

Abstract
Cardiac hypertrophy is triggered in response to mechanical stress and various neurohumoral factors, such as G-protein coupling receptor (GPCR) and gp130 cytokine receptor agonists. Recent studies have suggested cardiac Z-disc plays a pivotal role to regulate these cellular responses. Here, we demonstrate stimulations with GPCR agonists (norepinephrine, angiotensin II, and endothelin 1) and phorbol ester activated and translocated protein kinase D1 (PKD1) to the Z-discs in neonatal rat cardiomyocytes in a protein kinase C (PKC)-dependent manner, whereas gp130 agonist did not. Especially, upon the α-adrenergic receptor agonist stimulations, following the PKCε–PKD1 complex formation, PKCε-dependent activation of PKD1 was essential to induce hypertrophic responses. Constitutively active mutant of either PKD1 or PKCε also induced cardiac hypertrophy ex vivo. Taken together, the PKCε–PKD1 complex at Z-discs could play a pivotal role in the cardiac hypertrophy induced by GPCR agonists, at least α-adrenergic receptor agonist.
Keywords: PKD1, PKC, Hypertrophy, Cardiomyocyte, Z-disc, G-protein coupling receptor, Gp130, α-Adrenergic receptor, Phorbol ester
In heart, hypertrophy is a cellular response to hemodynamic stress, including arterial hypertension. Prolonged stress can lead to heart failure, resulting in the substantial increase in morbidity, and mortality of diseased human population [1]. Cardiac hypertrophy has been extensively analyzed using cultured neonatal rat cardiomyocytes, in which reactions are elicited by various stimuli, such as cytokines and neurohumoral factors [2]. Each stimulus triggers various and specific signaling pathways. Therefore, the elucidation of these pathways in cardiomyocytes is crucial for understanding the molecular mechanism of cardiac hypertrophy development.
Protein kinase D1 (PKD1) is a serine/threonine kinase [3,4], which can be activated by diacylglycerol, phorbol esters, and growth factors through a protein kinase C (PKC)-dependent signaling pathway [4,5]. The mechanism of PKD1 activation involves the phosphorylation of two serine residues (Ser 744 and Ser 748) within an activation loop in the C-terminal catalytic domain of PKD1 [6]. Various PKC isoforms (PKCη, PKCε, PKCθ, and PKCβI) are able to phosphorylate the activation loop of PKD1 in vitro or in vivo, indicating that PKD1 is a kinase acting downstream of PKC [7]. PKD2, a homologue of PKD1, which shares similar mechanisms of activation, has also been cloned [8]. PKD1 is also expressed in cardiomyocytes, and its expression is dependent on the heart's developmental stage, and α-adrenergic stimulation and phorbol esters activate PKD1 in a PKC-dependent manner [9]. However, the physiological significance of cardiac PKD1 has remained unsettled [10]. Here, we show that PKD1 is activated and translocates to the Z-discs after stimulations with phorbol ester or GPCR agonists such as α-adrenergic agent (norepinephrine, NE), angiotensin II (AngII), and endothelin I (ET1) in neonatal rat cardiomyocytes. The translocation and the activity of PKD1 are dependent on the activation of PKC (presumably PKCε). Constitutively active mutant of either PKD1 or PKCε spontaneously induces the Z-disc assembly, and increased expression of atrial natriuretic factor (ANF), an embryonic gene marker in cardiomyocytes. Upon norepinephrine stimulation, PKCε and PKD1 are assembled into a signaling complex at the Z-discs. This complex could participate in the development of cardiac hypertrophy induced by GPCR agonists (at least α-adrenergic receptor agonist).
Materials and methods
Plasmid constructions
A plasmid for the C-terminally HA-tagged PKCβI-DN (a dominant negative mutant of PKCβI) was generated by replacing Lys 371 with Met by site-directed mutagenesis. Plasmids for PKCζ-DN (K281M, HA-tagged) and PKCε-DN (K440R, HA-tagged) were described previously [11]. The plasmid for PKCε-CA (a constitutively active mutant of PKCε) was kindly provided by Prof. P.J. Parker [12]. Plasmids for the N-terminally GFP (green fluorescent protein)-fused PKD1-CA and PKD1-DN were constructed by mutagenesis (S744E/S748E and K618N, respectively) as described previously [13].
Antibodies
An anti-PKD1/2 monoclonal antibody (LC Laboratories), an anti-PKCε polyclonal antibody (Santa Cruz), an anti-sarcomeric α-actinin monoclonal antibody (Sigma, clone EA-53), an anti-H3 histone monoclonal antibody (Upstate Biotechnology), and an anti-ANF polyclonal antibody (BACHEM) were obtained commercially. An anti-active PKD1 polyclonal antibody was raised in rabbits by immunization with a phosphorylated C-terminal peptide of PKD1 [amino acid residues (aa) 912–918; SERVpSIL, where pS is phosphoserine] as described previously [14]. Secondary antibodies linked with horseradish peroxidase or fluorophores Cy2 and Cy3 were purchased from Amersham–Pharmacia.
Cell culture
Neonatal rat ventricular myocytes were prepared as described previously [15]. Briefly, hearts collected from 1- to 3-day-old Wistar rats were washed with phosphate-buffered saline (PBS) and treated three times with 1% (w/v) collagenase type I (Wako Pure Chemicals) at 37 °C for 20 min to disperse the cells. The cells were transferred to culture plates, and the fibroblasts were removed by adhesion onto the plates for at least 1 h. The cardiomyocytes were then plated overnight and maintained in DMEM containing 0.45% glucose, 10% (v/v) fetal bovine serum (FBS), 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin (Sigma). Plasmids were introduced into the cells either by using a transfection reagent, Duo Fect (Q-Biogen), or by electroporation using the Amaxa electroporator with the Rat Cardiomyocyte-Neonatal Nucleofector kit according to the manufacturer's instructions (Amaxa GmbH). Protein expression was verified 48 h after transfection.
Immunoblotting
Cells were treated with lysis buffer A [50 mM Tris (pH 7.4), 150 mM NaCl, 1.3% (v/v) Triton X-100, 1 mM EDTA, 1 mM EGTA, 1 mM dithiothreitol, 50 mM NaF, 1 mM Na3VO4, and protease inhibitor cocktail tablet (Roche)]. Total cell lysates were resolved by 10% SDS–PAGE, transferred to PVDF membranes (Immobilon-P, Millipore), and immunoblotted with the indicated antibodies. For detection, blots were visualized by enhanced chemiluminescence (ECL plus; Amersham–Pharmacia) using ECL films (Amersham–Pharmacia).
Immunofluorescence
Neonatal rat cardiomyocytes expressing GFP and GFP-PKD1-CA were grown on the poly-l-lysine-coated, 35-mm glass-bottomed dish (MATSUNAMI). Cells were washed twice with PBS, fixed with 4% (w/v) paraformaldehyde at room temperature for 30 min, and washed again twice with PBS. The fluorescence of GFP was observed using a confocal laser microscope LSM5 Pascal (Carl Zeiss). For immunodetection, the fixed cells were permeabilized in PBS containing 0.25% (v/v) Triton X-100 at 4 °C for 30 min, and then incubated in blocking buffer [PBS containing 3% (w/v) bovine serum albumin, 2% (v/v) FBS, 1% (v/v) normal goat serum, and 0.03% (v/v) Triton X-100]. The cells were incubated at 4 °C overnight in the blocking buffer containing the primary antibody. Dilutions used for the primary antibodies were as follows: 1/80 for anti-active and anti-inactive PKD1 polyclonal; and 1/100 for anti-ANF polyclonal and anti-sarcomeric α-actinin monoclonal. The cells were washed twice with PBS, and then incubated with either one or both of 0.3% (v/v) Cy2- and Cy3-conjugated secondary antibodies in PBS at room temperature for 1 h. The fluorescence of cells was observed under a confocal microscope.
Immunoprecipitation kinase assay
Cells were lysed using lysis buffer A and incubated with an anti-PKD monoclonal antibody at 4 °C for 60 min. Proteins were then adsorbed onto Sepharose 4B beads bound with protein G The beads were collected by brief centrifugation, washed once with lysis buffer A and twice with lysis buffer A without NaF, and re-suspended in 50 μl of the buffer. A 10-μl aliquot of the suspension was incubated for 5 min at 30 °C with an assay mixture (40 μl), containing 37.5 mM Tris, 37.5 mM MgCl2, 0.1 mM ATP, 5.8 μCi [γ-32P]ATP, and 40 μg of a synthetic peptide Syntide-2 (APLARTLSVAGLPGKK), a specific substrate for PKD1. The reaction mixture was spotted on pieces of P-81 phosphocellulose filter paper. After washing the paper in 75 mM phosphoric acid, the bound radioactivity was measured using a liquid scintillation counter.
Results
Expression and intracellular localization of PKD1 in phorbol ester-treated neonatal rat cardiomyocyte
We first studied the expression of PKD1 and PKD2 in the cultured neonatal rat cardiomyocytes by Western blotting using an anti-PKD1/2 monoclonal antibody. The cells were treated with a phorbol ester (TPA, 12-O-tetradecanoylphorbol-13-acetate) for 20 min to activate the signaling pathways involving PKC and PKD enzymes. As shown in Fig. 1A, PKD1 (about 115 kDa) was detected in cytoplasm of both TPA-treated and untreated cells, while PKD2 (about 105 kDa) was not detected. The membrane fractions also contained PKD1 but not PKD2 in both TPA-treated and untreated cells. The amount of PKD1 in the membrane fraction was higher in the TPA-treated cells than in the untreated cells, suggesting rapid translocation of PKD1 to cell membranes after TPA-induced activation. Furthermore, the amount of the active, phosphorylated form of PKD1 significantly increased in the membrane fraction upon TPA treatment (2nd panel), whereas that of the inactive, dephosphorylated form of PKD1 did not change (not shown). Thus, upon TPA treatment, the active PKD1 translocates from cytoplasm to membrane in cardiomyocytes.
Fig. 1.
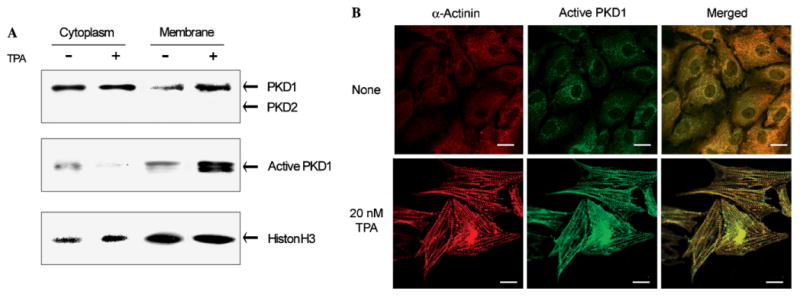
Intracellular localization of PKD1 in TPA-treated neonatal rat cardiomyocytes. (A) The cultured neonatal rat cardiomyocytes were treated with 20 nM TPA for 20 min in a serum-free medium. Cytoplasmic and membrane fractions were prepared using a standard procedure. Cytoplasmic and membrane fractions of the lysates of untreated (−) and TPA-treated (+) cells were subjected to SDS-PAGE (15 μg protein/lane), and then analyzed by an immunoblotting with the corresponding antibodies. Histone H3 was used as a control for calibration. (B) The cells were treated with 20 nM TPA for 18 h, and the TPA-treated and untreated cells were doubly stained for α-actinin with a Cy3-conjugated secondary antibody (emission at 570 nm) and for active PKD1 with a Cy2-conjugated secondary antibody (emission at 506 nm). Scale bar: 10 μm.
We next investigated by immunofluorescence the intracellular localization of PKD1 in neonatal rat cardiomyocytes. Before TPA treatment, no Z-disc is observed and less PKD1 is detected as the active form that localizes in the perinuclear region (Fig. 1B). When the cells were stimulated with TPA, the active PKD1 clearly localized at the newly formed Z-discs, as shown in Fig. 1B. These results demonstrate that in neonatal rat cardiomyocytes, stimulation with a phorbol ester induces acute translocation of active PKD1 to membrane (within 20 min) and then chronically to the Z-discs (after 18 h).
NE, ET1, and AngII, but not leukemia inhibitory factor, induce activation and translocation of PKD1 to Z-discs
Because stimulations with GPCR agonists such as NE, ET1, AngII, and a gp130 cytokine receptor agonist leukemia inhibitory factor (LIF) are known to provoke cardiac hypertrophy [2], we were interested to study the translocation of PKD1 in the context of cardiac hypertrophy treated with these agonists. Neonatal rat cardiomyocytes were stimulated for at least 38 h with either (1) NE (100 μM) in combination with propranolol (2 μM), a β-adrenergic blocker, to selectively stimulate the α-adrenergic receptors, (2) AngII (100 nM), (3) ET1 (10 nM), or (4) LIF (20 nM) that induces cardiac hypertrophy through the gp130 receptor. After stimulation with NE, AngII, and ET1, the active PKD1 colocalized with α-actinin that formed the sarcomeric Z-discs (Fig. 2). Interestingly, neither activation of PKD1 nor its translocation to the Z-discs was observed upon stimulation of the gp130 receptor with LIF.
Fig. 2.
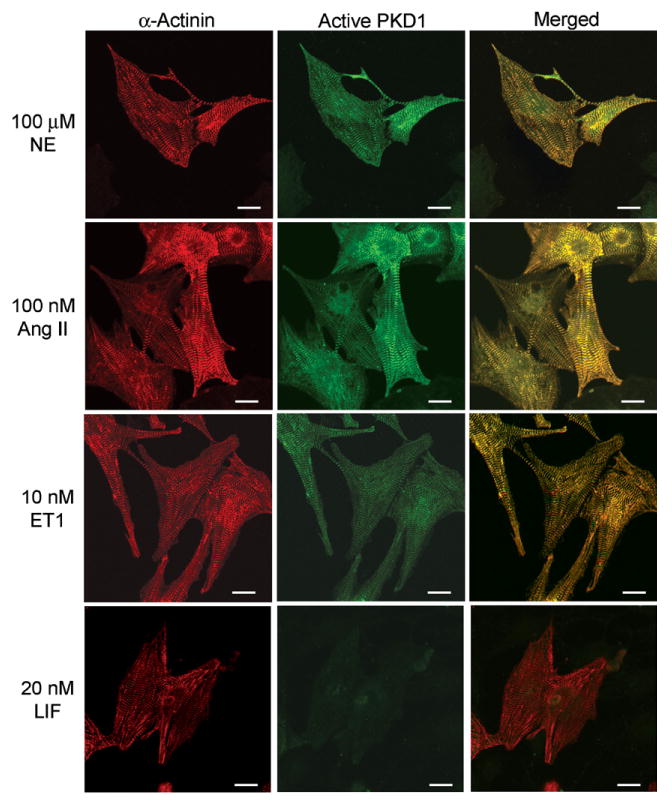
Translocation of active PKD1 to Z-discs by stimulation with neurohumoral factors. Neonatal rat cardiomyocytes were stimulated with either 100 μM NE plus 2 μM propranolol, 100 nM AngII, 10 nM ET1, or 20 nM LIF for 38 h. Immunofluorescence detection was done as described in Fig. 1B. Scale bar: 10 μm.
Since PKD1 activation generally depends on the phosphorylation of its active loop by PKC isoforms [16], we next examined whether PKC is involved in the activation of PKD1 in neonatal rat cardiomyocytes by using GF109203X, a specific inhibitor for conventional PKC isoforms (PKCα, βI, βII, and γ) and novel PKC isoforms (PKCδ, ε, η, and θ) [17]. When cardiomyocytes were stimulated with NE in the presence of propranolol and GF109203X, hypertrophic responses were strongly suppressed and the active PKD1 was scarcely detectable without localizing at poorly formed Z-discs (Fig. 3). We observed the similar results using the combination of TPA and GF109203X. In marked contrast, GF109203X treatment did not suppress LIF-stimulated sarcomeric Z-disc formation and the active PKD1 was undetectable in hypertrophic cardiomyocytes. Taken together, our results strongly suggest that stimulations with NE, TPA, AngII, and ET1 (but not LIF) induce the translocation of active PKD1 to the Z-discs in a PKC-dependent manner.
Fig. 3.
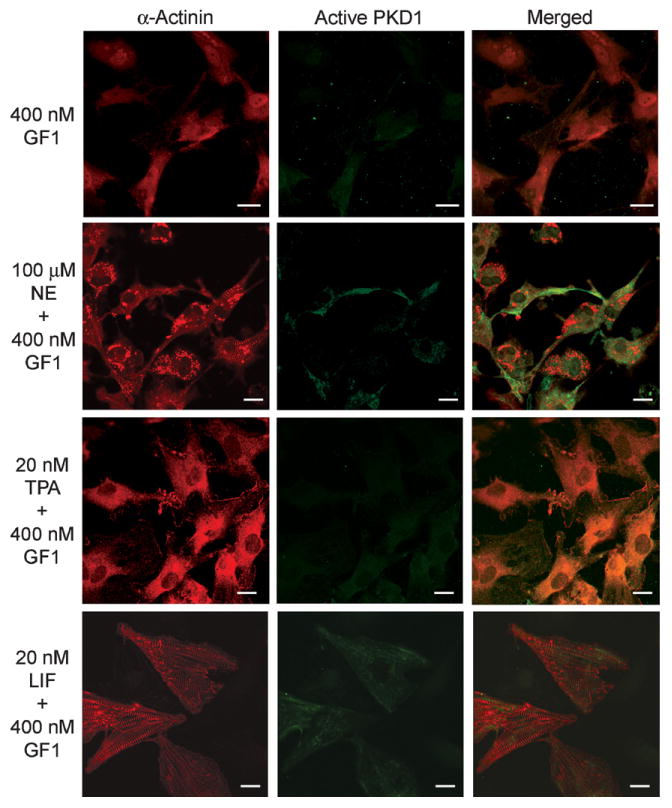
Effects of PKC inhibitor on Z-disc formation and translocation of active PKD1. Neonatal rat cardiomyocytes were stimulated with either 20 nM TPA, 100 μM NE plus 2 μM propranolol, or 20 nM LIF in the presence of 400 nM GF109203X (GF1) for 38 h. Immunofluorescence detection was done as described in Fig. 1B. Scale bar: 10 μm.
To examine if NE and TPA not only induce translocation of PKD1 but also activate it, we assayed in vitro the activity of endogenous PKD1 immunoprecipitated from the lysates of neonatal rat cardiomyocytes using a PKD1-specific peptide substrate, Syntide-2 [6] (Fig. 4A). Both TPA and NE rapidly and strongly enhanced the kinase activity of PKD1, compared to the non-stimulated conditions. When the cells were pre-treated with GF109203X, the phosphorylation activity was markedly reduced to the control level, even after TPA and NE stimulation. Clearly, LIF did not activate PKD1 irrespectively of the presence of GF109203X. Thus, in neonatal rat cardiomyocytes, PKD1 is activated by TPA and NE treatments in a PKC-dependent manner.
Fig. 4.
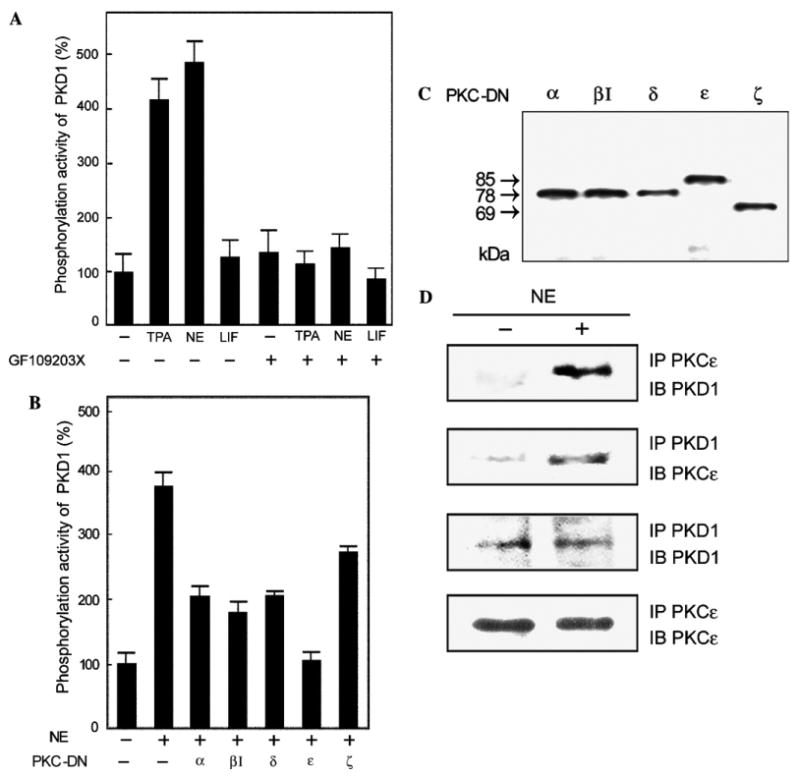
PKC-dependent activation of PKD1, effect of dominant negative PKC isoforms, and association of PKD1 and PKCε. (A) Neonatal rat cardiomyocytes were treated with either 20 nM TPA, 100 μM NE plus 2 μM propranolol, or 20 μM LIF with (+) or without (−) 400 nM GF1 for 20 min. PKD1 in the cell lysates was immunoprecipitated using an anti-PKD1/2 monoclonal antibody and its phosphorylation activity was measured as described in Materials and methods. The mean percentage of PKD1 activity compared to that of the control cells is shown with the standard deviation (n = 9). (B) The cells were transfected with plasmids for various PKC-DN isoforms (α, βI, δ, ε, and ζ) by using Duo Fect and stimulated with 100 μM NE plus 2 μM propranolol for 20 min (+). PKD1 was then immunoprecipitated and subjected to kinase assay as above (n = 9). (C) The cell lysates used in (B) were subjected to 10% SDS–PAGE and then analyzed by immunoblotting using an anti-HA monoclonal antibody (12CA5). (D) The cells were stimulated with 100 μM NE plus 2 μM propranolol for 20 min (+). Immunoprecipitates for PKCε or PKD1 were subjected to 10% SDS–PAGE, and then analyzed by immunoblotting with an anti-PKCε or anti-PKD1 antibody.
α-Adrenergic stimulation induces binding of PKCε with PKD1
Previous studies using cultured cell lines (non-cardiomyocytes) have demonstrated that PKD1 is phosphorylated and activated by two PKC isoforms, PKCε and PKCη [16]. These enzymes are the direct upstream kinases for PKD1 within various signaling pathways. Similarly, some specific PKC isoforms may be involved in the PKD1 activation in neonatal rat cardiomyocytes. To examine this possibility, the cells overexpressing dominant negative mutants of PKC isoforms (PKC-DN) were treated with NE, and the immunoprecipitated PKD1 was assayed in vitro (Fig. 4B). Western blot showed each PKC mutant was expressed equally in cardiomyocytes (Fig. 4C). Overexpression of PKCα-DN, PKCβ I-DN, PKCδ-DN, and PKCζ-DN only partially repressed the PKD1 activity in cardiomyocytes, presumably caused by a nonspecific repression (see Discussion), while overexpression of PKC-ε-DN almost completely inhibited the PKD1 activity induced by NE stimulation. This result strongly suggests that PKCε is an upstream activator of PKD1 in neonatal rat cardiomyocytes.
Similar to active PKD1 in TPA- and NE-treated cells (see above), active PKCε has been shown to translocate to the Z-discs when adult cardiomyocytes are treated with TPA or arachidonic acid [18,19]. Thus, both PKD1 and PKCε are localized at the Z-discs, suggesting possible interaction between them. Indeed, endogenous PKCε was co-immunoprecipitated with PKD1 and vice versa, particularly in the NE-stimulated cells (Fig. 4D). Similar results were also obtained upon stimulation with TPA (not shown), demonstrating that the interaction of PKD1 and PKCε is upregulated by the TPA- and NE-stimulation. This interaction corroborates the idea that PKCε is a direct upstream activator of PKD1 in neonatal rat cardiomyocytes.
Constitutively active mutant of PKD1 or PKCε induces cardiomyocytes hypertrophy and localizes at Z-discs
PKC-dependent activation of PKD1 and its translocation to the Z-discs suggest that PKD1 potentially participates in the development of cardiac hypertrophy. Therefore, a constitutively active mutant of PKD1 N-terminally fused with the green fluorescent protein (GFP-PKD1-CA) was transiently expressed in neonatal rat cardiomyocytes and its expression was monitored under a confocal microscope. As shown in Fig. 5A, GFP-PKD1-CA was able to increase the cell size and to form Z-discs without any stimuli. Moreover, GFP-PKD1-CA spontaneously localized at the Z-discs. When only GFP was transiently expressed as a control, neither the changes in cell size nor the formation of Z-discs was observed. Thus, the active PKD1 alone is sufficient to provoke an increase in cell size and the formation of Z-discs in neonatal rat cardiomyocytes. Also when PKCε-CA was transiently expressed, we could observe the formation of Z-discs along with the translocation of PKCε at the Z-discs (Fig. 5A).
Fig. 5.
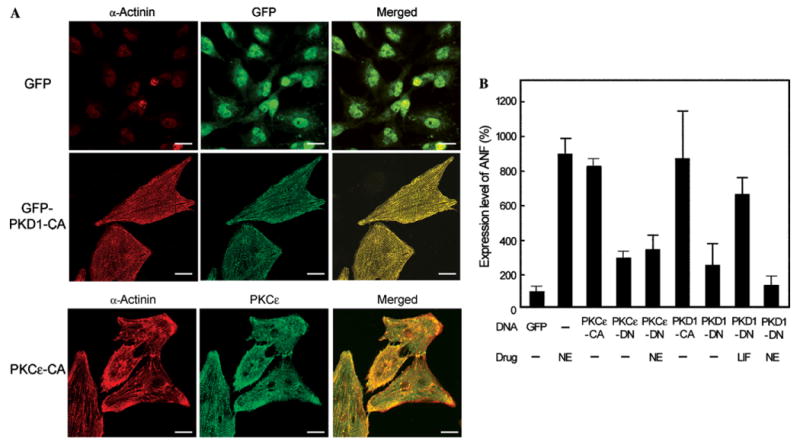
Induction of cardiac hypertrophy by overexpression of PKD1-CA or PKCε-CA. (A) Neonatal rat cardiomyocytes were transfected with the expression plasmid for GFP, GFP-PKD1-CA, or PKC ε-CA by electroporation. GFP-derived fluorescence was observed for the control GFP and PKD1-CA. Immunofluorescence was also observed for α-actinin and PKCε. (B) The cells were transfected with the expression plasmid for PKCε-CA, PKCε-DN, PKD1-CA, or PKD1-DN and stimulated with 100 μM NE plus 2 μM propranolol or 20 nM LIF for 38 h as indicated. The cells were then immunostained for ANF and the expression level of ANF was quantified on the basis of the Cy2-derived fluorescence in each cell. The mean percentage of expression level of ANF compared to that of the control cells (transfected with GFP) is shown with standard deviation (n = 40).
In addition to the increase in cell size and the organization of Z-discs, the expression levels of several embryonic proteins such as ANF, MHC-α (myosin heavy chain-α), and c-fos usually increase in the cardiac hypertrophy, and hence are considered as hypertrophy marker proteins [20]. To examine if the transient expression of GFP-PKD1-CA upregulates the expression of hypertrophic markers, we measured the expression of ANF by immunofluorescence using an anti-ANF polyclonal antibody (Fig. 5B). Transfection of either PKCε-CA or PKD1-CA strongly enhanced the ANF expression in the similar level to that attained by the NE treatment. In contrast, transfection of either PKCε-DN or PKD1-DN considerably prevented the ANF expression induced by the NE treatment but not that induced by the LIF treatment. These results collectively show that PKD1 activated by PKCε is sufficient to induce the cell size increase, Z-disc formation, and ANF expression in neonatal rat cardiomyocytes, and strongly suggest that the PKCε–PKD1 complex is involved in the cardiomyocyte signal transduction pathway that leads to the development of cardiac hypertrophy.
Discussion
PKCε binds and activates PKD1 at Z-discs
The absence of specific inhibitors makes it difficult to study the roles of PKD1 in cell signaling. Nevertheless, it has been demonstrated that cellular functions of PKD1 depend on its specific intracellular localization and also on the cell type [13,21,22]. Reinforcing the concept that PKD1 acts at specific locations, our study shows that active PKD1 is translocated to the cardiomyocyte Z-discs after stimulations with TPA, NE, AngII, and ET1 (Fig. 2). The Z-disc is a complex protein network that is not only involved in the maintenance of the sarcomere structure but also appears to be critical for cardiomyocyte signal transmission [23,24]. In cardiomyocytes, the translocation of active PKD1 to the Z-discs occurs only upon GPCR-derived stimulation, while LIF stimulation does not induce the PKD1 translocation. Upon LIF binding to the gp130 receptors, the JAK-STAT and MAP kinase pathways are principally activated, therefore explaining the absence of PKD1 activation and translocation [2].
It has been shown previously that upon phorbol ester activation PKCε also localizes at the Z-discs in neonatal and adult rat cardiomyocytes [18,19]. Our immunofluorescence experiments confirm and extend these findings, showing that not only PKCε but also PKD1 localizes at the Z-discs in neonatal rat cardiomyocytes upon stimulation (Figs. 1B, 2, 5A). The immunoprecipitation assay revealed that both enzymes actually interacted after NE stimulation, suggesting that PKCε modulates the activity of PKD1 once they are located at the Z-discs (Fig. 4D). The activation of PKD1 by PKCε is mainly dependent on the phosphorylation of a specific residue (Ser 744) and the autophosphorylation of a second one (Ser 748) [7]. This suggestion is supported by the immunoprecipitation kinase assay in which only the expression of PKCε-DN is able to completely inhibit the kinase activity of PKD1 stimulated by NE, whereas the other PKC-DN isoforms only partially repress the NE-induced activation (Fig. 4B). This partial repression is probably due to the high level of overexpression of the PKC-DN isoforms, resulting in nonspecific inhibition of the endogenous PKCε activity. Moreover, as shown in Fig. 5B, the NE-mediated ANF expression was significantly repressed by coexpression of PKCε-DN in cardiomyocyte. Thus, PKD1 is a direct target of PKCε in the α-adrenergic agonist-mediated signaling cascade, presumably in the GPCR agonist-mediated signaling cascade, leading to cardiomyocyte hypertrophy. To our knowledge, the present study is the first demonstration that PKCε is a direct upstream molecule of PKD1 in α-adrenergic agonist signaling cascade in cardiomyocytes. Recently, PKCε and δ were shown to activate PKD1 upon the AngII and ET1 stimulations in cardiomyocytes [25], and PKCη was also shown to activate PKD1 upon phorbol ester stimulation in cardiomyocytes [26]. These data suggest that each agonist activates PKD1 through the activation of distinct PKC isoforms in neonatal rat cardiomyocytes.
PKD1 is a key component of the cardiac hypertrophy signaling pathway
Several intracellular signaling pathways are involved in the development of cardiomyocyte hypertrophy and their activation principally depends on the nature of the stimulus. Our results show that PKD1 is present in neonatal rat cardiomyocytes and translocates to the Z-discs in response to various stimulations. Moreover, several lines of evidence show that PKD1 is a key element in the signaling cascade of cardiac hypertrophy. In neonatal rat cardiomyocytes, the expression of GFP-PKD1-CA was sufficient to provoke reorganization of α-actinin in sarcomeric Z-discs without any stimulation (Fig. 5A). The ANF expression, a marker of cardiac hypertrophy [20], was strongly enhanced by the expression of PKD1-CA, whereas that of PKD1-DN completely inhibited the ANF expression elicited by NE but not by the cytokine LIF (Fig. 5B). These results led us to conclude that active PKD1 plays a central role in the GPCR-mediated hypertrophy signaling in cardiomyocytes.
In the cardiomyocytes, Z-discs constitute the lateral boundaries of sarcomere and an anchoring site for the sarcomeric proteins [23]. However, Z-disc is not only an architectural structure but is also a complex protein network functioning as a biomechanical sensor and transducer of various cellular signals. PKCε has previously been shown to localize at the Z-discs upon TPA stimulation. However, the mechanism by which PKCε anchors to the Z-disc remained unknown. In this study, we demonstrated that active PKD1 also colocalizes at Z-discs along with the complex formation with PKCε. Recently, another scaffold protein, AKAP-Lbc, has been found to contribute to the PKD1 activation in two ways: it recruits PKCη and coordinates PKA phosphorylation events that release activated PKD1 in cardiomyocytes [26]. Since AKAP-Lbc neither localizes at Z-discs nor interacts with PKCε and activated PKD1, the AKAP-Lbc protein might be involved in distinct signaling cascade for inducing cardiac hypertrophy. It is highly likely that unidentified scaffold protein tethers the PKCε–PKD1 complex at Z-discs.
In conclusion, our study reveals that PKD1 plays a central role in several important GPCR signaling pathways of cardiomyocytes and that this enzyme is very likely involved in the development of cardiac hypertrophy. Moreover, we have shown that the PKCε–PKD1 complex is localized at the newly formed Z-discs for promoting the hypertrophy. If the more detailed role of the PKCε–PKD1 complex in the pathogenesis of cardiac hypertrophy is uncovered, the PKCε–PKD1 complex could become potential drug targets for the prevention and treatment of cardiac hypertrophy and heart failure.
Acknowledgments
This study was supported by research grants from the Circle for the Promotion of Science and Engineering, and the 21st Century Center of Excellence Program “Towards Creating New Industries Based on Inter-Nanoscience” (to M.I.), from “Structural and Functional Proteomics Consortium for Research on the Proteins Working in Brain and Nervous System” (to S.K.), and from “Fonds voor Wetenschappelijk Onderzoek-Vlaanderen,” the Belgian Government (IUAP P5/12), the Association for International Cancer Research, the Belgian Federation against Cancer, INTAS (Grant No. 2118), and the Flemish Government (GOA and BWTS) (to J.V.L. and J.R.V.). A.M. is a recipient of a Postdoctoral Fellowship from the Japan Society for Promotion of Science.
References
- 1.Frey N, Olson EN. Cardiac hypertrophy: the good, the bad, and the ugly. Annu Rev Physiol. 2003;65:45–79. doi: 10.1146/annurev.physiol.65.092101.142243. [DOI] [PubMed] [Google Scholar]
- 2.Molkentin JD, Dorn GW., II Cytoplasmic signaling pathways that regulate cardiac hypertrophy. Annu Rev Physiol. 2001;63:391–426. doi: 10.1146/annurev.physiol.63.1.391. [DOI] [PubMed] [Google Scholar]
- 3.Valverde AM, Sinnett-Smith J, Van Lint J, Rozengurt E. Molecular cloning and characterization of protein kinase D: a target for diacylglycerol and phorbol esters with a distinctive catalytic domain. Proc Natl Acad Sci USA. 1994;91:8572–8576. doi: 10.1073/pnas.91.18.8572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van Lint J, Sinnett-Smith J, Rozengurt E. Expression and characterization of PKD, a phorbol ester and diacylglycerol-stimulated serine protein kinase. J Biol Chem. 1995;270:1455–1461. doi: 10.1074/jbc.270.3.1455. [DOI] [PubMed] [Google Scholar]
- 5.Zugaza JL, Sinnett-Smith J, Van Lint J, Rozengurt E. Protein kinase D (PKD) activation in intact cells through a protein kinase C-dependent signal transduction pathway. EMBO J. 1996;15:6220–6230. [PMC free article] [PubMed] [Google Scholar]
- 6.Iglesias T, Waldron RT, Rozengurt E. Identification of in vivo phosphorylation sites required for protein kinase D activation. J Biol Chem. 1998;273:27662–27667. doi: 10.1074/jbc.273.42.27662. [DOI] [PubMed] [Google Scholar]
- 7.Waldron RT, Rozengurt E. Protein kinase C phosphorylates protein kinase D activation loop Ser744 and Ser748 and releases autoinhibition by the pleckstrin homology domain. J Biol Chem. 2003;278:154–163. doi: 10.1074/jbc.M208075200. [DOI] [PubMed] [Google Scholar]
- 8.Sturany S, Van Lint J, Müller F, Wilda M, Hameister H, Höcker M, Brey A, Gern U, Vandenheede J, Gress T, Adler G, Seufferlein T. Molecular cloning and characterization of the human protein kinase D2. A novel member of the protein kinase D family of serine threonine kinases. J Biol Chem. 2001;276:3310–3318. doi: 10.1074/jbc.M008719200. [DOI] [PubMed] [Google Scholar]
- 9.Haworth RS, Goss MW, Rozengurt E, Avkiran M. Expression and activity of protein kinase D/protein kinase C μin myocardium: evidence for α 1-adrenergic receptor- and protein kinase C-mediated regulation. J Mol Cell Cardiol. 2000;32:1013–1023. doi: 10.1006/jmcc.2000.1143. [DOI] [PubMed] [Google Scholar]
- 10.Brooks G, Goss MW, Rozengurt E, Galinanes M. Phorbol ester, but not ischemic preconditioning, activates protein kinase D in the rat heart. J Mol Cell Cardiol. 1997;29:2273–2283. doi: 10.1006/jmcc.1997.0466. [DOI] [PubMed] [Google Scholar]
- 11.Kuroda S, Tokunaga C, Kiyohara Y, Higuchi O, Konishi H, Mizuno K, Gill GN, Kikkawa U. Protein–protein interaction of zinc finger LIM domains with protein kinase C. J Biol Chem. 1996;27:31029–31032. doi: 10.1074/jbc.271.49.31029. [DOI] [PubMed] [Google Scholar]
- 12.Schönwasser DC, Marais RM, Marshall CJ, Parker PJ. Activation of the mitogen-activated protein kinase/extracellular signal-regulated kinase pathway by conventional, novel, and atypical protein kinase C isotypes. Mol Cell Biol. 1998;18:790–798. doi: 10.1128/mcb.18.2.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liljedahl M, Maeda Y, Colanzi A, Ayala I, Van Lint J, Malhotra V. Protein kinase D regulates the fission of cell surface destined transport carriers from the trans-Golgi network. Cell. 2001;104:409–420. doi: 10.1016/s0092-8674(01)00228-8. [DOI] [PubMed] [Google Scholar]
- 14.Matthews SA, Iglesias T, Rozengurt E, Cantrell D. Spatial and temporal regulation of protein kinase D (PKD) EMBO J. 2000;19:2935–2945. doi: 10.1093/emboj/19.12.2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakagawa N, Hoshijima M, Oyasu M, Saito N, Tanizawa K, Kuroda S. ENH, containing PDZ and LIM domains, heart/skeletal muscle-specific protein, associates with cytoskeletal proteins through the PDZ domain. Biochem Biophys Res Commun. 2000;272:505–512. doi: 10.1006/bbrc.2000.2787. [DOI] [PubMed] [Google Scholar]
- 16.Rykx A, DeKimpe L, Mikhalap S, Vantus T, Seufferlein T, Vandenheede JR, Van Lint J. Protein kinase D: a family affair. FEBS Lett. 2003;546:81–86. doi: 10.1016/s0014-5793(03)00487-3. [DOI] [PubMed] [Google Scholar]
- 17.Gschwendt M, Dieterich S, Rennecke J, Kittstein W, Mueller HJ, Johannes FJ. Inhibition of protein kinase C μby various inhibitors. Differentiation from protein kinase c isoenzymes. FEBS Lett. 1996;392:77–80. doi: 10.1016/0014-5793(96)00785-5. [DOI] [PubMed] [Google Scholar]
- 18.Huang XP, Pi Y, Lokuta AJ, Greaser ML, Walker JW. Arachidonic acid stimulates protein kinase C-epsilon redistribution in heart cells. J Cell Sci. 1997;110:1625–1634. doi: 10.1242/jcs.110.14.1625. [DOI] [PubMed] [Google Scholar]
- 19.Robia SL, Ghanta J, Robu VG, Walker JW. Localization and kinetics of protein kinase C ε anchoring in cardiac myocytes. Biophys J. 2001;80:2140–2151. doi: 10.1016/S0006-3495(01)76187-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dorn GW, II, Robbins J, Sugden PH. Phenotyping hypertrophy: eschew obfuscation. Circ Res. 2003;92:1171–1175. doi: 10.1161/01.RES.0000077012.11088.BC. [DOI] [PubMed] [Google Scholar]
- 21.Storz P, Toker A. Protein kinase D mediates a stress-induced NF-κB activation and survival pathway. EMBO J. 2003;22:109–120. doi: 10.1093/emboj/cdg009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marklund U, Lightfoot K, Cantrell D. Intracellular location and cell context-dependent function of protein kinase D. Immunity. 2003;19:491–501. doi: 10.1016/s1074-7613(03)00260-7. [DOI] [PubMed] [Google Scholar]
- 23.Clark KA, McElhinny AS, Beckerle MC, Gregorio CC. Striated muscle cytoarchitecture: an intricate web of form and function. Annu Rev Cell Dev Biol. 2002;18:637–706. doi: 10.1146/annurev.cellbio.18.012502.105840. [DOI] [PubMed] [Google Scholar]
- 24.Knöll R, Hoshijima M, Chien K. Cardiac mechanotransduction and implications for heart disease. J Mol Med. 2003;81:750–756. doi: 10.1007/s00109-003-0488-x. [DOI] [PubMed] [Google Scholar]
- 25.Vega RB, Harrison BC, Meadows E, Roberts CR, Papst PJ, Olson EN, McKinsey TA. Protein kinases C and D mediate agonist-dependent cardiac hypertrophy through nuclear export of histone deacetylase 5. Mol Cell Biol. 2004;24:8387–8395. doi: 10.1128/MCB.24.19.8374-8385.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carnegie GK, Smith FD, McConnachie G, Langeberg LK, Scott JD. AKAP-Lbc nucleates a protein kinase D activation scaffold. Mol Cell. 2004;15:889–899. doi: 10.1016/j.molcel.2004.09.015. [DOI] [PubMed] [Google Scholar]