

Abstract
The microtubule-associated protein tau exists as six isoforms created through the splicing of the second, third, and tenth exons. The isoforms are classified by their number of N-terminal exons (0N, 1N or 2N) and by their number of microtubule-binding repeat regions (3R or 4R). Hyperphosphorylated isoforms accumulate in insoluble aggregates in Alzheimer’s disease and other tauopathies. These neurodegenerative diseases can be categorized based on the isoform content of the aggregates they contain. Hyperphosphorylated tau has the general characteristics of an upward electrophoretic shift, decreased microtubule binding, and an association with aggregation. Previously we have shown that a combination of seven pseudophosphorylation mutations at sites phosphorylated by GSK-3β, referred to as 7-Phos, induced several of these characteristics in full-length 2N4R tau and led to the formation of fewer but longer filaments. We sought to determine whether the same phosphorylation pattern could cause differential effects in the other tau isoforms, possibly through varied conformational effects. Using in vitro techniques, we examined the electrophoretic mobility, aggregation properties and microtubule stabilization of all isoforms and their pseudophosphorylated counterparts. We found that pseudophosphorylation affected each isoform, but in several cases certain isoforms were affected more than others. These results suggest that hyperphosphorylation of tau isoforms could play a major role in determining the isoform composition of tau aggregates in disease.
Tau is a microtubule-associated protein typically found in neurons with a primary function of assisting in the assembly and stabilization of microtubules (1). The protein also interacts with the cellular membrane through components such as annexin A2 (2). In addition, tau associates with certain cellular kinases and has been shown to affect the localization of some, including fyn (3, 4). The protein, tau, exists as six isoforms created through alternative mRNA splicing of the second, third, and tenth exons (5). The second and third exons reside near the N-terminus of the protein and isoforms can be classified based on the presence or absence of these exons. If both exons are present the protein is referred to as 2N, if only the second exon is present it is known as 1N, and if neither is present it is classified as 0N. Exon 10 contains one of four microtubule-binding repeat regions (MTBR). If this exon is present in the isoform the protein is referred to as 4R and if it is absent the protein is classified as 3R (6).
In some disease states tau dissociates from microtubules and aggregates inside of neurons, leading to neurodegeneration (7, 8). This group of neurodegenerative disorders is collectively known as tauopathies. Tauopathies can be classified based on the isoform content and phosphorylation pattern found in tau aggregates (7). While aggregates from Class I tauopathies contain all six isoforms, Class II aggregates are characterized by a larger ratio of 4R to 3R tau, and Class III tauopathies are associated with aggregates containing mostly 3R isoforms. The Class III tauopathies are also characterized by a lack of phosphorylation at the 262 and 356 serine residues (9, 10). Aggregates from the fourth class of tauopathies typically contain only 0N isoforms (7). The various tauopathies can give rise to vastly different pathological effects and the changes in isoform content seen in the aggregates could play a role in affecting these variations. While the root causes of differential isoform inclusion into pathological aggregates are unknown, it is possible that phosphorylation of the isoforms could differently alter their function and activity sufficiently to contribute to this phenomenon.
Several links between phosphorylation of tau and the pathology of Alzheimer’s disease (AD) and other tauopathies have been described. A potential important factor is the increase in hyperphosphorylated tau found in AD-affected brains. Hyperphosphorylated tau can be identified by an upward electrophoretic mobility shift, even in the presence of SDS, and a decreased ability to stabilize microtubule assembly (11, 12). It is probable that several kinases and phosphatases are involved in generating hyperphosphorylated tau, but one kinase likely to be involved is glycogen synthase kinase 3β (GSK-3β) due to its association with the formation of neurofibrillary tangles and paired-helical filaments found in AD (13–15). In addition, several sites commonly found to be phosphorylated in pathogenic tau are known to be phosphorylated by GSK-3β (16, 17).
While links to phosphorylation and certain kinases are known, it can be difficult to study the effects that specific phosphorylation sites can have on the function and in vitro aggregation of the protein because it is nearly impossible to control the level and specific location of kinase activity on tau due to the large number of phosphorylation sites and the relative promiscuity of kinases toward tau (18–21). This can complicate collection and interpretation of data. One method used to circumvent this issue is to utilize site-directed mutagenesis to create pseudophosphorylation mutants by changing a serine or threonine to an aspartic acid or glutamic acid in order to mimic the size and the shape of a phosphate molecule (22, 23). Although the effects of pseudophosphorylation may not completely replicate the effects of actual phosphorylation events, the technique has previously been utilized effectively to examine conformational changes in the protein and behavioral effects that may arise from these changes (24–30).
A combination of pseudophosphorylation mutations, referred to as 7-Phos for the seven specific sites mutated, were previously created in 2N4R full-length tau (27). The 7-Phos mutation alters the following residues, numbered according to their position in 2N4R tau: S199E, S202E, T205E, T231E, S235D, S396E, and S404E. Five of these sites are phosphorylated by GSK-3β when the kinase is incubated in vitro with arachidonic acid-induced pre-polymerized tau and the S202 and S235 sites are often phosphorylated in conjunction with the S199 and T231 sites (13, 27). All of the selected sites are phosphorylated at elevated levels in AD and the combination of these mutations replicated several aspects of pathogenic tau (21). The 7-Phos mutations, in 2N4R tau, were shown to induce an electrophoretic mobility shift similar to that of the hyperphosphorylated protein seen in AD. Additionally, the 7-Phos mutation caused a decrease in the 2N4R tau isoform’s affinity for microtubules and inducer-initiated nucleation of tau polymerization (27).
As described above, the pathology of tauopathies can vary based on the isoform content of their aggregates and tau phosphorylation patterns differ between the four classes of the diseases as well. The six isoforms have differences in their primary structures which could place the same phosphorylation sites in different structural contexts. Therefore, phosphorylation at the same sites, but in different isoforms, could have widely varied effects on their conformation. These conformational changes could be sufficient to cause some isoforms to dissociate from microtubules and aggregate while others maintain their normal function, as seen in some tauopathy classes.
In this study we demonstrate that the 7-Phos mutation does affect the isoforms differently. The SDS-resistant upward shift in electrophoretic mobility was present in all isoforms but differed in magnitude. The mutation enhanced the polymerization of isoforms such as 0N4R while greatly inhibiting the aggregation of the 3R isoforms. The mutation also decreased the microtubule stabilization function of all isoforms but affected 0N4R less than the other five. These results suggest that phosphorylation patterns generated by the same set of kinases or phosphatases could be sufficient to increase the propensity of some isoforms to aggregate while reducing the aggregation of others, resulting in the differential isoform inclusion in pathological tau aggregates.
EXPERIMENTAL PROCEDURES
Chemicals and reagents
All wild-type and pseudophosphorylated mutant proteins were expressed and purified as described previously (31). The mutations were made utilizing the Quikchange site-directed mutagenesis kit from Stratagene (La Jolla, CA). The following mutations were made in each of the 6 isoforms in order to create the “7-Phos” mutants: S199E, S202E, T205E, T231E, S235D, S396E, and S404E. Polymerization was induced using arachidonic acid (ARA) from Cayman Chemicals (Ann Arbor, MI).
SDS-PAGE
Each protein sample was incubated in buffer containing 2% SDS and 1% β-mercaptoethanol and then boiled for five minutes. One μg of each protein was loaded onto a 10% SDS-PAGE and resolved through electrophoresis. The urea-denatured protein samples were incubated in buffer containing 2% SDS, 1% β-mercaptoethanol, and 6 M urea (27). The protein samples were resolved on a 10% SDS-PAGE with 6 M urea and stained with Coomassie Brilliant Blue R-250.
ARA-induced Polymerization
Recombinant tau protein was incubated in polymerization buffer containing 10 mM HEPES (pH 7.64), 5 mM DTT, 100 mM NaCl, 0.1 mM EDTA, and 3.75% ethanol. Arachidonic acid was used as an inducer of tau polymerization. The effects of altering protein or ARA levels were found by varying final protein concentration from 0 to 4 μM with 75 μM ARA or varying the final ARA concentration from 0 to 150 μM with 2 μM protein (14). Based on these results a combination of 2 μM protein and 75 μM ARA were used in subsequent experiments in order to approach maximum polymerization for each tau isoform and its corresponding 7-Phos mutant.
Thioflavin S fluorescence
Polymerization was measured by utilizing staining with thioflavin S (ThS) from Sigma-Aldrich (St. Louis, MO). Polymerization reactions were incubated for 18 hours at 25 °C. ThS was then added to the reactions in 96-well plates to a final concentration of 20 μM. The shift in fluorescence was measured with an excitation wavelength of 440 nm and an emission wavelength of 520 nm using a Cary Eclipse Fluorescence Spectrophotometer (Varian Analytical Instruments, Walnut Valley, CA). The PMT voltage was set to 650 V but the procedure was otherwise performed as described earlier (31).
Right-angle Laser Light Scattering
Polymerization reactions were transferred to 5 mm x 5 mm optical glass fluorometer cuvettes (Starna Cells, Atascadero, CA). The cuvettes were placed in the path of a 12 mW solid state laser at λ = 532 nm and operating at 7.6 mW (B&W Tek, Inc., Newark, DE). The level of polymerization was measured by capturing images of light scattered perpendicular to the beam using a SONY XC-ST270 digital camera. The images were captured at an aperture setting of f5.6–8. The intensities of the images were quantified using Adobe Photoshop CS5 version 12.0.1 (32, 33).
ARA-Induced Polymerization Kinetics
Polymerization reactions with 2 μM protein and 75 μM ARA, at the same reaction conditions described earlier, were mixed in 5 mm x 5 mm optical glass fluorometer cuvettes. Polymerization was measured at regular time intervals through right-angle laser light scattering (LLS) until an apparent steady-state was reached (27).
Transmission Electron Microscopy
The polymerization reaction samples were diluted 1:10 in polymerization buffer with glutaraldehyde added to a final concentration of 2%. After the five minute fixation with glutaraldehyde, 10 μL of each sample was added to a formvar carbon-coated grid for one minute. The grid was blotted on filter paper, washed with water, blotted, washed with 2% uranyl acetate, and blotted dry again. The grid was then stained with 2% uranyl acetate for one minute and blotted dry on the filter paper again. The grids were examined with a TECNAI G2 20 electron microscope (FEI Co., Hillsboro, OR) and images were taken with the Gatan Digital Micrograph imaging system. The images were collected at a magnification of 3600X. The filament numbers and areas were quantified through the use of Image-Pro Plus 6.0 (27).
Tubulin Polymerization Assay
Polymerization of tubulin was measured by utilizing the Tubulin Polymerization Assay kit from Cytoskeleton, Inc. (Denver, CO). The reactions were measured in 96-well Costar black polystyrene flat-bottom plates (Corning, Inc., Corning, NY). Each well contained GTP at 1 mM and tubulin at 2 mg/mL in an 80 mM PIPES buffer (pH 6.9) with 2 mM MgCl2 and 0.5 mM EGTA. Each of the wild-type or 7-Phos isoforms was added to a different well at a final concentration of 1 μM. Another well contained 1 μM paclitaxel in order to act as a positive control for polymerization and as a standard for normalizing the results for each of the other reactions. The two negative control reactions contained tubulin with no additional compound added. Upon addition of the protein or control reagent to the other wells, the plate was placed at 37 °C and shaken for 5 seconds in a FlexStation II Fluorometer (Molecular Devices Corporation, Sunnyvale, CA). The fluorescence was measured every minute for one hour with a λex of 355 nm and a λem of 455 nm. The resulting data were fit to the Gompertz equation as described in (27). The maximum amount of polymerization is found as a, the time from the initiation of the reaction to when polymerization is measured, or lag time, is calculated as ti − b, and kapp is equal to 1/b and proportional to the maximum rate of polymerization. These parameters were used to describe the characteristics of the polymerization curve.
RESULTS
Generation of 7-Phos Mutants for All Tau Isoforms
Pseudophosphorylation mutants were generated in all six tau isoforms by making S199E, S202E, T205E, T231E, S235D, S396E, and S404E mutations (Figure 1A). The mutants were analyzed by SDS-PAGE and found to exhibit an upward mobility shift as had been previously described for the 2N4R isoform (27). All 7-Phos isoforms displayed a decrease in electrophoretic mobility when compared to their corresponding wild-type protein (Figure 1B). However, upon addition of 6 M urea, the upward mobility shift in the 7-Phos proteins was still present but largely decreased for all isoforms (Figure 1C). The electrophoretic shifts, in the absence of urea, were similar for all isoforms but slightly increased with the presence of additional N-terminal exons (Figure 1D). Because the wild-type and 7-Phos proteins have more similar migration distances in the presence of 6 M urea, but not in its absence, it is likely that the 7-Phos mutations are inducing an SDS-resistant conformational change in the proteins that can be interrupted with urea. This pseudophosphorylation-induced conformational change is the cause of the slowed electrophoretic migration and is similar to the retardation evident in hyperphosphorylated tau associated with disease.
Figure 1. 7-Phos induces an SDS-resistant electrophoretic shift in all tau isoforms.

(A) Six isoforms of tau are created through alternative mRNA splicing of the second, third and tenth exons. The latter contains the second of four microtubule-binding regions. Pseudophosphorylation mutations were made at seven sites commonly phosphorylated in Alzheimer’s disease. This combination of mutations is referred to as 7-Phos. (B) 1 μg of the wild-type and 7-Phos variants for each of the six isoforms separated via 10% SDS-PAGE. MW refers to the molecular weight markers. The isoforms are labeled above the lanes with WT indicating wild-type protein and 7P indicating 7-Phos protein for each isoform. (C) The same amount of protein loaded onto a 10% SDS-PAGE with 6 M urea. (D) The mean values of the electrophoretic shifts (y-axis, in kDa) for each isoform (labeled below x-axis) separated via 10% SDS-PAGE. These values were calculated by subtracting the apparent molecular weight of wild-type isoforms from the apparent molecular weight of the corresponding 7-Phos isoforms. Bars represent the mean shift from three gels with the standard deviation.
Pseudohyperphosphorylation differentially affects ARA-induced polymerization of isoforms
In vitro experiments were performed to determine the effects of the 7-Phos mutation on the ARA-induced polymerization of the six isoforms. The concentration of the inducer was held constant and the protein concentration varied from 0–4 μM. The amount of polymerization generally increased with increases in protein concentration (Figures 2 and 3). The 7-Phos mutants containing only three MTBRs, or 3R isoforms, polymerized less than their corresponding wild-type 3R isoforms when measured by ThS staining (Figure 2A-C) and right-angle LLS (Figure 3A-C). In contrast, the 1N4R and 2N4R 7-Phos isoforms polymerized more similarly to their corresponding 1N4R and 2N4R wild-type proteins (Figures 2E-F and 3E-F) as compared to the 3R isoforms (compare Figures 2A-C, E-F and 3A-C, E-F). Unlike any of the other isoforms, the 0N4R 7-Phos protein polymerized more than its corresponding wild-type for most protein concentrations (Figures 2D and 3D).
Figure 2. Polymerization of varied tau concentrations measured by thioflavin S staining.

Wild-type (closed circles •) and 7-Phos tau isoforms (open squares □) polymerization reactions with 0 to 4 μM protein (x-axis) and 75 μM arachidonic acid were measured by thioflavin S fluorescence (y-axis) after 18 hours incubation at 25 °C. Individual graphs are shown for (A) 0N3R, (B) 1N3R, (C) 2N3R, (D) 0N4R, (E) 1N4R, and (F) 2N4R isoforms. Bars represent the mean from 3 experiments ± standard deviation.
Figure 3. Polymerization of varied tau concentrations measured by right-angle laser light scattering.

Wild-type (closed circles •) and 7-Phos tau isoform (open squares □) polymerization reactions with 0 to 4 μM protein (x-axis) and 75 μM arachidonic acid were measured by intensity of right-angle laser light scattering (y-axis) after 18 hours incubation at 25 °C. Individual graphs are shown for (A) 0N3R, (B) 1N3R, (C) 2N3R, (D) 0N4R, (E) 1N4R, and (F) 2N4R isoforms. Data are the mean from 3 experiments and bars represent ± s.d.
To determine whether the 7-Phos modification altered the response to different concentrations of the inducer, the protein concentration was held constant and the concentration of inducer was varied from 0 μM to 150 μM. Data of this nature generally has a biphasic shape, with increasing polymerization up to a maximum level as the inducer concentration increases. After this maximum of polymerization is reached, any further addition of inducer decreases the amount of polymerization compared to the peak. The biphasic nature of inducer-mediated tau polymerization has been previously reported (14, 34). The polymerization for each of the 3R 7-Phos isoforms was lower than their corresponding non-modified 3R proteins, as measured by ThS staining (Figure 4A-C). Additionally, the 3R 7-Phos isoforms required a higher inducer concentration to reach maximum polymerization than the 3R wild-type isoforms. When the polymerization was measured by LLS, very little polymerization was detected for any of the 3R 7-Phos isoforms while the wild-type 3R isoforms also displayed the biphasic curve but with a sharp increase at the highest inducer concentrations (Figure 5A-C). Because these increases in light scattering, primarily seen at the 150 μM ARA concentration, are not replicated when measured by ThS staining (Figure 4A-F) or transmission electron microscopy (data not shown) they are likely due to formation of ARA micelles and not increased tau polymerization. Additionally, in Figures 4 and 5, we are greatly altering the ARA to protein ratio which has been shown to affect ThS and LLS readings differently (14).
Figure 4. Polymerization of 2 μM tau with 0 to 150 μM ARA measured by thioflavin S fluorescence.

Wild-type (closed circles •) and 7-Phos tau isoform (open squares □) polymerization reactions with 2 μM protein and 0 to 150 μM arachidonic acid (x-axis) were measured by thioflavin S fluorescence (y-axis) after 18 hours incubation at 25 °C. The fluorescence value of the 0 μM ARA reaction was subtracted from the other values. Individual graphs are shown for (A) 0N3R, (B) 1N3R, (C) 2N3R, (D) 0N4R, (E) 1N4R, and (F) 2N4R isoforms. Bars represent ± s.d. from 3 experiments.
Figure 5. Polymerization of 2 μM tau induced with varied ARA concentration (0 μM to 150 μM) measured by right-angle laser light scattering.

Wild-type (closed circles •) and 7-Phos tau isoform (open squares □) polymerization reactions with 2 μM protein (x-axis) and 0 to 75 μM arachidonic acid were measured by intensity of right-angle laser light scattering (y-axis) after 18 hours incubation at 25 °C. Individual graphs are shown for (A) 0N3R, (B) 1N3R, (C) 2N3R, (D) 0N4R, (E) 1N4R, and (F) 2N4R isoforms. Bars represent ± s.d. from 3 experiments.
The polymerization of 0N4R 7-Phos was greater than its corresponding wild-type at the higher inducer concentrations tested when measured by ThS fluorescence (Figure 4D). However, the LLS measurements indicated that polymerization of the 0N4R 7-Phos isoform was greater than that of the 0N4R wild-type isoform at 50 and 100 μM ARA but less than wild-type at the higher concentrations tested. The polymerization curves for 1N4R and 2N4R wild-type and 7-Phos variants were more similar to one another than what was observed for the other isoforms (Figure 4E-F and Figure 5E-F).
Any discrepancies between ThS staining and LLS readings could be due to subtle differences in how measurements from the two methods are affected by changes in length or other morphological characteristics in the population of filaments (14, 35–37). Fewer, but longer, filaments may decrease the amount of light scattered but could have little change in ThS binding if the total amount of polymerization is similar. In addition, changes in morphology, such as a change in the β-sheet structure or the formation of straight filaments rather than paired helical filaments, could also affect the affinity of ThS binding. Factors other than morphology changes, such as the formation of ARA micelles, could also have an effect on LLS values. Because of these challenges we believe that utilizing both methods, as well as transmission electron microscopy, will give us a clearer picture of alterations in the polymerization ability of these tau variants.
Visual inspection of the above curves revealed that most proteins at 2 μM concentration had a peak in polymerization either at or close to 75 μM ARA. As these polymerization conditions are routinely used for in vitro tau polymerization reactions, they were chosen for further analysis of the effects of the 7-Phos modification on the tau isoforms (27, 38, 39). The amounts of polymerization were determined after 18 hour incubation under these reaction conditions. The amounts of 7-Phos protein polymerization were normalized to their corresponding wild-type values to obtain a percent change from wild-type (Figure 6A and 6B). The 7-Phos pseudohyperphosphorylation mutation affected the 3R isoforms differently than it affected the 4R isoforms. The three 3R isoforms all showed a decrease in polymerization measured by ThS staining (Figure 6A) and a greater than 50% decrease in polymerization measured by LLS intensity (Figure 6B). In contrast, the 4R isoforms all had increased polymerization levels with the 7-Phos mutation according to ThS fluorescence (Figure 6A) and an increase or very little change in LLS values (Figure 6B).
Figure 6. Percent change in polymerization of 2 μM 7-Phos protein and 75 μM ARA polymerization reactions as compared to wild-type protein.

Polymerization reactions were prepared with 2 μM tau and 75 μM ARA and incubated for 18 hours at 25 °C. Polymerization was measured by (A) ThS fluorescence and (B) right-angle laser light scattering as described earlier. Results for each isoform are labeled on X-axis. Bars represent the average from 3 experiments ± s.d.
Pseudohyperphosphorylation affects the number and the morphology of tau filaments differently among the six isoforms
Samples of polymerization reactions under near optimal conditions (Figure 6) were examined by transmission electron microscopy to determine whether the 7-Phos mutation affected filament morphology (Figure 7). The mutation had varying effects on both the morphology and number of filaments present, although no changes were apparent in filament width or structure at higher magnification (data not shown). In general, filaments for the 7-Phos proteins were fewer in number, larger in size, and formed similar or less total mass of filaments (Figure 8). The exceptions were 0N4R 7-Phos (more filaments, more total mass) (Figure 8A and 8C), and 2N3R (shorter filaments) (Figure 8B). It should be mentioned that part of the effect seen on the number and morphology of 0N4R tau filaments is likely due to uneven distribution of wild type filaments, which is lessened by GSK-3β phosphorylation and is therefore likely to be decreased for 7-Phos 0N4R (33).
Figure 7. Electron Micrographs of 2 μM protein and 75 μM ARA polymerization reactions.
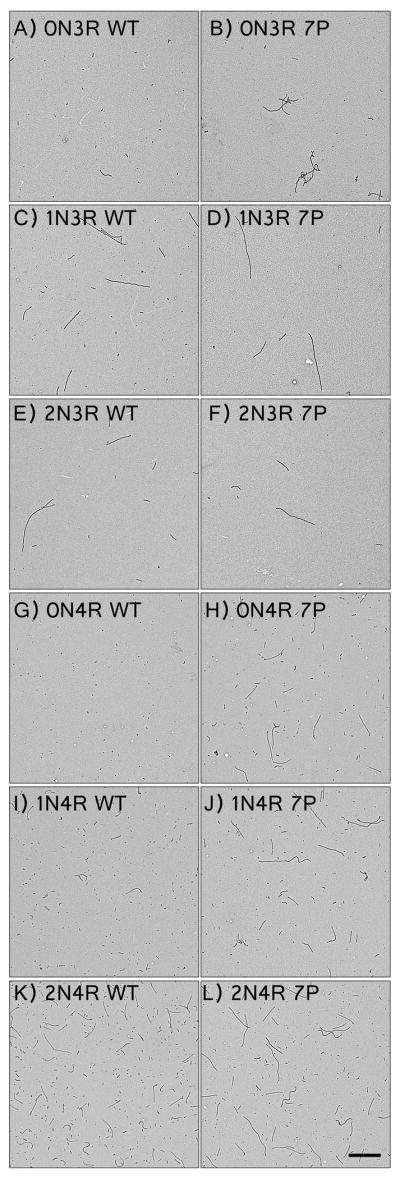
Representative electron micrographs of polymerization reactions of wild-type isoforms (left): (A) 0N3R, (C) 1N3R, (E) 2N3R, (G) 0N4R, (I) 1N4R, and (K) 2N4R; and corresponding 7-Phos isoforms (right): (B) 0N3R, (D) 1N3R, (F) 2N3R, (H) 0N4R, (J) 1N4R, and (L) 2N4R. The images were taken at a magnification of 3600x and the scale bar in (L) represent 1 μm and applies to all images.
Figure 8. Quantitation of filaments from transmission electron micrographs.
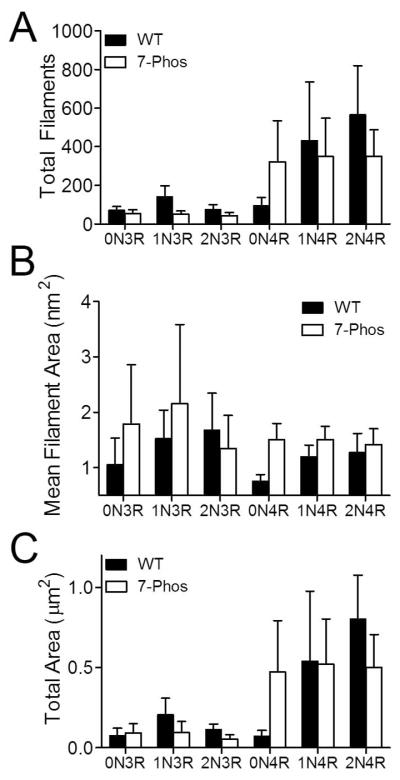
Quantitation of transmission electron micrographs from Figure 7 were performed as described in Materials and Methods. Values for (A) total number of filaments, (B) average filament length (μm), and (C) total length (number x average length) are shown for wild-type (black bars) and 7-Phos (white bars) for each isoform (labeled on x-axis). Data represent average values from 3 separate experiments ± s.d.
7-Phos affects the kinetics of polymerization
Polymerization reactions were monitored by LLS over time to measure the kinetics of polymerization. Wild-type 3R isoforms had very long lag times before polymerization was detected (Figure 9A-C), while the 4R isoforms had shorter lag times and reached maximum polymerization within approximately 80 minutes (Figure 9D-F).
Figure 9. Kinetics of tau polymerization.

Polymerization reactions were monitored by right-angle laser light scattering (arbitrary units, y-axis) at specific time points (minutes, x-axis) until the amount of polymerization approached an apparent steady-state. The experiment was repeated three times for the wild-type (closed circles •) and 7-Phos (open squares □) of (A) 0N3R, (B) 1N3R, (C) 2N3R, (D) 0N4R, (E) 1N4R, and (F) 2N4R. The equations were fit to the Gompertz equation and (G) the maximum amount of polymerization, (H) the lag time, and (I) the polymerization rate (kapp) for each was calculated. Isoforms are labeled on the x-axis. Data is shown as normalized values to the amount of wild-type polymerization (percent change from wild-type_ for three separate trials. Error bars represent the s.d. from 3 experiments. Data is not included for the 3R isoforms in (H) and (I) because the data could not be satisfactorily fit to the kinetics equation.
The maximum amount of polymerization was greatly decreased for the 3R 7-Phos isoforms compared to their wild-type counterparts (Figure 9A-C). As expected, very little polymerization was detected for the 3R 7-Phos isoforms by LLS and the data could not be fit to a kinetics equation (see Figure 5A-C). In contrast, the maximum amount of polymerization for the 4R 7-Phos isoforms were similar to or increased over their wild-type versions (Figure 9G) although their lag times before significant polymerization were longer (Figure 9H). The rate of polymerization was also slower (Figure 9I).
The ability of the protein to stabilize tubulin polymerization is affected by pseudohyperphosphorylation mutations
One of the normal functions of tau is the stabilization of microtubule assembly (1, 40–42). Phosphorylation of the protein can decrease its affinity for microtubules as well as its ability to induce the polymerization of tubulin dimers into microtubules (43–46). An in vitro microtubule polymerization assay was used to measure the effect of the 7-Phos pseudohyperphosphorylation mutation on the ability of each isoform to stabilize tubulin polymerization (Figure 10A-F).
Figure 10. Tau isoform stabilization of microtubule polymerization.

The ability of wild-type (•) and 7-Phos (□) tau isoforms to stabilize the polymerization of tubulin into microtubules was measured using a fluorescence assay. The relative fluorescence units (y-axis) are plotted against time (x-axis) for (A) 0N3R, (B) 1N3R, (C) 2N3R, (D) 0N4R, (E) 1N4R, and (F) 2N4R. Data points represent 3 separate trials ± s.d. The data was normalized to paclitaxel controls (crosshatches) that were performed in addition to tau reactions. Tau/tubulin reactions did not contain paclitaxel. Kinetics parameters were calculated from each of the three trials and the means of the percent change from wild-type with the addition of 7-Phos for (G) the kapp, or polymerization rate, (H) lag time, or the time from initiation of the reaction until polymerization is detected, and (I) the maximum polymerization after 60 minutes are shown ± s.d.
The 7-Phos mutation inhibited the ability of all six tau isoforms to stabilize the polymerization of tubulin. The rates of tubulin polymerization were decreased (Figure 10G), the lag times required to initiate microtubule polymerization were increased (Figure 10H), and the amounts of overall tubulin polymerization were reduced (Figure 10I). Of these, 7-Phos 0N4R seemed to be the least affected in all aspects.
DISCUSSION
The major common feature among neurodegenerative tauopathies is the polymerization of the tau protein into filamentous aggregates. While these aggregates are present in all tauopathies, the diseases can differ in the combination of tau isoforms included. The various combinations of isoforms present within the tangles can be used to classify the neurodegenerative tauopathies (7), indicating aggregation selectivity of certain isoforms in particular tauopathies. The causes of selective aggregation of tau isoforms are unknown but it is possible that phosphorylation plays an important role.
Phosphorylation of tau has long been associated with aggregation of tau in neurodegenerative tauopathies. The pathological tau found in neurofibrillary tangles is often hyperphosphorylated (47, 48). Kinases, such as GSK-3β, may display increased activity in Alzheimer’s disease (49) and are associated with neurofibrillary tangles (50). In addition, the activities of phosphatases, such as PP2A, are decreased in many tauopathies (51, 52). The phosphorylation of tau can have profound effects on its conformation. Although tau typically exists as a natively unfolded protein, it has been shown to adopt a global hairpin conformation with the N- and C-termini folding over to interact with its MTBR (24). Pseudophosphorylation at certain sites can affect the compaction of the protein (30, 53). Because the isoforms of tau differ from each other by up to 89 amino acids (20–25%), it is reasonable that similar levels or similar patterns of phosphorylation could have significantly different effects on the isoform conformations and functions.
Pseudophosphorylation can provide a useful way to explore the effects of site-specific phosphorylation on the conformation and function of a protein. However, we also recognize that it may not exactly replicate an actual phosphorylation event and all of its effects. While limitations exist and caution should be used in interpreting implications of the results, pseudophosphorylation can be a valuable tool to explore site-specific effects of phosphorylation and, indeed, has been used in multiple in vitro and cell culture studies of tau (23, 53–55).
In one such study, it has previously been reported that pseudophosphorylation mutations at seven sites on full-length 2N4R tau replicated certain characteristics associated with hyperphosphorylated tau (27). One of these characteristics was an SDS-resistant electrophoretic mobility shift, likely due to a conformational change in the protein altering the accessibility of the protein backbone to SDS. The pseudophosphorylation mutations at the seven sites also negatively affected the normal interaction of the protein with microtubules compared to wild-type 2N4R tau (27). In addition, the 7-Phos mutation affected the in vitro polymerization of the isoform by inducing longer, but less frequent, filament formation (27). We therefore sought to determine whether the same level and pattern of pseudophosphorylation mutations would have different effects on the function of the other tau isoforms.
In this study we have shown that pseudophosphorylation at seven identical sites on the backbone of the six tau isoforms resulted in an SDS-resistant electrophoretic shift in each protein, indicating that the conformation of the proteins had been altered by the 7-Phos mutation. However, the magnitude of these changes was not equal. Previous studies have shown that pseudophosphorylation of 2N4R tau at the S199, S202, S205, T212, S214, S396, and S404 sites induce conformational changes that increase the reaction of the protein with the conformational-dependent antibody MC1, an antibody that recognizes pathological tau associated with electrophoretic shifts (53, 56). However, there are conflicting results as to whether this pseudophosphorylation is causing a tightening or a loosening of the hairpin conformation (30, 53). Our results could coincide with either model but it seems likely that a global compaction of tau conformation would result in regions of tau becoming inaccessible, thereby reducing SDS binding and slowing electrophoretic migration. Our results would suggest that the level of compaction caused by pseudophosphorylation is not the same for the six isoforms, indicating that similar levels and sites of phosphorylation have different effects on the conformation of tau isoforms. These differential effects on conformation could lead to changes in the other properties of the protein.
In support of this, pseudohyperphosphorylation affected the ARA induction of tau polymerization differently among the isoforms. The 7-Phos 3R isoforms did not polymerize as well as their wild-type counterparts while 7-Phos 4R isoforms polymerized as well or better than wild-type 4R. This, combined with the differences in electrophoretic mobility, suggests the possibility that the MTBR of the 3R isoforms may be less accessible to ARA when they are in a conformation induced by the 7-Phos mutation. Previous results showing that the MTBR of tau isoforms differ from one another both structurally and mechanistically support this supposition (57). It is possible that different results could be obtained using different inducer molecules such as heparin, planar aromatic dyes, polyanionic microspheres, mRNA or alkyl sulfate detergents. While intriguing, we feel that these analyses are beyond the scope of the current manuscript and are more appropriate for future experimentation.
Because hyperphosphorylated tau is associated with aggregation in AD and other tauopathies we might expect that pseudophosphorylation would lead to increased polymerization in our in vitro reactions. However, for most of the isoforms, polymerization was similar or decreased compared to the corresponding wild-type isoforms. This could be due to the limitations of pseudophosphorylation as a model or the homogenous reactions give different results than a mixture of phosphorylated forms of tau. However, it is also possible that similar phosphorylation patterns could lead to different conformational effects on the isoforms. The varied conformational effects could then lead to increased aggregation for some isoforms while decreasing the likelihood of aggregation for others. Variable conformational changes could help explain why the isoform content of aggregates can vary by tauopathy.
Compaction of the carboxy-terminus toward the MTBR would be expected to inhibit tau aggregation (58, 59), while compaction of the amino-terminus toward the MTBR would be expected to enhance tau aggregation (38, 53). If this is the case, inhibition caused by compaction of the carboxy-terminus would likely be relatively similar among the isoforms since that region does not differ between them. However, the compaction of the amino terminus caused by phosphorylation would be expected to be different between the isoforms due to the presence or absence of exons 2 and 3, and we would therefore expect this compaction of the amino terminus to have more varied effects on polymerization of the isoforms. Indeed, polymerization of the 0N 7-Phos isoforms seems to be less inhibited compared to its wild-type counterparts than was observed for the 1N and 2N isoforms. In fact, polymerization of the 0N4R 7-Phos in enhanced compared to wild-type 0N4R tau under certain conditions. The absence of exons 2 and 3 could lead to a tighter compaction of the amino-terminus when these sites in the proline-rich region are phosphorylated. This tighter compaction could then lead to increased tau aggregation.
In general, the four-repeat isoforms seem to be less affected by pseudo-phosphorylation and compaction than the three-repeat isoforms, presumably due to the additional MTBR. This additional repeat reduces the impact of conformational-dependent enhancement or inhibition of tau aggregation leading to similar aggregation for wild-type and 7-Phos of each isoforms. It is likely that the additional repeat either results in a structural conformation that protects the MTBR from the amino and carboxy terminals, or that the additional repeat provides a second site for inducer interactions, thereby overcoming inhibition or enhancement by mass action.
Other differences in the polymerization of 7-Phos isoforms existed at the optimal ratio of 2 μM protein and 75 μM ARA. All of the 7-Phos isoforms polymerized more slowly than wild-type and fewer, but longer, filaments were formed for many of the 7-Phos isoforms. This also suggests that compaction due to phosphorylation could negatively affect the ARA-induced nucleation of filaments but could also stabilize filament elongation. This compaction could be differentially affecting the accessibility of the MTBR as discussed above, resulting in less nucleation and slower growth, causing the differences in morphology observed between isoforms. The effect was greatest in 0N4R tau which displayed an increase in polymerization, number of filaments formed, and length of filaments with the 7-Phos pseudophosphorylation mutation. Part of this effect could be due to increased or improved filament distribution on the electron microscopy grids (33) but the agreement between the SDS-PAGE migration, quantitative electron microscopy, LLS, and ThS fluorescence data suggest that the 0N4R 7-Phos has a more accessible MTBR.
Differences in accessibility of the MTBR would be expected to affect the ability of tau to stabilize microtubule polymerization as well. The 7-Phos mutation caused a reduction in the ability of all six isoforms to stabilize microtubules. The overall amount of tubulin polymerization was decreased, the lag times were increased, and the rate of polymerization was decreased. The degrees of changes were not equal among the isoforms. For example, 0N4R tau had much less severe defects in its ability to stabilize microtubules. Additionally, the 0N3R 7-Phos stabilized microtubule growth more readily than the other 3R 7-Phos isoforms did. This indicates that phosphorylation of 0N isoforms could cause the MTBR to be more accessible than in 1N or 2N isoforms. In general, pseudohyperphosphorylated 4R isoforms had more similar aggregation and microtubule stabilization characteristics as compared to their wild-type counterparts than the 3R isoforms. This indicates that compaction of tau due to phosphorylation may be obscuring the MTBRs more for 3R isoforms than 4R isoforms.
In summary, our results are consistent with similar levels and patterns of phosphorylation having substantially different effects on tau isoforms. This result is important considering efforts to target phosphorylation as a therapeutic intervention for tauopathies. Our results suggest that such therapies will have different effects on the six isoforms of tau which could lead to complications. They also suggest that animal models used to study the effects of phosphorylation on the toxicity of tau would be affected by the choice of isoforms. Future studies will be required to determine whether these results are specific to these sites and this inducer or whether similar results will be obtained using alternative inducers or different sites to generate alternative pseudohyperphosphorylated versions of tau isoforms.
Acknowledgments
We thank Akosua Kernizan for and James Odum for their assistance in generating the 7-Phos constructs of tau isoforms.
Abbreviations
- AD
Alzheimer’s disease
- ARA
arachidonic acid
- GSK-3β
glycogen synthase kinase-3β
- LLS
right-angle laser light scattering
- MTBR
microtubule-binding repeat region
- ThS
thioflavin S
Footnotes
Support was provided by the National Institutes of Health grants AG022428 (T.C.G.) and AG025898 (T.C.G.) and the J.R. and Inez Jay Fund (T.C.G.)
References
- 1.Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW. A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A. 1975;72:1858–1862. doi: 10.1073/pnas.72.5.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gauthier-Kemper A, Weissmann C, Golovyashkina N, Sebo-Lemke Z, Drewes G, Gerke V, Heinisch JJ, Brandt R. The frontotemporal dementia mutation R406W blocks tau’s interaction with the membrane in an annexin A2-dependent manner. J Cell Biol. 2011;192:647–661. doi: 10.1083/jcb.201007161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wolfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Gotz J. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- 4.Lee G, Newman ST, Gard DL, Band H, Panchamoorthy G. Tau interacts with src-family non-receptor tyrosine kinases. J Cell Sci. 1998;111(Pt 21):3167–3177. doi: 10.1242/jcs.111.21.3167. [DOI] [PubMed] [Google Scholar]
- 5.Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron. 1989;3:519–526. doi: 10.1016/0896-6273(89)90210-9. [DOI] [PubMed] [Google Scholar]
- 6.Himmler A, Drechsel D, Kirschner MW, Martin DW., Jr Tau consists of a set of proteins with repeated C-terminal microtubule-binding domains and variable N-terminal domains. Mol Cell Biol. 1989;9:1381–1388. doi: 10.1128/mcb.9.4.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sergeant N, Delacourte A, Buee L. Tau protein as a differential biomarker of tauopathies. Biochim Biophys Acta. 2005;1739:179–197. doi: 10.1016/j.bbadis.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 8.Goedert M, Wischik CM, Crowther RA, Walker JE, Klug A. Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as the microtubule-associated protein tau. Proc Natl Acad Sci U S A. 1988;85:4051–4055. doi: 10.1073/pnas.85.11.4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mailliot C, Sergeant N, Bussiere T, Caillet-Boudin ML, Delacourte A, Buee L. Phosphorylation of specific sets of tau isoforms reflects different neurofibrillary degeneration processes. FEBS Lett. 1998;433:201–204. doi: 10.1016/s0014-5793(98)00910-7. [DOI] [PubMed] [Google Scholar]
- 10.Probst A, Tolnay M, Langui D, Goedert M, Spillantini MG. Pick’s disease: hyperphosphorylated tau protein segregates to the somatoaxonal compartment. Acta Neuropathol (Berl) 1996;92:588–596. doi: 10.1007/s004010050565. [DOI] [PubMed] [Google Scholar]
- 11.Iqbal K, Alonso Adel C, Chen S, Chohan MO, El-Akkad E, Gong CX, Khatoon S, Li B, Liu F, Rahman A, Tanimukai H, Grundke-Iqbal I. Tau pathology in Alzheimer disease and other tauopathies. Biochim Biophys Acta. 2005;1739:198–210. doi: 10.1016/j.bbadis.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 12.Lindwall G, Cole RD. Phosphorylation affects the ability of tau protein to promote microtubule assembly. J Biol Chem. 1984;259:5301–5305. [PubMed] [Google Scholar]
- 13.Rankin CA, Sun Q, Gamblin TC. Pre-assembled tau filaments phosphorylated by GSK-3β form large tangle-like structures. Neurobiol Dis. 2008;31:368–377. doi: 10.1016/j.nbd.2008.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carlson SW, Branden M, Voss K, Sun Q, Rankin CA, Gamblin TC. A complex mechanism for inducer mediated tau polymerization. Biochemistry. 2007;46:8838–8849. doi: 10.1021/bi700403a. [DOI] [PubMed] [Google Scholar]
- 15.Balaraman Y, Limaye AR, Levey AI, Srinivasan S. Glycogen synthase kinase 3beta and Alzheimer’s disease: pathophysiological and therapeutic significance. Cell Mol Life Sci. 2006;63:1226–1235. doi: 10.1007/s00018-005-5597-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Song JS, Yang SD. Tau protein kinase I/GSK-3 beta/kinase FA in heparin phosphorylates tau on Ser199, Thr231, Ser235, Ser262, Ser369, and Ser400 sites phosphorylated in Alzheimer disease brain. J Protein Chem. 1995;14:95–105. doi: 10.1007/BF01888367. [DOI] [PubMed] [Google Scholar]
- 17.Ishiguro K, Shiratsuchi A, Sato S, Omori A, Arioka M, Kobayashi S, Uchida T, Imahori K. Glycogen synthase kinase 3 beta is identical to tau protein kinase I generating several epitopes of paired helical filaments. FEBS Lett. 1993;325:167–172. doi: 10.1016/0014-5793(93)81066-9. [DOI] [PubMed] [Google Scholar]
- 18.Ishiguro K, Takamatsu M, Tomizawa K, Omori A, Takahashi M, Arioka M, Uchida T, Imahori K. Tau protein kinase I converts normal tau protein into A68-like component of paired helical filaments. J Biol Chem. 1992;267:10897–10901. [PubMed] [Google Scholar]
- 19.Imahori K, Uchida T. Physiology and pathology of tau protein kinases in relation to Alzheimer’s disease. J Biochem (Tokyo) 1997;121:179–188. [PubMed] [Google Scholar]
- 20.Kobayashi S, Ishiguro K, Omori A, Takamatsu M, Arioka M, Imahori K, Uchida T. A cdc2-related kinase PSSALRE/cdk5 is homologous with the 30 kDa subunit of tau protein kinase II, a proline-directed protein kinase associated with microtubule. FEBS Lett. 1993;335:171–175. doi: 10.1016/0014-5793(93)80723-8. [DOI] [PubMed] [Google Scholar]
- 21.Mi K, Johnson GV. The role of tau phosphorylation in the pathogenesis of Alzheimer’s disease. Curr Alzheimer Res. 2006;3:449–463. doi: 10.2174/156720506779025279. [DOI] [PubMed] [Google Scholar]
- 22.Leger J, Kempf M, Lee G, Brandt R. Conversion of serine to aspartate imitates phosphorylation-induced changes in the structure and function of microtubule-associated protein tau. J Biol Chem. 1997;272:8441–8446. doi: 10.1074/jbc.272.13.8441. [DOI] [PubMed] [Google Scholar]
- 23.Eidenmuller J, Fath T, Hellwig A, Reed J, Sontag E, Brandt R. Structural and functional implications of tau hyperphosphorylation: information from phosphorylation-mimicking mutated tau proteins. Biochemistry. 2000;39:13166–13175. doi: 10.1021/bi001290z. [DOI] [PubMed] [Google Scholar]
- 24.Jeganathan S, von Bergen M, Brutlach H, Steinhoff HJ, Mandelkow E. Global hairpin folding of tau in solution. Biochemistry. 2006;45:2283–2293. doi: 10.1021/bi0521543. [DOI] [PubMed] [Google Scholar]
- 25.Necula M, Kuret J. Site-specific pseudophosphorylation modulates the rate of tau filament dissociation. FEBS Lett. 2005;579:1453–1457. doi: 10.1016/j.febslet.2005.01.047. [DOI] [PubMed] [Google Scholar]
- 26.Necula M, Kuret J. Pseudophosphorylation and glycation of tau protein enhance but do not trigger fibrillization in vitro. J Biol Chem. 2004;279:49694–49703. doi: 10.1074/jbc.M405527200. [DOI] [PubMed] [Google Scholar]
- 27.Sun Q, Gamblin TC. Pseudohyperphosphorylation causing AD-like changes in tau has significant effects on its polymerization. Biochemistry. 2009;48:6002–6011. doi: 10.1021/bi900602h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fischer D, Mukrasch MD, Biernat J, Bibow S, Blackledge M, Griesinger C, Mandelkow E, Zweckstetter M. Conformational changes specific for pseudophosphorylation at serine 262 selectively impair binding of tau to microtubules. Biochemistry. 2009;48:10047–10055. doi: 10.1021/bi901090m. [DOI] [PubMed] [Google Scholar]
- 29.Ding H, Matthews TA, Johnson GV. Site-specific phosphorylation and caspase cleavage differentially impact tau-microtubule interactions and tau aggregation. J Biol Chem. 2006;281:19107–19114. doi: 10.1074/jbc.M511697200. [DOI] [PubMed] [Google Scholar]
- 30.Bibow S, Ozenne V, Biernat J, Blackledge M, Mandelkow E, Zweckstetter M. Structural Impact of Proline-Directed Pseudophosphorylation at AT8, AT100, and PHF1 Epitopes on 441-Residue Tau. J Am Chem Soc. 2011 doi: 10.1021/ja205836j. [DOI] [PubMed] [Google Scholar]
- 31.Rankin CA, Sun Q, Gamblin TC. Pseudo-phosphorylation of tau at Ser202 and Thr205 affects tau filament formation. Brain Res Mol Brain Res. 2005 doi: 10.1016/j.molbrainres.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 32.Gamblin TC, King ME, Dawson H, Vitek MP, Kuret J, Berry RW, Binder LI. In vitro polymerization of tau protein monitored by laser light scattering: method and application to the study of FTDP-17 mutants. Biochemistry. 2000;39:6136–6144. doi: 10.1021/bi000201f. [DOI] [PubMed] [Google Scholar]
- 33.Voss K, Gamblin TC. GSK-3β phosphorylation of functionally distinct tau isoforms has differential, but mild effects. Mol Neurodegener. 2009;4:1–12. doi: 10.1186/1750-1326-4-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Friedhoff P, Schneider A, Mandelkow EM, Mandelkow E. Rapid assembly of Alzheimer-like paired helical filaments from microtubule-associated protein tau monitored by fluorescence in solution. Biochemistry. 1998;37:10223–10230. doi: 10.1021/bi980537d. [DOI] [PubMed] [Google Scholar]
- 35.Berne BJ. Interpretation of the light scattering from long rods. J Mol Biol. 1974;89:755–758. doi: 10.1016/0022-2836(74)90049-7. [DOI] [PubMed] [Google Scholar]
- 36.Santa-Maria I, Perez M, Hernandez F, Avila J, Moreno FJ. Characteristics of the binding of thioflavin S to tau paired helical filaments. J Alzheimers Dis. 2006;9:279–285. doi: 10.3233/jad-2006-9307. [DOI] [PubMed] [Google Scholar]
- 37.Barghorn S, Mandelkow E. Toward a unified scheme for the aggregation of tau into Alzheimer paired helical filaments. Biochemistry. 2002;41:14885–14896. doi: 10.1021/bi026469j. [DOI] [PubMed] [Google Scholar]
- 38.Gamblin TC, Berry RW, Binder LI. Tau polymerization: role of the amino terminus. Biochemistry. 2003;42:2252–2257. doi: 10.1021/bi0272510. [DOI] [PubMed] [Google Scholar]
- 39.Wilson DM, Binder LI. Free fatty acids stimulate the polymerization of tau and amyloid beta peptides. In vitro evidence for a common effector of pathogenesis in Alzheimer’s disease. Am J Pathol. 1997;150:2181–2195. [PMC free article] [PubMed] [Google Scholar]
- 40.Witman GB, Cleveland DW, Weingarten MD, Kirschner MW. Tubulin requires tau for growth onto microtubule initiating sites. Proc Natl Acad Sci U S A. 1976;73:4070–4074. doi: 10.1073/pnas.73.11.4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Esmaeli-Azad B, McCarty JH, Feinstein SC. Sense and antisense transfection analysis of tau function: tau influences net microtubule assembly, neurite outgrowth and neuritic stability. J Cell Sci. 1994;107:869–879. doi: 10.1242/jcs.107.4.869. [DOI] [PubMed] [Google Scholar]
- 42.Drubin DG, Kirschner MW. Tau protein function in living cells. J Cell Biol. 1986;103:2739–2746. doi: 10.1083/jcb.103.6.2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Biernat J, Gustke N, Drewes G, Mandelkow EM, Mandelkow E. Phosphorylation of Ser262 strongly reduces binding of tau to microtubules: distinction between PHF-like immunoreactivity and microtubule binding. Neuron. 1993;11:153–163. doi: 10.1016/0896-6273(93)90279-z. [DOI] [PubMed] [Google Scholar]
- 44.Cho JH, Johnson GV. Glycogen synthase kinase 3beta phosphorylates tau at both primed and unprimed sites. Differential impact on microtubule binding. J Biol Chem. 2003;278:187–193. doi: 10.1074/jbc.M206236200. [DOI] [PubMed] [Google Scholar]
- 45.Lu PJ, Wulf G, Zhou XZ, Davies P, Lu KP. The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature. 1999;399:784–788. doi: 10.1038/21650. [DOI] [PubMed] [Google Scholar]
- 46.Yoshida H, Ihara Y. Tau in paired helical filaments is functionally distinct from fetal tau: assembly incompetence of paired helical filament-tau. J Neurochem. 1993;61:1183–1186. doi: 10.1111/j.1471-4159.1993.tb03642.x. [DOI] [PubMed] [Google Scholar]
- 47.Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A. 1986;83:4913–4917. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee VM, Balin BJ, Otvos L, Jr, Trojanowski JQ. A68: a major subunit of paired helical filaments and derivatized forms of normal tau. Science. 1991;251:675–678. doi: 10.1126/science.1899488. [DOI] [PubMed] [Google Scholar]
- 49.Leroy K, Yilmaz Z, Brion JP. Increased level of active GSK-3beta in Alzheimer’s disease and accumulation in argyrophilic grains and in neurones at different stages of neurofibrillary degeneration. Neuropathol Appl Neurobiol. 2007;33:43–55. doi: 10.1111/j.1365-2990.2006.00795.x. [DOI] [PubMed] [Google Scholar]
- 50.Pei JJ, Braak E, Braak H, Grundke-Iqbal I, Iqbal K, Winblad B, Cowburn RF. Distribution of active glycogen synthase kinase 3beta (GSK-3beta) in brains staged for Alzheimer disease neurofibrillary changes. J Neuropathol Exp Neurol. 1999;58:1010–1019. doi: 10.1097/00005072-199909000-00011. [DOI] [PubMed] [Google Scholar]
- 51.Gong CX, Shaikh S, Wang JZ, Zaidi T, Grundke-Iqbal I, Iqbal K. Phosphatase activity toward abnormally phosphorylated tau: decrease in Alzheimer disease brain. J Neurochem. 1995;65:732–738. doi: 10.1046/j.1471-4159.1995.65020732.x. [DOI] [PubMed] [Google Scholar]
- 52.Gong CX, Singh TJ, Grundke-Iqbal I, Iqbal K. Phosphoprotein phosphatase activities in Alzheimer disease brain. J Neurochem. 1993;61:921–927. doi: 10.1111/j.1471-4159.1993.tb03603.x. [DOI] [PubMed] [Google Scholar]
- 53.Jeganathan S, Hascher A, Chinnathambi S, Biernat J, Mandelkow EM, Mandelkow E. Proline-directed pseudo-phosphorylation at AT8 and PHF1 epitopes induces a compaction of the paperclip folding of Tau and generates a pathological (MC-1) conformation. J Biol Chem. 2008;283:32066–32076. doi: 10.1074/jbc.M805300200. [DOI] [PubMed] [Google Scholar]
- 54.Haase C, Stieler JT, Arendt T, Holzer M. Pseudophosphorylation of tau protein alters its ability for self-aggregation. J Neurochem. 2004;88:1509–1520. doi: 10.1046/j.1471-4159.2003.02287.x. [DOI] [PubMed] [Google Scholar]
- 55.Fath T, Eidenmuller J, Brandt R. Tau-mediated cytotoxicity in a pseudohyperphosphorylation model of Alzheimer’s disease. J Neurosci. 2002;22:9733–9741. doi: 10.1523/JNEUROSCI.22-22-09733.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jicha GA, Bowser R, Kazam IG, Davies P. Alz-50 and MC-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. J Neurosci Res. 1997;48:128–132. doi: 10.1002/(sici)1097-4547(19970415)48:2<128::aid-jnr5>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 57.Goode BL, Chau M, Denis PE, Feinstein SC. Structural and functional differences between 3-repeat and 4-repeat tau isoforms. Implications for normal tau function and the onset of neurodegenerative disease. J Biol Chem. 2000;275:38182–38189. doi: 10.1074/jbc.M007489200. [DOI] [PubMed] [Google Scholar]
- 58.Abraha A, Ghoshal N, Gamblin TC, Cryns V, Berry RW, Kuret J, Binder LI. C-terminal inhibition of tau assembly in vitro and in Alzheimer’s disease. J Cell Sci. 2000;113(Pt 21):3737–3745. doi: 10.1242/jcs.113.21.3737. [DOI] [PubMed] [Google Scholar]
- 59.Berry RW, Abraha A, Lagalwar S, LaPointe N, Gamblin TC, Cryns VL, Binder LI. Inhibition of tau polymerization by its carboxy-terminal caspase cleavage fragment. Biochemistry. 2003;42:8325–8331. doi: 10.1021/bi027348m. [DOI] [PubMed] [Google Scholar]