the dimensionality of the DEER background factor to three (compared to approximately 2 in proteoliposomes) (Figure 1E). The use of Nanodiscs is facilitated by an order of magnitude increase in DEER sensitivity achieved at Q-band frequency (Ghimire et al., 2009) relative to the commonly used X-band frequency. The synergistic convergence of these two technologies overcomes the bottlenecks for widespread application of DEER to sample-limited membrane proteins.
Protein fluctuation dynamics: contribution to DEER distance distributions
Equilibrium fluctuation dynamics refers to thermally driven protein motion occurring on multiple time scales with different amplitudes (Mittermaier and Kay, 2009). It reflects protein excursions between local energy minima and is manifested by dynamic modes of side chain isomerization on the ps-ns scale, ns excursion of flexible loops, and all the way to movement of secondary structures or domains in the μs-s regime. In the solid state (where DEER experiments are carried out), this conformational sampling results in static disorder, provided the freezing process does not trap fluctuating structural elements in a single energy minimum. To the extent that conformations have different average distance between spin labels, the static disorder contributes directly to the width of the distance distribution (σ). It is noted that the solid state nature of the measurement implies that distance distributions contain contributions from all protein dynamic modes regardless of their time scales.
In the absence of protein fluctuations, the intrinsic width of the distance distribution arises from the flexibility of the spin label side chain. The most commonly used spin label, 1-oxyl-2,2,5,5-tetramethylpyrroline-3-methyl-methanethiosulfonate (MTSSL), allow rotations around four internal bonds linking the nitroxide ring to the protein backbone (Mchaourab et al., 1996). Crystal structures of spin labeled T4 lysozyme defined a subset of spin label rotamers some of which are resolved to the nitroxide rings (Langen et al., 2000). Transition between these rotamers can change the distance between the labels thus contributing to the width of the distance distribution. The structures also reveal the potential for direct interaction between the ring and neighboring side chain and main chain atoms potentially biasing the rotamer population and making prediction of the intrinsic width more complex.
Therefore, interpretation of the width of the distance distribution requires untangling the intrinsic contribution from that of protein dynamics. In principle, it is possible to use atomistic molecular dynamics (MD) simulations to sample the rotamer distribution for each label in a pair and obtain the distribution width in the absence of protein dynamics. Despite a number of successful reports (Borovykh et al., 2006; Fajer et al., 2007; Sompornpisut et al., 2008), long computation times, particularly when considered for multiple label pairs, in conjunction with potential imprecision in spin label parametrization hinder routine application of this approach. Moreover, trajectories as long as 100 ns may not be sufficient to efficiently sample the spin label rotamer space (Sezer et al., 2008).
To overcome this problem, Jeschke and co-workers (Polyhach et al., 2011) created a spin label rotamer library from a long MD trajectory thereby circumventing the need for repeated MD simulations for each pair. Instead, the rotamers are evaluated in the protein of interest for their relative energies calculated from a modified Lennard-Jones potential. A simulated distance distribution is thus generated from the pair-wise distances between rotamers weighted by their relative population. This approach has been successfully applied to determine the dimer arrangement of the Na+/H+ exchanger (Hilger et al., 2007) and a transmembrane segment in the proline symporter PutP (Hilger et al., 2009). However, extensive benchmarking is needed to assess whether the rotamer library provides a complete representation of the spin label conformational space.
We have adopted an empirical approach to obtain an estimate of the intrinsic distribution width. For this purpose, ~60 pairs of spin labels were introduced at surface sites in T4 lysozyme (T4L) focusing on the helical C-terminal domain and avoiding regions of the protein affected by the hinge bending motion in solution (Figure 2A) (Kazmier et al., 2010 and unpublished data). Each of the resulting distance distributions was parametrized by rav and σ. The histogram in Figure 2B displays the frequency of a given standard deviation binned every 0.5 Å. Although the sites were selected to be solvent-exposed, the distance distributions are generally narrow, consistent with previous models of limited-amplitude motion of the MTSSL spin label at such sites (Columbus et al., 2001; Mchaourab et al., 1996). Ideally, a similar benchmarking exercise would establish the intrinsic distribution width for a membrane protein model system. However, we note that the spin label mobility is not altered at lipid-facing exposed sites strongly suggesting that the intrinsic distribution width for membrane proteins will have a similar shape to Figure 2B (Altenbach et al., 1990; Dong et al., 2005).
Figure 2. Empirically-determined intrinsic width of distance distributions.

(A) Structure of T4L highlighting representative pairs used for distance measurements between spin labels.
(B) Sigma (σ) values calculated from experimental distance distributions from T4L are shown as a histogram binned at intervals of 0.5Å.
DEER readout of triggered conformational changes
Membrane protein functional cycles require the interconversion between distinct conformations or shifts in preexisting conformational equilibria. Typically, segments of the protein undergo defined motions in response to energy input such as changes in transmembrane voltage, binding or hydrolysis of ATP or binding of ligand or substrates. To the extent that these movements result in changes in residue environment, they also alter the mobility and accessibility of spin labels. In most cases however, these parameters cannot be quantitatively interpreted to reveal the nature and magnitude of the underlying structural changes.
In contrast, protein motions are directly manifested by changes in the average distance and/or the shape and width of the distribution (Illustration in Figure 3). The former reports the amplitude of movement between two most probable conformations of the protein while the latter reflects changes in the underlying conformational ensemble as illustrated below. The simplest interpretation of the DEER data in terms of structural changes requires that the set of spin label rotamers remains unchanged between the different protein conformations. Repacking of the label can lead to changes in rav and/or affect the width of the distance distribution. In general, judicious selection of unconstrained, exposed sites for spin labeling circumvents this confounding factor. The room temperature spin label mobility can be used to confirm the lack of spin label repacking as a result of conformational changes.
Figure 3. DEER detection of triggered conformational changes.

(A) Hypothetical motion of a transmembrane helix (orange) during the transition from State A to State B alters the average distance (rav, arrows) between spin labels. The rotameric ensemble of each label, represented by white sticks, was generated from a rotamer library using the program MMM (Polyhach et al., 2011).
(B) If states A and B are distinct conformers of different energies, the conformational shift will manifest primarily as a change in rav, evident as an increased period of the spin echo decay (inset).
(C) If states A and B represent two conformations present in equilibrium, altering the biochemical conditions will alter the contribution of each distinct conformation (dashed curves) to the distance distribution (green curve).
Strategies for detection and interpretation of conformational changes by spin labeling and EPR will be illustrated through specific examples on transporters, receptors and channels. Despite the relatively young age of biological DEER spectroscopy, there are numerous examples in the literature highlighting the power of this approach to both water-soluble and membrane proteins. Notable among them are studies of membrane associated α-synuclein (Georgieva et al., 2008; Jao et al., 2008), myosin dynamics (Klein et al., 2008; Vileno et al., 2011) and SNARE protein mechanisms (Tong et al., 2009).
Alternating Access of Active Transporters
Active membrane transporters handle vectorial traffic moving energetically uphill against concentration gradients and play critical roles in synaptic transmission, maintenance of ion homeostasis, and the phenomenon of multidrug resistance. The thermodynamics of the problem are rendered favorable through coupling of substrate transport to the direct use of ATP or the discharge of electrochemical gradients. In the parlance of active transport, the conversion of energy input to the work of substrate translocation proceeds through transporter conformational motion that switches the accessibility of substrate binding sites. The spectrum and amplitudes of transporters’ conformational motion and the basis of substrate specificity are exciting frontiers.
Detection of large amplitude conformational changes: ATP-binding cassette transporters
ATP-binding cassette (ABC) transporters, one of the largest families of transporters, harness ATP energy to power the import and export of solutes that range in size from small molecules to entire protein domains (Higgins, 2001). Eukaryotic ABC transporters predominantly extrude hydrophobic molecules and are implicated in resistance to chemotherapy (Higgins, 2001). The functional unit of an ABC transporter consists of two nucleotide-binding domains (NBDs) that bind and hydrolyze ATP each linked to a transmembrane domain (TMD) that provides a transport pathway across the bilayer. Transport entails the transduction of ATP energy to alternately expose the transport pathway to opposite sides of the membrane.
The lipid flippase MsbA from Escherichia coli has emerged as the model system for defining the structural dynamics of ABC exporters. In parallel to crystallographic studies (Ward et al., 2007), extensive spin labeling and EPR analysis illustrates the distinct yet complementary perspective of the two approaches. EPR has also been used to explore the structure and dynamics of the bacterial ABC maltose importer yielding novel insight into its mechanism (Orelle et al., 2010).
Initial spin labeling of MsbA (Dong et al., 2005) targeted the accessibilities and relative proximities of three transmembrane (TM) helices—2, 5, and 6—and adjacent regions of the intracellular domain and periplasmic loops (112 residues). The spin labels revealed the presence of an asymmetric, water-exposed chamber that is open to the cytoplasm in the absence of bound nucleotides. Comparison of mobility and accessibility parameters obtained in the apo, ATP-bound, and in the transition state of ATP hydrolysis revealed that in liposomes, MsbA undergoes substantial conformational changes that occlude the chamber to the cytoplasm and increase hydration in the periplasm (Figure 4A). Although proximity analyses between symmetry-related spin labels were consistent with distance changes of opposite signs on either side of the transporter, the short-range nature of the CW-EPR data did not allow determination of the movement amplitude. Moreover, the EPR constraints indicated that the liposome structure is incompatible with the apo MsbA crystal structure and provided yet another line of evidence for fundamental distortion in the original structures (Chang and Roth, 2001), which were subsequently retracted and corrected. The magnitude of MsbA conformational changes in detergent and liposomes was subsequently determined from analysis of distance changes by DEER. An initial set of mutants strategically distributed in the three domains of MsbA revealed that ATP hydrolysis in the NBD, which engenders a 30-40Å distance change between the two domains, is propagated to the TMD thereby closing the cytoplasmic side and concomitantly opening the periplasmic side (Borbat et al., 2007).
Figure 4. ATP-driven alternating access in MsbA: large distance changes between two distinct conformers.

(A) Spin label accessibility (Π) to NiEDDA was probed at 201 positions (spheres) on TMs 2-6. The change in accessibility (ΔΠ = ΠADP+Vi − Πapo) between the apo and ADP+Vi bound states is depicted by a gradient from red (decreased) to white (no change) to blue (increased) and mapped onto the apo MsbA structure (NBDs and helices not probed are removed for clarity). Inset: Plot of Π values for TM3 residues 136-165 in the apo (black) and ADP+Vi bound (maroon) states.
(B) Representative spin label pairs mapped on the apo and AMP-PNP bound structures. The distance distribution of each pair measured by DEER in the absence of nucleotide (black) or the presence of ADP+Vi (maroon).
The (corrected) MsbA structures reveal that the apo and nucleotide-bound conformations (Ward et al., 2007) are related by a geometric transformation described as alternating access with a twist (structures in Figure 4B). Apo MsbA has an open structure where the two NBDs are separated by 50 Å. The TM helices are arranged in two wings that form a V-shaped chamber open to the cytoplasm and the inner leaflet of the bilayer in agreement with accessibility analysis of spin labels (Dong et al., 2005; Zou and Mchaourab, 2009). In the AMPPNP-bound structure, hereafter referred to as the closed structure, the two NBDs form the canonical ATP dimer while the two TM wings of the transporter pack in the cytoplasm and split in an outward-facing conformation at the extracellular side. To create this opening, a twisting motion repacks the TM helices, changing the identity of the swapped helices between the two monomers.
Extensive DEER analysis compared the pattern of distance changes to that predicted from the open/closed crystallographic transformation with the goal of experimentally verifying its main features and placing the crystal structures in a defined mechanistic context (Zou et al., 2009). Changes in the average distance between spin labels are consistent with the scale of conformational changes implied by the open to closed transition (Figure 4B). However, detailed quantitative comparison was hindered by the low resolution of the open structure which introduces a 5-10Å uncertainty in the predicted distances. Distance distributions at sites in the NBD and coupling loops that transmit the signal of ATP binding/hydrolysis are broad in the apo conformation and their widths decrease in the post-hydrolysis intermediate. The reverse effect is observed on the extracellular side with an increase in conformational flexibility in the post-hydrolysis intermediate. This led Zou et al. (2009) to conjecture that flexibility may be required to accommodate the diverse size and chemical identities of substrates. The systematic analysis of MsbA illustrates the use of spin labeling to discover and map conformational dynamics as well as test specific models inspired by crystal structures.
Conformational equilibria from DEER distance distribution: Na+-coupled leucine transporter
LeuT is a sodium-coupled amino acid transporter from Aquifex aeolicus. Originally thrust into the limelight as a model for human neurotransmitter:sodium symporters (NSS), the frequent observation of its fold in transporters with no obvious evolutionary relationships (Krishnamurthy et al., 2009) suggests that the architecture represents a general mechanism for ion-coupled transport.
The crystal structure of LeuT, determined to remarkably high resolution by Gouaux and coworkers (Yamashita et al., 2005), revealed that leucine is bound in an occluded and dehydrated cavity located near the middle of the bilayer. It is stabilized by two Na+ ions and backbone carbonyls contributed by unwound regions of helices 1 and 6. Approximately 20 Å of highly packed protein block access from the intracellular side constituting a “thick gate”, while the extracellular side is closed off by a “thin gate” consisting of few residues. Despite its high resolution, the mechanistic significance of LeuT crystal structure has been challenged based on the observation that the crystallization detergent, OG, inhibits its functional reconstitution (Quick et al., 2009).
To determine how leucine accesses its binding site from the extracellular milieu, Claxton et al. (2010) placed spin labels at residues in and around the extracellular vestibule and putative permeation pathway. The residues were predicted to be optimal for sensing ion/substrate-induced movement from MD simulations. Patterns of accessibility changes to NiEDDA revealed increased hydration of the extracellular vestibule and Leu permeation pathway in the Na+-bound intermediates (Figure 5A, right). Subsequent Leu binding induces opposite accessibility changes indicating a closing of the extracellular pathway to the transporter (Figure 5A, left).
Figure 5. Shifts in conformational equilibria: Na+ and Leu binding to LeuT.

(A) Spin label accessibility (Π) to NiEDDA was probed at positions (side chains shown) on the extracellular side. The change in accessibility (ΔΠ) between the apo and Na+-bound states or the Na+- and Na+/Leu-bound states is depicted by a gradient from red (decreased) to white (no change) to blue (increased) and mapped onto the Na+/Leu-bound structure of LeuT.
(B) Distance changes between spin labels at positions 309 in EL4 (yellow) and 480 reveal changes in the conformational ensemble in the apo (black), Na+-bound (green), and Na+/Leu-bound (red) states.
The substrate-induced movement in the extracellular vestibule is also manifested by distance changes in spin label pairs. However, in contrast to MsbA, Na+ and/or Na+/Leu binding did not induce large changes in the weighted average distance. In fact, distance distributions in the loops initially targeted by Claxton et al. (2010), illustrated in Figure 5B for the pair 309/480, were broad or consisted of multiple populations. Their width (σ, Figure 5B) indicated fluctuation dynamics beyond the flexibility of the spin labels (Figure 2B); the asymmetric shape suggested contributions from an ensemble of conformations of the extracellular loop EL4a where residue 309 is located.
The equilibrium dynamics of EL4a were established by comparing the shapes and widths in three biochemical conditions. Relative to apo LeuT, Na+ binding broadens the distribution by increasing the probability of LeuT conformers with large separation between the two labels. In contrast, Na+/Leu binding quenches these dynamic fluctuations, presumably arising from EL4a, leading to a closer distance between the labels. The Na+-induced opening of the extracellular vestibule and its subsequent closing by Leu binding is consistent with the pattern of NiEDDA accessibility (Figure 5A). Thus to rationalize the distance distribution, at least two conformations of LeuT in equilibrium have to be postulated. Conceptualization of the problem by an average structure with a corresponding average distance in each intermediate would obscure critical aspects of the mechanism. Moreover, the significance of the average distance is diminished for broad distributions because any one distance only represents a small fraction of the population.
An alternating access mechanism postulates movement on the packed intracellular side of LeuT to create an exit pathway. To define the structural elements involved in substrate release, we introduced spin label pairs positioned to monitor distances between structural elements of the LeuT thick gate (Kazmier and Mchaourab, unpublished data). The pair 7/86, reporting on the distance between the N-terminus and intracellular loop 1 (IL1), reveals clear evidence of fluctuation dynamics on the intracellular side. The bimodal distance distribution in the apo intermediate reflects two distinct LeuT conformations differing by 20 Å in the distance between the spin labels (Figure 6). In one conformation, the average distance is consistent with the crystal structure. Outward movement of the N-terminus relative to IL1 is required to account for the second population in the distance distribution thus defining structural rearrangements in the second LeuT conformation. The widths for both components reflect the expected flexibility of these loops. Ion/substrate binding does not affect the widths; rather it shifts the ratio of the two populations/conformations confirming that they are in equilibrium.
Figure 6. Conformational fluctuations of LeuT intracellular side revealed by DEER and FRET.

Cys at positions 7 and 86 on the intracellular side of LeuT were modified with the Cy-5/Cy-3 FRET pair or with spin labels. Distance distributions from DEER (upper panel) or FRET efficiency histograms from single-molecule FRET (lower panel) were measured in the apo (black), Na+-bound (red), and Na+/Leu-bound (blue) states.
The EPR evidence for conformational equilibrium reinforces conclusions obtained from single molecule fluorescence-resonance energy transfer (FRET) analysis of fluorophores introduced at the same residues (Zhao et al., 2010) (Figure 6). Not only is there a correspondence between low and high FRET states and large and short distances respectively, but the dependence of the population ratio on ion/substrate binding is remarkably similar. While the ergodic theorem establishes correspondence between ensemble and time-averaged properties, the agreement in Figure 6 suggests that “non-ergodic” differences between the two experiments, such as different probes and different temperatures, do not significantly distort the equilibrium. Integrated use of the two techniques should provide new opportunities to spatially (EPR) and temporally (single molecule) characterize conformational fluctuations.
Detection of transport intermediates. Doubly-occluded conformation of Lactose permease
Lactose permease (LacY), a sugar:H+ symporter found in the inner membrane of E. coli, is the paradigm of the major facilitator superfamily (MFS) of secondary transporters, and one of the most thoroughly studied membrane proteins (Guan and Kaback, 2006). Early applications of EPR to LacY used nitroxide scanning of loops to assign the position of the membrane boundary on the TM helices (e.g. Ujwal et al., 1995) and employed dipolar coupling between two spin labels (Wu et al., 1996) or between a spin label and an engineered metal binding site (Voss et al., 2001) to define the pairwise interactions between helices. These qualitative, local distance constraints were generally consistent with later crystal structures of LacY in the open-in state (Abramson et al., 2003).
The greatest mechanistic insight was gained from DEER analysis of sugar-induced conformational changes. Smirnova et al. (2007) introduced nine spin-labeled Cys pairs on the cytoplasmic and periplasmic ends of TM helices predicted to undergo the largest magnitude distance change upon transport turnover. In the presence of a galactoside, distances between spin labels on the cytoplasmic side decreased while distances between spin labels on the periplasmic side increased. This conformational change was interpreted as the transition to an open-out conformation of LacY. Interestingly, distance distributions of label pairs on the cytoplasmic side also revealed a third population of structures consistent with a conformation where the cytoplasmic pathway is partially closed. Overall the DEER data complemented the open-in crystal structure and provided a specific model for alternating access of LacY. The detection of three distinct populations demonstrated the power of this technique to define intermediates in the transport cycle.
Distortion of BtuB Dynamics by Osmolytes and Crystal Contacts
BtuB, the outer membrane E. coli vitamin B12 (CNCbl) transporter, is perhaps the most thoroughly studied example of differences between crystal structures and EPR constraints in lipid bilayers. Substrate translocation is energized by the inner membrane proton potential through coupling to the transperiplasmic protein TonB via a highly conserved N-terminal region on the periplasmic side of BtuB, termed the Ton box. The origin of the controversy is the observation by spin labeling EPR that the N-terminal Ton box undergoes a substrate-dependent conformational transition to generate an unfolded or disordered protein segment (Fanucci et al., 2003). Distance measurements between pairs of spin labels indicate that this transition extends the Ton box 20-30 Å into the periplasmic space (Xu et al., 2006). In contrast, the Ton box remains folded within the transporter in crystal structures of BtuB either in the presence or absence of substrate (Freed et al., 2010). Given its role in energy transduction, defining the conformational dynamics of this segment is critical for a mechanistic understanding of BtuB transport.
To address this controversy, the laboratory of David Cafiso carried out a detailed analysis of the effects of solutes (Kim et al., 2008) and osmolytes (Flores Jimenez et al., 2010) on the dynamics of BtuB, particularly at the Ton box. They also determined crystal structures of a BtuB mutant spin labeled at site 10 in the Ton box (Freed et al., 2010). They found that the EPR-detected, substrate-induced unfolding of this mutant Ton box is blocked in the crystal but not in lipid bilayers (Figure 7). The stabilization of the folded Ton box conformation in the crystal has its origin in an equal energetic contribution from the solutes and osmolytes in the crystallization buffer and from lattice forces. This example narrates a cautionary tale of the ambiguity associated with crystal structures of flexible segments or molecules and provides a compelling rationale for using dynamic spectroscopic approaches to bridge the divide between static structure and function.
Figure 7. Suppression of flexibility in crystals of BtuB.
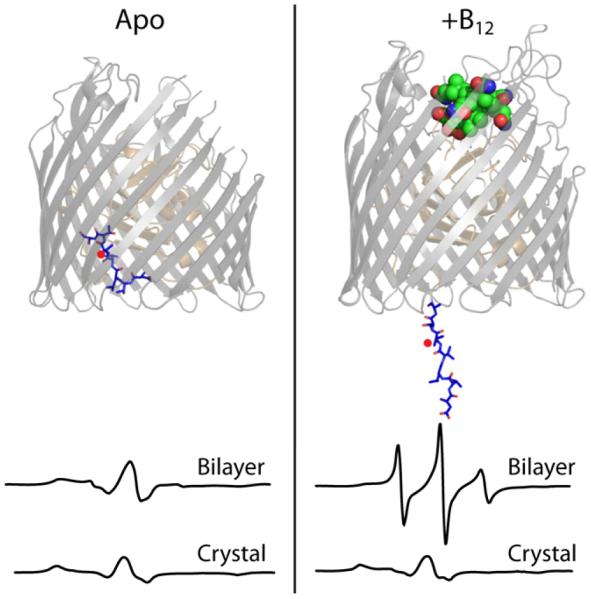
The structural model of BtuB illustrates the unpacking of the N-terminal Ton box (blue sticks) upon binding of vitamin B12 (green spheres). The CW-EPR spectra of a spin label at position 12 of BtuB (red dot) show that B12 binding markedly increased mobility of this peptide in the context of a lipid bilayer (top spectra) but caused no change in the context of a crystal (bottom spectra).
Small Helical Movements in GPCR: Photoactivation of Rhodopsin
Rhodopsin is not only the first membrane protein studied by EPR, but the studies also motivated much of the development and benchmarking of SDSL. Visual Rhodopsin is a G-protein coupled receptor present in vertebrate retina rod cells. It is composed of seven TM helices and contains a stably bound 11-cis-retinal. Absorption of a photon causes retinal to isomerize to all-trans, which begins a chain reaction of structural changes resulting in the activation of transducin, the associated G-protein.
The initial work consisted of extensive nitroxide scanning that targeted loop regions to simultaneously define the membrane boundary and map light-induced conformational changes (Reviewed in Hubbell et al., 2003). Changes in spin label mobility, and thus tertiary contacts, indicated the location, direction, and type of movements that underlie photoactivation—most significantly a rigid-body motion of TM6 away from the bulk of the protein. To better define the magnitude of this and other helical motions, Hubbell and colleagues measured distances by dipolar coupling between spin labels. In all of the short-range studies, spin labels had to be introduced on the inside of the protein in order to ensure sufficient proximity for dipolar coupling. Labeling buried positions likely biased the rotamers adopted by the label and possibly altered the local structure hindering a quantitative interpretation of the helix movement.
The EPR constraints were in reasonable agreement with the crystal structure of Rhodopsin in the inactivated state (Palczewski et al., 2000). Subsequently, Salom et al. (2006) reported the crystal structure of the putative photoactivated intermediate, achieved by the illumination of dark state crystals. Surprisingly, this structure was very close to the dark state with minimal movement of helices, initiating a controversy that continued until recently. However, the authors’ selection of crystallization conditions that preserved the crystalline lattice upon illumination likely suppressed the conformational flexibility of rhodopsin.
In a culminating study, Altenbach et al. (2008) measured long-range distances between 16 pairs of spin labels on the surface of rhodopsin in the ground and photoactivated states using DEER. The quantitative distance changes were consistent with the qualitative conclusions of previous work and showed that TM 6 undergoes a ~5 Å (from the center of rhodopsin) rigid body motion (Figure 8). From these distances a model of activation was derived. A recent crystal structure of metarhodopsinII (Choe et al., 2011) demonstrates that a rigid body movement of TM6 is the primary structural change associated with photoactivation and G-protein binding, consistent with the conclusion from EPR analysis.
Figure 8. Structural changes on the cytoplasmic side of Rhodopsin upon activation.

(A) The probabilities of spin label locations are shown for sites 74, 225, 252, and 308 as contour maps overlaid on the dark state structure where TM helices are labeled 1-8. The contours are based on a global analysis of 16 different pairwise distance distributions obtained by DEER.
(B) The probability contours after light activation clearly show location changes, most prominently an outward movement of site 252. Dotted circles indicate the original locations.
(C) Cross-sections of the contours along the dotted lines shown in (A) and (B) show the relative radial movements from the dark state (solid lines) to the activated state (dotted lines). Sites 74 and 225 are static. Site 252 shows a large outward movement while site 308 shows a small inward movement.
Insight into K+ Channel Gating from Spin Label Reporters
Similar to Rhodopsin, K+ channels constituted the proving ground for the EPR tool kit in the pre-DEER age. These channels play a central role in signaling by electrically excitable membranes involved in many physiological processes, most notably the propagation of an action potential along a neuron. The crystal structure of KcsA from Streptococcus lividans (Doyle et al., 1998) revolutionized the K+ channel field by revealing the structural basis of ion conductivity and selectivity. KcsA is composed of two TM helices that oligomerize into a functional tetramer with the conductive pore and selectivity filter at its center lined by the inner helices (TM2). Perozo and colleagues accomplished a SDSL “tour de force” by nitroxide scanning almost the entire length of KcsA (Perozo et al., 1999). The extensive mobility, accessibility, and dipolar coupling data revealed the membrane topology and helical packing of the functional tetramer. Moreover, data collected at multiple pH values characterized the acid-induced opening of the inner gate, formed by the four inner helices of the tetramer (Figure 9).
Figure 9. Conformational transitions that define activation gating in K+ channels.

(A) Location of KcsA cysteine mutations (colored spheres) showing strong dipolar coupling in the closed state (neutral pH). Two diagonally-related subunits (red cylinders) shown for clarity.
(B) The semi-quantitative interaction parameter (calculated relative to the under-labeled spectra) illustrates the nature of the conformational motion in TM2. Data is shown for the closed conformation (open bars) and the open conformation (filled bars). The length of the arrow represents the difference in spin-spin interaction parameter between the two conformations.
(C) Mechanistic representation of the opening of the internal vestibule during K+ channel gating from the crystal structures of closed KcsA and open MthK, an archaeal K+ channel.
In subsequent mechanistic studies, spin labels at select positions on the inner helix and the selectivity filter were used as reporters to define the interplay between two types of gating occurring in KcsA (Cordero-Morales et al., 2007). The extent of dipolar coupling at specific sites reported the status of the gate formed by the inner helices. Opening of this gate was pH dependent and mostly independent of the “C-type inactivation” caused by collapse of the selectivity filter, simultaneously monitored by dipolar coupling at a local site. The key finding was that conditions stabilizing the filter also raised the pH of activation of the inner gate, and conditions favoring an open inner gate destabilize the conductive conformation of the selectivity filter. A structural basis of this effect was later identified from crystallographic analysis of targeted KcsA mutants demonstrating the integrated use of the two techniques (Cuello et al., 2010). Thus SDSL and EPR studies complemented KcsA crystallographic analysis advancing the understanding of the multi-part gating mechanism responsible for regulating channel activity.
Modeling protein structure and conformational dynamics from EPR restraints
Despite the success of spin labeling in identifying and mapping conformational changes, evidenced by work highlighted so far, transformation of EPR distances between spin labels to corresponding restraints between Cα carbons is challenging. For spin labeling EPR to become a platform for discovery, computational methods for structural and dynamic interpretation of EPR parameters need to be developed. As extrinsic probes, spin labels shape the methodology and interpretation of EPR in fundamental ways. Not only is there the potential for functional and structural perturbation but the spin label linking arm introduces intrinsic uncertainties to models constructed from EPR restraints. In contrast to the determination of EPR parameters, which is firmly established in rigorous treatment of the spin Hamiltonian, structural interpretation of the data necessitates a model of the spin label relative to the backbone, an internally consistent transfer function that links spectral and structural parameters. An additional consequence of using reporter groups is the sparseness of EPR data sets. Limited by experimental throughput, the number of EPR restraints per residue is typically many fewer than that used in NMR structure determination. Importantly, as discussed below, in the absence of a crystal structure, the restraints are not necessarily optimal or of uniform value for modeling structure and dynamics.
Although a benchmarked strategy for EPR-based modeling of structures is not yet available, simplifying approximations and assumptions have been applied to the systems reviewed here, and elsewhere in the literature, to successfully model structure and conformational changes with outcomes subsequently verified by other techniques. In most cases, the sampling of conformational space was restricted by prior data that suggested particular folds or motifs (Koteiche et al., 1998; Poirier et al., 1998), by structural simplicity such as small size and high symmetries (Cortes et al., 2001), or by restriction on the type of motion to rigid body or simple helix rotation (Altenbach et al., 2008; Perozo et al., 1999).
The more general question of whether EPR restraints restrict conformational space to a set of convergent models of acceptable resolution or enable detailed description of structural rearrangements starting from a high resolution structure has only been recently addressed. Alexander et al. (2008) carried out a systematic feasibility analysis of de novo protein structure determination from EPR restraints in T4L. This study also aimed to directly define the information content of EPR restraints and the impact of the sparse data on model quality. The distance between spin labels was converted into a distance range between β-carbons using a simple “motion on a cone” model, treating the spin label as an average vector relative to the β-carbon. Because of an assumed isotropic distribution of the label in this model, the function relating the distance between the two spin labels to that between the corresponding β-carbon was relatively broad, i.e. the derived restraint has large uncertainty. This study made two novel contributions. First, it heralded the use of the Rosetta folding algorithm (Das and Baker, 2008) as an alternative computation platform to MD simulations. Second, it demonstrated that a detailed model of the spin label conformations at each site may not be required. Even with a simple boundary function to interpret the restraints (Figure 10), 25 EPR restraints were sufficient to generate models with the correct fold. Subsequent high resolution refinement yielded structures that are within 1 Å RMSD from the crystal structure. This remarkable outcome was rationalized by the robust Rosetta knowledge-based energy function, which captures the principle of protein assembly encoded in known structures, compensating for the sparseness of EPR restraints. In turn, the EPR restraints efficiently restrict conformational space enabling Rosetta to find the global energy minimum.
Figure 10. Structure Determination by EPR and Rosetta.

Overview of hypothetical de novo modeling of a polytopic membrane protein guided by EPR restraints. Three restraints are highlighted for simplicity but a larger number is required even for a small 3-helix protein. In this scheme, secondary structural element (SSE) definitions inform optimized selection of label pairs for restraints. Analysis of DEER measurements returns distance distributions, which are transformed into probabilistic boundary functions to describe the distance between β-carbons (dCβ) of the label pairs. Restraint violation scores measure model agreement with these functions and guide Monte Carlo modeling trajectories. Selecting for models with both low energy and low restraint violations have been shown to effectively limit model pools to low RMSD models (as shown in the 3D plot). These models proceed to all-atom, high resolution refinement with explicit modeling of the restraints, resulting in a best model.
This study set the stage for an analysis of the information content of EPR restraints. Alexander et al. demonstrated the importance of high information content (defined as the ratio between sequence separation and Euclidean distance) as a criterion for restraint quality. The improvement in model quality by Rosetta folding was attributed to 8 (of 25) restraints with the highest information content. Because throughput of EPR methods and the ensuing restraint sparseness is defined by the number of pairs to be constructed, a rational approach for the selection of spin labeled sites is required. Kazmier et al. (2010) developed an algorithm for selection of distance restraints with optimized information content. An optimal algorithm was generated, and its applicability was experimentally established through prediction of a set of pairs for spin label incorporation, experimental determination of the distances and then restrained Rosetta modeling of the T4L C-terminal domain. Improvement in model quality required a limited number of restraints determined by the pairwise connectivity of T4L α-helices (21 restraints for 7 helices). Finally this study introduced a practical criterion for identifying the “native-like” model out of the thousands generated by Rosetta. These models have simultaneously low Rosetta energy and restraint violation scores.
The ultimate goal of the RosettaEPR project (Hirst et al., 2011) is to establish a suite of algorithms that guide experimentalists in the selection of labeling sites and provide a platform for structural interpretation of the data. Its extension to membrane proteins (illustrated in Figure 10), currently under development, will tackle the challenges of the less robust Rosettamembrane energy function (implemented in Rosettamembrane) (Yarov-Yarovoy et al., 2006). A parallel effort is underway to establish methodology for restrained modeling of conformers starting from a high resolution structure.
Concluding Remarks
The examples described above, along with numerous other studies, illustrate the contribution of modern spin labeling and EPR to bridging structure and mechanism. They emphasize systematic application of the methodology to refute or confirm structures and/or models and assess their mechanistic significance, identify regions of distortion due to crystal contacts and/or detergent solubilization, and determine or test models of conformational dynamics. Although EPR data sets can be used to determine models of unknown structures, the availability of a crystal structure of one or more intermediates simplifies the selection of sites and increases experimental throughput. The next frontier is to develop benchmarked computational approaches for EPR restraint-modeling of conformers starting from crystal structures. Similarly, continued development of spectroscopic tools to quantitatively measure and analyze EPR parameters and to increase absolute sensitivity will expand its applicability to systems of limited quantities such as eukaryotic membrane proteins.
Acknowledgements
We thank Richard Stein for providing data on σ distribution in T4L and for assistance in figure preparation. Daniel Terry provided the single-molecule FRET data from LeuT. Christian Altenbach, David Cafiso and Eduardo Perozo contributed figures summarizing data on Rhodopsin, BtuB and KcsA, respectively. We also thank Hanane Koteiche, Shruti Sharma and Brandy Verhalen for critical reading of the manuscript. The authors’ laboratory is supported by National Institutes of Health grants U54 GM087519 and R01 GM077659 to HSM.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abramson J, Smirnova I, Kasho V, Verner G, Kaback HR, Iwata S. Structure and mechanism of the lactose permease of Escherichia coli. Science. 2003;301:610–615. doi: 10.1126/science.1088196. [DOI] [PubMed] [Google Scholar]
- Alexander N, Bortolus M, Al-Mestarihi A, Mchaourab H, Meiler J. De novo high-resolution protein structure determination from sparse spin-labeling EPR data. Structure. 2008;16:181–195. doi: 10.1016/j.str.2007.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altenbach C, Froncisz W, Hemker R, Mchaourab H, Hubbell WL. Accessibility of nitroxide side chains: absolute Heisenberg exchange rates from power saturation EPR. Biophys J. 2005;89:2103–2112. doi: 10.1529/biophysj.105.059063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altenbach C, Kusnetzow AK, Ernst OP, Hofmann KP, Hubbell WL. High-resolution distance mapping in rhodopsin reveals the pattern of helix movement due to activation. Proc Natl Acad Sci U S A. 2008;105:7439–7444. doi: 10.1073/pnas.0802515105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altenbach C, Marti T, Khorana HG, Hubbell WL. Transmembrane protein structure: spin labeling of bacteriorhodopsin mutants. Science. 1990;248:1088–1092. doi: 10.1126/science.2160734. [DOI] [PubMed] [Google Scholar]
- Bayburt TH, Sligar SG. Membrane protein assembly into Nanodiscs. FEBS Lett. 2010;584:1721–1727. doi: 10.1016/j.febslet.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borbat PP, Mchaourab HS, Freed JH. Protein structure determination using long-distance constraints from double-quantum coherence ESR: study of T4 lysozyme. J Am Chem Soc. 2002;124:5304–5314. doi: 10.1021/ja020040y. [DOI] [PubMed] [Google Scholar]
- Borbat PP, Surendhran K, Bortolus M, Zou P, Freed JH, Mchaourab HS. Conformational motion of the ABC transporter MsbA induced by ATP hydrolysis. PLoS Biol. 2007;5:e271. doi: 10.1371/journal.pbio.0050271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borovykh IV, Ceola S, Gajula P, Gast P, Steinhoff HJ, Huber M. Distance between a native cofactor and a spin label in the reaction centre of Rhodobacter sphaeroides by a two-frequency pulsed electron paramagnetic resonance method and molecular dynamics simulations. J Magn Reson. 2006;180:178–185. doi: 10.1016/j.jmr.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Bowie JU. Stabilizing membrane proteins. Curr Opin Struct Biol. 2001;11:397–402. doi: 10.1016/s0959-440x(00)00223-2. [DOI] [PubMed] [Google Scholar]
- Chang G, Roth CB. Structure of MsbA from E. coli: a homolog of the multidrug resistance ATP binding cassette (ABC) transporters. Science. 2001;293:1793–1800. doi: 10.1126/science.293.5536.1793. [DOI] [PubMed] [Google Scholar]
- Chiang YW, Borbat PP, Freed JH. The determination of pair distance distributions by pulsed ESR using Tikhonov regularization. Journal of Magnetic Resonance. 2005;172:279–295. doi: 10.1016/j.jmr.2004.10.012. [DOI] [PubMed] [Google Scholar]
- Choe HW, Kim YJ, Park JH, Morizumi T, Pai EF, Krauss N, Hofmann KP, Scheerer P, Ernst OP. Crystal structure of metarhodopsin II. Nature. 2011;471:651–655. doi: 10.1038/nature09789. [DOI] [PubMed] [Google Scholar]
- Claxton DP, Quick M, Shi L, de Carvalho FD, Weinstein H, Javitch JA, Mchaourab HS. Ion/substrate-dependent conformational dynamics of a bacterial homolog of neurotransmitter:sodium symporters. Nat Struct Mol Biol. 2010;17:822–829. doi: 10.1038/nsmb.1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Columbus L, Kalai T, Jeko J, Hideg K, Hubbell WL. Molecular motion of spin labeled side chains in alpha-helices: analysis by variation of side chain structure. Biochemistry. 2001;40:3828–3846. doi: 10.1021/bi002645h. [DOI] [PubMed] [Google Scholar]
- Cordero-Morales JF, Jogini V, Lewis A, Vasquez V, Cortes DM, Roux B, Perozo E. Molecular driving forces determining potassium channel slow inactivation. Nat Struct Mol Biol. 2007;14:1062–1069. doi: 10.1038/nsmb1309. [DOI] [PubMed] [Google Scholar]
- Cortes DM, Cuello LG, Perozo E. Molecular architecture of full-length KcsA: role of cytoplasmic domains in ion permeation and activation gating. J Gen Physiol. 2001;117:165–180. doi: 10.1085/jgp.117.2.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross TA, Sharma M, Yi M, Zhou HX. Influence of solubilizing environments on membrane protein structures. Trends Biochem Sci. 2011;36:117–125. doi: 10.1016/j.tibs.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuello LG, Jogini V, Cortes DM, Pan AC, Gagnon DG, Dalmas O, Cordero-Morales JF, Chakrapani S, Roux B, Perozo E. Structural basis for the coupling between activation and inactivation gates in K(+) channels. Nature. 2010;466:272–275. doi: 10.1038/nature09136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R, Baker D. Macromolecular modeling with rosetta. Annu Rev Biochem. 2008;77:363–382. doi: 10.1146/annurev.biochem.77.062906.171838. [DOI] [PubMed] [Google Scholar]
- Dong J, Yang G, Mchaourab HS. Structural basis of energy transduction in the transport cycle of MsbA. Science. 2005;308:1023–1028. doi: 10.1126/science.1106592. [DOI] [PubMed] [Google Scholar]
- Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- Fajer MI, Li H, Yang W, Fajer PG. Mapping electron paramagnetic resonance spin label conformations by the simulated scaling method. Journal of the American Chemical Society. 2007;129:13840–13846. doi: 10.1021/ja071404v. [DOI] [PubMed] [Google Scholar]
- Fanucci GE, Coggshall KA, Cadieux N, Kim M, Kadner RJ, Cafiso DS. Substrate-induced conformational changes of the periplasmic N-terminus of an outer-membrane transporter by site-directed spin labeling. Biochemistry. 2003;42:1391–1400. doi: 10.1021/bi027120z. [DOI] [PubMed] [Google Scholar]
- Flores Jimenez RH, Do Cao MA, Kim M, Cafiso DS. Osmolytes modulate conformational exchange in solvent-exposed regions of membrane proteins. Protein Sci. 2010;19:269–278. doi: 10.1002/pro.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed DM, Horanyi PS, Wiener MC, Cafiso DS. Conformational exchange in a membrane transport protein is altered in protein crystals. Biophys J. 2010;99:1604–1610. doi: 10.1016/j.bpj.2010.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgieva ER, Ramlall TF, Borbat PP, Freed JH, Eliezer D. Membrane-bound alpha-synuclein forms an extended helix: long-distance pulsed ESR measurements using vesicles, bicelles, and rodlike micelles. J Am Chem Soc. 2008;130:12856–12857. doi: 10.1021/ja804517m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghimire H, McCarrick RM, Budil DE, Lorigan GA. Significantly improved sensitivity of Q-band PELDOR/DEER experiments relative to X-band is observed in measuring the intercoil distance of a leucine zipper motif peptide (GCN4-LZ) Biochemistry. 2009;48:5782–5784. doi: 10.1021/bi900781u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouaux E, Mackinnon R. Principles of selective ion transport in channels and pumps. Science. 2005;310:1461–1465. doi: 10.1126/science.1113666. [DOI] [PubMed] [Google Scholar]
- Guan L, Kaback HR. Lessons from lactose permease. Annu Rev Biophys Biomol Struct. 2006;35:67–91. doi: 10.1146/annurev.biophys.35.040405.102005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha Y. Structural principles of intramembrane proteases. Curr Opin Struct Biol. 2007;17:405–411. doi: 10.1016/j.sbi.2007.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins CF. ABC transporters: physiology, structure and mechanism--an overview. Research in Microbiology. 2001;152:205–210. doi: 10.1016/s0923-2508(01)01193-7. [DOI] [PubMed] [Google Scholar]
- Hilger D, Polyhach Y, Jung H, Jeschke G. Backbone structure of transmembrane domain IX of the Na+/proline transporter PutP of Escherichia coli. Biophys J. 2009;96:217–225. doi: 10.1016/j.bpj.2008.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilger D, Polyhach Y, Padan E, Jung H, Jeschke G. High-resolution structure of a Na+/H+ antiporter dimer obtained by pulsed electron paramagnetic resonance distance measurements. Biophys J. 2007;93:3675–3683. doi: 10.1529/biophysj.107.109769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst SJ, Alexander N, Mchaourab HS, Meiler J. RosettaEPR: an integrated tool for protein structure determination from sparse EPR data. J Struct Biol. 2011;173:506–514. doi: 10.1016/j.jsb.2010.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbell WL, Altenbach C, Hubbell CM, Khorana HG. Rhodopsin structure, dynamics, and activation: a perspective from crystallography, site-directed spin labeling, sulfhydryl reactivity, and disulfide cross-linking. Adv Protein Chem. 2003;63:243–290. doi: 10.1016/s0065-3233(03)63010-x. [DOI] [PubMed] [Google Scholar]
- Hubbell WL, Cafiso DS, Altenbach C. Identifying conformational changes with site-directed spin labeling. Nature Structural Biology. 2000;7:735–739. doi: 10.1038/78956. [DOI] [PubMed] [Google Scholar]
- Hubbell WL, Mchaourab HS, Altenbach C, Lietzow MA. Watching proteins move using site-directed spin labeling. Structure. 1996;4:779–783. doi: 10.1016/s0969-2126(96)00085-8. [DOI] [PubMed] [Google Scholar]
- Jao CC, Hegde BG, Chen J, Haworth IS, Langen R. Structure of membrane-bound alpha-synuclein from site-directed spin labeling and computational refinement. Proc Natl Acad Sci U S A. 2008;105:19666–19671. doi: 10.1073/pnas.0807826105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeschke G, Chechik V, Ionita P, Godt A, Zimmermann H, Banham J, Timmel CR, Hilger D, Jung H. DeerAnalysis2006 - a comprehensive software package for analyzing pulsed ELDOR data. Applied Magnetic Resonance. 2006;30:473–498. [Google Scholar]
- Jeschke G, Polyhach Y. Distance measurements on spin-labelled biomacromolecules by pulsed electron paramagnetic resonance. Phys Chem Chem Phys. 2007;9:1895–1910. doi: 10.1039/b614920k. [DOI] [PubMed] [Google Scholar]
- Jeschke G, Wegener C, Nietschke M, Jung H, Steinhoff HJ. Interresidual distance determination by four-pulse double electron-electron resonance in an integral membrane protein: the Na+/proline transporter PutP of Escherichia coli. Biophysical Journal. 2004;86:2551–2557. doi: 10.1016/S0006-3495(04)74310-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazmier K, Alexander NS, Meiler J, McHaourab HS. Algorithm for selection of optimized EPR distance restraints for de novo protein structure determination. J Struct Biol. 2010 doi: 10.1016/j.jsb.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Xu Q, Murray D, Cafiso DS. Solutes alter the conformation of the ligand binding loops in outer membrane transporters. Biochemistry. 2008;47:670–679. doi: 10.1021/bi7016415. [DOI] [PubMed] [Google Scholar]
- Klein JC, Burr AR, Svensson B, Kennedy DJ, Allingham J, Titus MA, Rayment I, Thomas DD. Actin-binding cleft closure in myosin II probed by site-directed spin labeling and pulsed EPR. Proc Natl Acad Sci U S A. 2008;105:12867–12872. doi: 10.1073/pnas.0802286105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobilka B, Schertler GF. New G-protein-coupled receptor crystal structures: insights and limitations. Trends Pharmacol Sci. 2008;29:79–83. doi: 10.1016/j.tips.2007.11.009. [DOI] [PubMed] [Google Scholar]
- Koteiche HA, Berengian AR, Mchaourab HS. Identification of protein folding patterns using site-directed spin labeling. Structural characterization of a beta-sheet and putative substrate binding regions in the conserved domain of alpha A-crystallin. Biochemistry. 1998;37:12681–12688. doi: 10.1021/bi9814078. [DOI] [PubMed] [Google Scholar]
- Krishnamurthy H, Piscitelli CL, Gouaux E. Unlocking the molecular secrets of sodium-coupled transporters. Nature. 2009;459:347–355. doi: 10.1038/nature08143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langen R, Oh KJ, Cascio D, Hubbell WL. Crystal structures of spin labeled T4 lysozyme mutants: implications for the interpretation of EPR spectra in terms of structure. Biochemistry. 2000;39:8396–8405. doi: 10.1021/bi000604f. [DOI] [PubMed] [Google Scholar]
- Mchaourab HS, Lietzow MA, Hideg K, Hubbell WL. Motion of spin-labeled side chains in T4 lysozyme. Correlation with protein structure and dynamics. Biochemistry. 1996;35:7692–7704. doi: 10.1021/bi960482k. [DOI] [PubMed] [Google Scholar]
- Milov AD, Ponomarev AB, Tsvetkov YD. Electron Electron Double-Resonance in Electron-Spin Echo - Model Biradical Systems and the Sensitized Photolysis of Decalin. Chemical Physics Letters. 1984;110:67–72. [Google Scholar]
- Mittermaier AK, Kay LE. Observing biological dynamics at atomic resolution using NMR. Trends Biochem Sci. 2009;34:601–611. doi: 10.1016/j.tibs.2009.07.004. [DOI] [PubMed] [Google Scholar]
- Ogawa S, McConnell HM. Spin-label study of hemoglobin conformations in solution. Proc Natl Acad Sci U S A. 1967;58:19–26. doi: 10.1073/pnas.58.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orelle C, Alvarez FJ, Oldham ML, Orelle A, Wiley TE, Chen J, Davidson AL. Dynamics of alpha-helical subdomain rotation in the intact maltose ATP-binding cassette transporter. Proc Natl Acad Sci U S A. 2010;107:20293–20298. doi: 10.1073/pnas.1006544107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- Pannier M, Veit S, Godt A, Jeschke G, Spiess HW. Dead-time free measurement of dipole-dipole interactions between electron spins. J Magn Reson. 2000;142:331–340. doi: 10.1006/jmre.1999.1944. [DOI] [PubMed] [Google Scholar]
- Perozo E, Cortes DM, Cuello LG. Structural rearrangements underlying K+-channel activation gating. Science. 1999;285:73–78. doi: 10.1126/science.285.5424.73. [DOI] [PubMed] [Google Scholar]
- Poirier MA, Xiao W, Macosko JC, Chan C, Shin YK, Bennett MK. The synaptic SNARE complex is a parallel four-stranded helical bundle. Nat Struct Biol. 1998;5:765–769. doi: 10.1038/1799. [DOI] [PubMed] [Google Scholar]
- Polyhach Y, Bordignon E, Jeschke G. Rotamer libraries of spin labelled cysteines for protein studies. Phys Chem Chem Phys. 2011;13:2356–2366. doi: 10.1039/c0cp01865a. [DOI] [PubMed] [Google Scholar]
- Quick M, Winther AM, Shi L, Nissen P, Weinstein H, Javitch JA. Binding of an octylglucoside detergent molecule in the second substrate (S2) site of LeuT establishes an inhibitor-bound conformation. Proc Natl Acad Sci U S A. 2009;106:5563–5568. doi: 10.1073/pnas.0811322106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabenstein MD, Shin YK. Determination of the distance between two spin labels attached to a macromolecule. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:8239–8243. doi: 10.1073/pnas.92.18.8239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees DC, Johnson E, Lewinson O. ABC transporters: the power to change. Nat Rev Mol Cell Biol. 2009;10:218–227. doi: 10.1038/nrm2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salom D, Lodowski DT, Stenkamp RE, Le Trong I, Golczak M, Jastrzebska B, Harris T, Ballesteros JA, Palczewski K. Crystal structure of a photoactivated deprotonated intermediate of rhodopsin. Proc Natl Acad Sci U S A. 2006;103:16123–16128. doi: 10.1073/pnas.0608022103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sezer D, Freed JH, Roux B. Parametrization, molecular dynamics simulation, and calculation of electron spin resonance spectra of a nitroxide spin label on a polyalanine alpha-helix. J Phys Chem B. 2008;112:5755–5767. doi: 10.1021/jp711375x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova I, Kasho V, Choe JY, Altenbach C, Hubbell WL, Kaback HR. Sugar binding induces an outward facing conformation of LacY. Proc Natl Acad Sci U S A. 2007;104:16504–16509. doi: 10.1073/pnas.0708258104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sompornpisut P, Roux B, Perozo E. Structural refinement of membrane proteins by restrained molecular dynamics and solvent accessibility data. Biophys J. 2008;95:5349–5361. doi: 10.1529/biophysj.108.142984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong J, Borbat PP, Freed JH, Shin YK. A scissors mechanism for stimulation of SNARE-mediated lipid mixing by cholesterol. Proc Natl Acad Sci U S A. 2009;106:5141–5146. doi: 10.1073/pnas.0813138106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ujwal ML, Jung H, Bibi E, Manoil C, Altenbach C, Hubbell WL, Kaback HR. Membrane topology of helices VII and XI in the lactose permease of Escherichia coli studied by lacY-phoA fusion analysis and site-directed spectroscopy. Biochemistry. 1995;34:14909–14917. doi: 10.1021/bi00045a036. [DOI] [PubMed] [Google Scholar]
- Vileno B, Chamoun J, Liang H, Brewer P, Haldeman BD, Facemyer KC, Salzameda B, Song L, Li HC, Cremo CR, et al. Broad disorder and the allosteric mechanism of myosin II regulation by phosphorylation. Proc Natl Acad Sci U S A. 2011;108:8218–8223. doi: 10.1073/pnas.1014137108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss J, Wu J, Hubbell WL, Jacques V, Meares CF, Kaback HR. Helix packing in the lactose permease of Escherichia coli: distances between site-directed nitroxides and a lanthanide. Biochemistry. 2001;40:3184–3188. doi: 10.1021/bi002333e. [DOI] [PubMed] [Google Scholar]
- Wahl P, Weber G. Fluorescence depolarization of rabbit gamma globulin conjugates. J Mol Biol. 1967;30:371–382. doi: 10.1016/s0022-2836(67)80045-7. [DOI] [PubMed] [Google Scholar]
- Ward A, Reyes CL, Yu J, Roth CB, Chang G. Flexibility in the ABC transporter MsbA: Alternating access with a twist. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:19005–19010. doi: 10.1073/pnas.0709388104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Voss J, Hubbell WL, Kaback HR. Site-directed spin labeling and chemical crosslinking demonstrate that helix V is close to helices VII and VIII in the lactose permease of Escherichia coli. Proc Natl Acad Sci U S A. 1996;93:10123–10127. doi: 10.1073/pnas.93.19.10123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Ellena JF, Kim M, Cafiso DS. Substrate-dependent unfolding of the energy coupling motif of a membrane transport protein determined by double electron-electron resonance. Biochemistry. 2006;45:10847–10854. doi: 10.1021/bi061051x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl--dependent neurotransmitter transporters. Nature. 2005;437:215–223. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- Yarov-Yarovoy V, Schonbrun J, Baker D. Multipass membrane protein structure prediction using Rosetta. Proteins. 2006;62:1010–1025. doi: 10.1002/prot.20817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Terry D, Shi L, Weinstein H, Blanchard SC, Javitch JA. Single-molecule dynamics of gating in a neurotransmitter transporter homologue. Nature. 2010;465:188–193. doi: 10.1038/nature09057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou P, Bortolus M, Mchaourab HS. Conformational cycle of the ABC transporter MsbA in liposomes: detailed analysis using double electron-electron resonance spectroscopy. J Mol Biol. 2009;393:586–597. doi: 10.1016/j.jmb.2009.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou P, Mchaourab HS. Alternating access of the putative substrate-binding chamber in the ABC transporter MsbA. J Mol Biol. 2009;393:574–585. doi: 10.1016/j.jmb.2009.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou P, Mchaourab HS. Increased sensitivity and extended range of distance measurements in spin-labeled membrane proteins: Q-band double electron-electron resonance and nanoscale bilayers. Biophys J. 2010;98:L18–20. doi: 10.1016/j.bpj.2009.12.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]