

Non-technical summary
High blood pressure is known to be caused by an increase in activity of the sympathetic nerves that constrict blood vessels in skeletal muscle and the gut. Experiments in the spontaneous hypertensive rat (SHR) suggest that the hypertension is brought about by an increase in coupling between respiration and sympathetic outflow. We tested whether this mechanism occurs in two models of elevated muscle sympathetic nerve activity (MSNA) in human subjects: essential hypertension (HT) and chronic obstructive pulmonary disease (COPD). Unlike the SHR model, respiratory modulation of MSNA was not increased in either HT or COPD. These results help us understand how the cardiovascular and respiratory systems interact in health and disease.
Abstract
Abstract
We examined cardiac and respiratory modulation of muscle sympathetic nerve activity (MSNA) in 13 patients with essential hypertension (HT) and 15 with chronic obstructive pulmonary disease (COPD), and compared these with a group of young healthy controls (YHC) and older healthy controls (OHC). There were no significant differences in age of the OHC and HT subjects. MSNA was recorded via a tungsten microelectrode inserted percutaneously into the common peroneal nerve. Respiration was recorded by a strain-gauge transducer around the chest and ECG recorded by surface electrodes. Cardiac and respiratory modulation of MSNA was quantified by fitting polynomials to the cross-correlation histograms constructed between the sympathetic spikes and ECG or respiration. Cardiac modulation was high across all groups, but was significantly lower in COPD (75.9 ± 4.4%) than in the HT (92.4 ± 3.0%), OHC (93.7 ± 1.3%) or YHC (89.1 ± 1.6%) groups. Across all groups, respiratory modulation was significantly lower than cardiac modulation. Respiratory modulation in HT (45.2 ± 5.7%) and COPD (37.5 ± 6.3%) was not higher than in the OHC (47.2 ± 5.4%) or YHC (49.5 ± 6.0%) groups. We have shown that respiratory modulation of MSNA is present in all groups, is consistently lower than the magnitude of cardiac modulation, and is not increased in HT or COPD, arguing against an amplified respiratory–sympathetic coupling in hypertension. Moreover, given that patients with COPD are chronically asphyxic, these data indicate that an increased chemical drive does not increase respiratory modulation of MSNA.
Introduction
Muscle sympathetic nerve activity (MSNA), as recorded via microelectrodes from peripheral nerves in awake human subjects (microneurography), is entrained to the cardiac cycle by the arterial baroreceptors. In addition, MSNA is modulated by respiration, the pulse-synchronous vasoconstrictor discharges tending to occur in expiration and decreasing in intensity during mid-inspiration (Hagbarth & Vallbo, 1968; Eckberg et al. 1985, 1988; Seals et al. 1990, 1993; Macefield & Wallin, 1995; Macefield et al. 2002). Elevated levels of muscle sympathetic nerve activity feature in many cardiovascular and cardiorespiratory diseases. MSNA is high in patients with congestive heart failure (Leimbach et al. 1986; Grassi et al. 1995; Macefield et al. 1999), essential hypertension (Grassi et al. 1998; Schlaich et al. 2004; Lambert et al. 2007), pregnancy-induced hypertension (Schobel et al. 1996; Fischer et al. 2004), renovascular hypertension (Johansson et al. 1999; Miyajima et al. 2001) and the hypertension associated with chronic kidney disease (Hausberg et al. 2002; Schlaich et al. 2009). MSNA is also greatly elevated in the obstructive sleep apnoea syndrome (Hedner et al. 1988; Carlsson et al. 1993; Somers et al. 1995; Narkiewicz et al. 1999; Elam et al. 2002) and in chronic obstructive pulmonary disease (Heindl et al. 2001; Raupach et al. 2008; Ashley et al. 2010). The mechanisms for the sympathoexcitation differ in each of these pathophysiological states and are generally poorly understood. Yet, given the importance of the physiological coupling between the cardiovascular and respiratory systems, it is surprising that little research has assessed how strong this coupling is in different disease states.
Based on recent findings in the spontaneously hypertensive rat, in which respiratory modulation of sympathetic vasoconstrictor drive was shown to be increased and a theory proposed that ascribes the hypertension to this increase in respiratory modulation (Czyzyk-Krzeska & Trzebski, 1990; Simms et al. 2009), we tested the hypothesis that the magnitude of respiratory modulation of sympathetic vasoconstrictor drive is likewise increased in essential hypertension in human subjects. We also tested the hypothesis that respiratory modulation of MSNA is also increased in other states of elevated MSNA without hypertension. Specifically, we assessed the magnitude of respiratory modulation in patients with chronic obstructive pulmonary disease (COPD): these patients suffer from marked airflow limitation, primarily in expiration, and a compromised gas exchange (Heindl et al. 2001; Raupach et al. 2008; Ashley et al. 2010). Interestingly, although the levels of MSNA seen in COPD are similar to those seen in the obstructive sleep apnoea syndrome (OSAS) – in which repeated episodes of nocturnal hypoxaemia cause an increase in MSNA and neurogenic hypertension (Hedner et al. 1988; Carlsson et al. 1993; Narkiewicz et al. 1999; Elam et al. 2002) – in COPD there is no hypertension (Heindl et al. 2001; Raupach et al. 2008; Ashley et al. 2010). This then provides a nice means of factoring out the increase in blood pressure per se from the increase in MSNA observed in both essential hypertension and COPD. Furthermore, in COPD the chronic hypoxaemia and hypercapnia lead to an increase in inspiratory drive: mean firing rates of motor units recorded from the diaphragm, scalene and parasternal inspiratory muscles are all increased in COPD (Gandevia et al. 1996; De Troyer et al. 1997). So it is not unreasonable to posit that respiratory modulation of MSNA will also be augmented. Indeed, an elevated resting level of MSNA and an elevated respiratory drive may well be expected to increase, by central neuronal means, the coupling between the cardiovascular and respiratory systems, which will be evidenced by a magnitude of respiratory modulation of MSNA which is higher than what we expect to see in essential hypertension.
Methods
Participants
Data were obtained from 22 healthy participants, subdivided into a younger group (n = 12; 8 male, 4 female) with a mean age (± SEM) of 29 ± 2 years and an older group (n = 10; 5 male, 5 female) with a mean age of 50 ± 3 years, and 10 male and 3 female patients with long-standing essential hypertension (age 58 ± 2 years). A retrospective analysis was performed on data obtained from 8 male and 7 female patients with chronic obstructive pulmonary disease (COPD) with a mean age 71 ± 2 years, a subset of data previously reported from our laboratory (Ashley et al. 2010). Both the hypertensive and COPD patient groups remained on their scheduled pharmacological and physiotherapeutic treatment during the recordings. The hypertensive patients were on combinations of Ca2+ channel blockers (n = 5), angiotensin II receptor antagonists (n = 5), β-adrenergic blockers (n = 4), angiotensin-converting enzyme (ACE) inhibitors (n = 2) and statins (n = 5). One COPD patient was being treated with an angiotensin II receptor antagonist, one with thyroxine and another was on supplemental oxygen (discontinued 1 h before the recording); inhaled respiratory medications included salbutamol, ipatropium, fluticasone propionate, salmeterol and beclomethasone dipropionate. Each participant provided informed written consent to the procedures, which conformed to the standards set by the Declaration of Helsinki and which were conducted under the approval of the Human Research Ethics Committees of the University of Western Sydney and the University of New South Wales.
General procedures
Participants lay semi-recumbent in a chair with their backs at 45 deg and their legs supported horizontally. Spontaneous MSNA was recorded from fascicles of the common peroneal nerve supplying the ankle and toe extensor and foot everter muscles via tungsten microelectrodes (Frederick Haer and Co., Bowdoinham, ME, USA) inserted percutaneously at the level of the fibular head; a nearby subdermal electrode with a larger uninsulated tip served as the reference electrode. The impedances of the microelectrodes (∼100–150 kΩ), favoured oligo-unitary and multi-unit, rather than unitary, recordings. Neural activity was amplified (gain 2 × 104, bandpass 0.3–5.0 kHz) using an isolated amplifier and headstage (NeuroAmpEX, ADInstruments, Sydney, Australia) and stored on computer (10 kHz sampling) using a computer-based data acquisition and analysis system (PowerLab 16SP hardware and LabChart 7 software; ADInstruments). ECG (0.3 Hz to 1.0 kHz) was recorded with Ag–AgCl surface electrodes on the chest and sampled at 2 kHz. Respiration (DC to 100 Hz) was recorded using a strain-gauge transducer (Pneumotrace, UFI, Morro Bay, CA, USA) wrapped around the chest and sampled at 100 Hz. Continuous non-invasive blood pressure was recorded using radial arterial tonometry (Colin 7000 NIBP, Colin Corp., Japan) or digital arterial plethysmography (Finometer, Finapres Medical Systems, The Netherlands), sampled at 400 Hz. Spontaneous MSNA, ECG, blood pressure and respiration were recorded for at least 30 min and periods of stable activity (>10 min) were sampled for analysis.
Analysis
MSNA was displayed as an RMS-processed (root mean square, moving average, time constant 200 ms) signal but the primary analysis was conducted on the raw, negative-going, sympathetic spikes to avoid any contamination from spikes generated by positive-going myelinated axons (such as spontaneously active muscle spindles) or motor units associated with spontaneous EMG activity. This approach has been described previously, and provides a more sensitive means of analysing sympathetic outflow than the standard method of simply counting the number of sympathetic bursts (Bent et al. 2006). Briefly, negative-going spikes in the neurogram (with a half-width of 0.2–0.5 ms) were detected using window discriminator software (Spike Histogram for Macintosh v2.2, ADInstruments), while the times of occurrence of the R-waves of the ECG and of the inspiratory peaks of the respiratory signal were computed using Peak Analysis software (ADInstruments). Autocorrelation histograms for the respiratory and cardiac signals, and cross-correlation histograms between the MSNA and ECG or respiration, were generated by the Spike Histogram software (50 ms bins). Discriminator levels of the neural activity were adjusted so that negative-going (C-fibre) spikes exhibited a robust cardiac modulation, as revealed by cross-correlation between the neural activity and the ECG. These same discriminator settings were used for construction of cross-correlograms between the MSNA and the positive peaks of the respiratory signal. Smoothed polynomial curves were fitted to the histogram data using a graphical analysis program; lower-order polynomials were used to fit curves to the slower respiratory cross-correlograms while higher-order polynomials were required to fit curves to the cardiac cross-correlograms (Prism 5.0, GraphPad Software, USA). Quantification of the modulation of MSNA was performed by measuring the difference in the number of spikes at the peak of the modulation and the number at the trough, expressed as a percentage: Modulation Index = [(Peak – Trough)/Peak]× 100. For calculating the modulation index the peak–trough difference extended over the same interval as the respiratory or cardiac periods. In addition, in each subject we calculated the number of sympathetic spikes in different phases of the respiratory cycle, as defined by the peak of inspiration: expiratory activity was calculated from –3.0 to –1.5 s of the inspiratory peak (time 0), inspiratory activity was calculated from –1.5 to 0 s and post-inspiratory activity from 0 to 1.5 s. For this analysis the neural activity was displaced back in time 1.2–1.3 s in each subject to account for the baroreflex delay. The total number of sympathetic spikes was divided by the number of minutes in the recording and presented as spikes per minute. Mean blood pressure was also calculated in each of these intervals to assess respiratory fluctuations in blood pressure. MSNA was also quantified according to standard time-domain analysis of the RMS-processed signal as burst frequency (bursts min−1) and burst incidence (bursts (100 heart beats)−1). Analysis of variance, coupled with Tukey's multiple comparisons test, was used to assess statistical significance across each group (Prism 5.0, GraphPad Software, USA). All values are expressed as means and standard errors of the mean, and P < 0.05 was considered statistically significant.
Results
Cardiovascular and respiratory details of the younger and older healthy controls (YHC and OHC) and the patients with essential hypertension (HT) and chronic obstructive pulmonary disease (COPD) are provided in Table 1. The mean age of the hypertensive patients was not significantly different from that of the older healthy control subjects, but was significantly higher than that of the younger healthy controls and significantly lower than that of the COPD patients. For convenience we have made statistical comparisons with the older healthy controls. It can be seen that there were no significant differences in heart rate or respiratory period across groups but, as expected, systolic and diastolic pressures (and hence mean arterial pressure) were significantly higher in the HT group. There were no differences in any of the blood pressure parameters across the YHC, OHC or COPD groups. As previously described, MSNA was significantly elevated in HT and COPD, with burst frequency (but not burst incidence) being significantly higher in COPD than in HT (P < 0.01). Burst incidence was significantly lower in the YHC group than in the OHC (P < 0.05), HT (P < 0.005) or COPD (P < 0.005) groups. Blood gas analysis in the COPD patients revealed significant hypoxaemia and hypercapnia, as reported in the original study from which the current data were obtained (Ashley et al. 2010).
Table 1.
Cardiorespiratory parameters of the young healthy controls (YHC), older healthy controls (OHC), patients with hypertension (HT) or obstructive pulmonary disease (COPD)
| YHC (n = 12) | OHC (n = 10) | HT (n = 13) | COPD (n = 15) | |
|---|---|---|---|---|
| Age (years) | 29 ± 2*** | 50 ± 3 | 58 ± 2 | 71 ± 2*** |
| Systolic BP (mmHg) | 126 ± 4*** | 130 ± 6 | 152 ± 4* | 133 ± 5 |
| Diastolic BP (mmHg) | 70 ± 4 | 75 ± 4 | 88 ± 2* | 74 ± 3 |
| Mean BP (mmHg) | 88 ± 4 | 90 ± 6 | 110 ± 2** | 95 ± 3 |
| Heart rate (beats min−1) | 72 ± 5 | 60 ± 3 | 63 ± 5 | 70 ± 3 |
| Resp. period (s) | 3.9 ± 0.2 | 4.0 ± 0.2 | 3.9 ± 0.3 | 3.5 ± 0.3 |
| Burst freq. (bursts min−1) | 22 ± 2 | 29 ± 3 | 51 ± 3*** | 62 ± 2*** |
| Burst incidence (bursts (100 hb)−1) | 33 ± 3* | 49 ± 6 | 80 ± 4*** | 86 ± 2*** |
Values presented are mean ± SEM. For clarity, only statistical comparisons are shown relative to data obtained in the OHC group, for which there was no significant age difference from the HT group:
P < 0.05
P < 0.01
P < 0.001.
See text for further comparisons. BP, blood pressure; hb, heart beats.
Experimental records from a normotensive healthy female control subject (OHC) are shown in Fig. 1; records from a female subject with essential hypertension are shown in Fig. 2. In both figures event markers are shown for the sympathetic spikes, the ECG and the peaks of inspiration. These event markers were used to construct the autocorrelation and cross-correlation histograms shown in Fig. 3. As illustrated in Fig. 3B, cross-correlation histograms between the times of occurrence of the sympathetic spikes and of the ECG show a very tight coupling of MSNA to the R-wave of the ECG, while the respiratory rhythmicity is weaker (Fig. 3A). Cardiac and respiratory modulation of MSNA was calculated from the smoothed polynomials fitted to the cross-correlation histograms.
Figure 1. Multi-unit recording of muscle sympathetic nerve activity from a 45-year-old normotensive female subject.
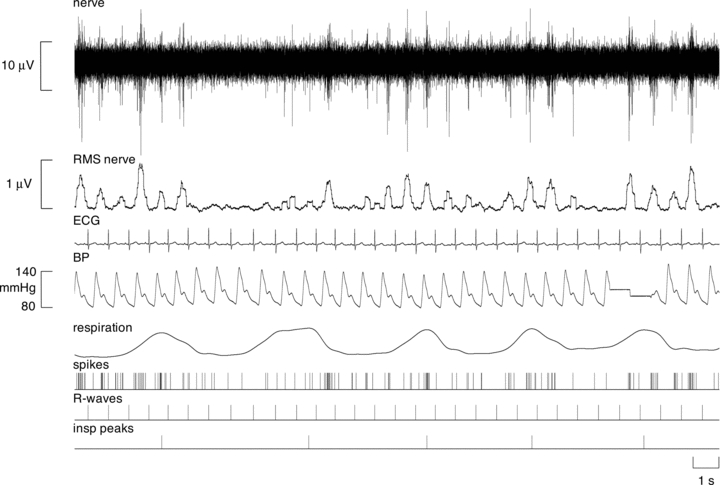
The mean-voltage neurogram is shown in the RMS-nerve trace; this was used to quantify the number of sympathetic bursts. Note that bursts of MSNA occurred in many, but not all, cardiac intervals in this subject. Discriminated spikes extracted from the nerve recording are illustrated as standard pulses (spikes). The times of occurrence of each heart beat (R-waves) and peaks of each breath (insp. peaks) are also shown as standard pulses. These timing events were used to generate the cross-correlation and auto-correlation histograms.
Figure 2. Multi-unit recording of muscle sympathetic nerve activity in a 64-year-old hypertensive female subject.

The mean-voltage neurogram is shown in the RMS-nerve trace. Note that bursts of MSNA occurred in most cardiac intervals in this subject, and that overall levels of MSNA were higher than in the control subject shown in Fig. 1.
Figure 3. Cross-correlation histograms (upper traces in each panel) and auto-correlation histograms (lower traces in each panel) between sympathetic spikes and respiration (A) and ECG (B).

Data obtained from the same subject illustrated in Fig. 1. Smoothed polynomials (thick lines) have been fitted to the histograms. The numbers on the y-axis refer to the numbers of spikes per 50 ms bin. Time zero, corresponding to the triggering event in the cross- or auto-correlograms, is indicated by the vertical dashed lines.
Mean data are shown graphically in Fig. 4. Cardiac modulation was high across all groups, but was significantly lower in COPD (75.9 ± 4.4%; P < 0.01) than in the HT (92.4 ± 3.0%), OHC (93.7 ± 1.3%) or YHC (89.1 ± 1.6%) groups. Across groups there was no difference in respiratory period, although respiratory rate tended to be higher (and respiratory period lower) in the COPD patients (presumably because of the elevated chemical drive to breathe). Respiratory modulation of MSNA was significantly lower than cardiac modulation across all groups. Interestingly, while we had predicted that respiratory modulation would be higher in HT, there was no significant difference between this group (45.2 ± 5.7%) and the YHC (49.5 ± 6.0%) and OHC (47.2 ± 5.4%) groups. Moreover, given the chronic asphyxia, we had predicted that respiratory modulation in COPD would be higher than in HT, but in fact it was lower (37.5 ± 6.3%) than in the HT, YHC and OHC groups, though this difference failed to reach statistical significance. The phase shift between the peak of inspiration and the peak of the respiratory modulation of MSNA was not significantly different across groups: 3.40 ± 0.3 s (YHC group), 3.48 ± 0.3 s (OHC), 3.53 ± 0.4 s (HT) and 2.50 ± 0.4 s (COPD). There were also no significant differences in the phase lag between the R-wave and the peak of cardiac modulation for the YHC (0.60 ± 0.1 s), OHC (0.54 ± 0.1 s), HT (0.55 ± 0.1 s) and COPD (0.52 ± 0.1 s) groups.
Figure 4. Mean cardiac and respiratory modulation indices across the four groups studied.
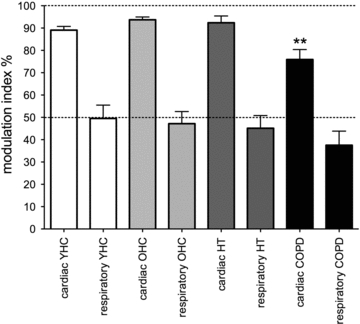
YHC, young healthy control (n = 12); OHC, older healthy control (n = 10); HT, patients with essential hypertension (n = 13); COPD, patients with chronic obstructive pulmonary disease (n = 15). Across groups, cardiac modulation indices were consistently higher than the respiratory modulation indices. Relative to the value in the OHC group, cardiac modulation was significantly lower in COPD; **P < 0.01.
Table 2 shows the total MSNA, calculated as the number of sympathetic spikes in a one-minute period across the different phases of the respiratory cycle (see Methods). Although MSNA tended to be higher during inspiration in the YHC, OHC and HT groups, but higher in expiration in the COPD group, these differences failed to reach statistical significance. There were also no significant respiratory variations in mean blood pressure within each group (Table 2).
Table 2.
Total MSNA, calculated as the number of sympathetic spikes in a one-minute period, and mean blood pressure (mmHg), across the different phases of the respiratory cycle in the four groups studied
| YHC (n = 12) | OHC (n = 10) | HT (n = 13) | COPD (n = 15) | |
|---|---|---|---|---|
| Expiratory MSNA | 71 ± 11 | 71 ± 13 | 63 ± 10 | 161 ± 55 |
| Inspiratory MSNA | 86 ± 17 | 85 ± 15 | 77 ± 16 | 154 ± 49 |
| Post-inspiratory MSNA | 66 ± 9 | 68 ± 16 | 64 ± 12 | 153 ± 47 |
| Expiratory mean BP | 89.6 ± 5.7 | 93.1 ± 4.1 | 109.8 ± 3.1 | 82.3 ± 4.4 |
| Inspiratory mean BP | 88.8 ± 5.4 | 92.6 ± 4.1 | 109.8 ± 3.0 | 82.1 ± 4.4 |
| Post-inspiratory mean BP | 88.5 ± 5.7 | 93.3 ± 4.0 | 110.2 ± 3.1 | 82.7 ± 4.2 |
Values presented are mean ± SEM. There were no significant differences in MSNA or mean BP across the respiratory cycle in any group.
Discussion
This is the first study to compare the magnitude of respiratory modulation in two pathophysiological states of increased muscle sympathetic nerve activity – essential hypertension and chronic obstructive pulmonary disease – with that of healthy subjects. Contrary to our expectations, compared to age-matched healthy subjects, respiratory modulation was not increased in the hypertensive group, despite the clear evidence of a significantly elevated blood pressure. This would argue against an amplified respiratory–sympathetic coupling as being a causative factor in the generation of hypertension. Moreover, given that patients with chronic obstructive pulmonary disease are chronically asphyxic, our observations of a slightly lower (or at least statistically unchanged) respiratory modulation in COPD indicates that an increased chemical drive does not increase respiratory modulation of MSNA. That cardiac rhythmicity of MSNA was significantly lower in COPD suggests perhaps that the sympathetic bursts are not completely inhibited by the arterial baroreceptors in COPD; indeed, we know that the sensitivity of the sympathetic baroreflex is reduced in COPD (Raupach et al. 2008).
Respiratory modulation of MSNA
In humans, MSNA is inhibited in mid-inspiration (Hagbarth & Vallbo, 1968; Eckberg et al. 1985, 1988; Seals et al. 1990, 1993; Macefield & Wallin, 1995; Macefield et al. 2002), whereas in the cat vasoconstrictor outflow occurs during inspiration (Bainton et al. 1985; Cohen & Gootman, 1970; Koizumi et al. 1971; Barman & Gebber, 1976; Boczek-Funke et al. 1992). Why muscle (or splanchnic) vasoconstrictor drive is synchronized to expiration in humans but inspiration in cats is not clear, but Boczek-Funke et al. (1992) have shown that although sympathetic activity increases during inspiration it is initially depressed in early inspiration and reaches a nadir in post-inspiration. Experiments in anaesthetized animals are typically performed during neuromuscular blockade and artificial ventilation, conditions in which central (phrenic discharge) and peripheral (lung inflation) respiratory activities may become dissociated, with central inspiration occuring during peripheral expiration. Because of this entrainment of the central respiratory rhythm to the ventilator, the respiratory changes in arterial pressure also become dissociated. Indeed, Boczek-Funke et al. (1992) identified two peaks in the modulation of vasoconstrictor activity: one (direct) the result of central inspiratory drive, and the other (indirect) the result of mechanically induced unloading of arterial baroreceptors. In animals with intact vagus nerves, however, the inspiratory peak is replaced by an expiratory peak (as in humans), attributed to unloading of baroreceptors. While the incidence of MSNA is inversely related to diastolic pressure in humans (Sundlöf & Wallin, 1978), a relationship that holds throughout the respiratory cycle (Eckberg et al. 1985), we now know that the respiratory modulation is independent of the changes in blood pressure: the phase relationship between MSNA burst amplitude and tidal volume is preserved when respiration is brought about either by negative-pressure or positive-pressure ventilation and the inspiratory changes in diastolic pressure reversed (Seals et al. 1993; Macefield & Wallin, 1995). Moreover, the finding that for a given diastolic pressure the level of MSNA is lower during inspiration than during expiration supports the idea that the sympathetic inhibition is largely independent of the influence of the arterial baroreceptors (Seals et al. 1990, 1993). In the current study, we found no significant respiratory fluctuations in blood pressure in any group. Interestingly, unlike the cat, the rat shows a pattern of respiratory modulation of sympathetic activity that is broadly similar to that of humans: inhibition of sympathetic activity commences at the onset of inspiration, reaches a minimum at mid-inspiration, and maximal activity occurs in the post-inspiratory phase and, occasionally, in late expiration (Czyzyk-Krzeska & Trzebski, 1990).
Sympathoexcitation in HT and COPD
In our recent paper, in which we compared the firing properties of single muscle vasoconstrictor neurons in COPD (Ashley et al. 2010) with those obtained in the obstructive sleep apnoea syndrome (Elam et al. 2002), we pointed out that it is highly likely that the ongoing hypoxaemia and hypercapnia associated with severe COPD (but not OSAS) are responsible for the augmented MSNA. We believe the peripheral chemoreceptors contribute to the elevated MSNA in COPD because oxygen administration has been shown to lower MSNA (Heindl et al. 2001) and slow, deep breathing has also been shown to reduce MSNA in COPD, presumably by improving gas exchange (Raupach et al. 2008). Conversely, the aetiology of the elevated MSNA in essential hypertension is (by definition) unknown, but it is generally accepted that the hypertension is neurogenic (Grassi et al. 1998; Schlaich et al. 2004; Lambert et al. 2007). A provocative hypothesis has been proposed to explain the hypertension: an increase in respiratory coupling of sympathetically mediated vasoconstriction (Czyzyk-Krzeska & Trzebski, 1990; Simms et al. 2009). In the normotensive Wistar-Kyoto rat (WKY), the lowest level of cervical and renal sympathetic activity occurs in mid-inspiration and the highest in the post-inspiratory phase; in the spontaneously hypertensive rat (SHR), on the other hand, the lowest sympathetic activity occurs during the post-inspiratory period and the highest activity occurs in mid-inspiration and mid-expiration (Czyzyk-Krzeska & Trzebski, 1990). Using the working heart–brainstem preparation (in which the animal is decerebrated, its lungs removed and the heart and brainstem supported by retrograde artificial perfusion via the descending aorta; Paton, 1996) Simms et al. (2009) showed that perfusion pressure (and hence vascular resistance) and thoracic sympathetic nerve activity were elevated in the neonatal and young (3–5 weeks) SHR but not in the normotensive WKY rat (Simms et al. 2009). Moreover, phrenic-nerve-triggered averaging revealed a specific increase in respiratory-related activity of the sympathetic vasoconstrictor drive, with sympathetic outflow occurring in the post-inspiratory phase in the WKY rat but with the peak occurring progressively earlier in inspiration in the 3-week- and 5-week-old SHR rats. Given that this survived denervation of the arterial baroreceptors (and peripheral chemoreceptors), and is independent of inputs from the lungs (which had been removed), the authors conclude that this reflects an increase in coupling between the inspiratory and vasoconstrictor drives, and that this contributes to the generation of hypertension.
So, why do we not see an increase in respiratory rhythmicity of MSNA in human essential hypertension, or indeed in patients with COPD? Obviously, the awake human subject is not the same as the anaesthetized rat, and especially the highly reduced working heart–brainstem preparation, but it is not unreasonable to think that respiratory coupling of MSNA would be increased in human hypertension and especially in COPD. However, we do know that another state of sympathoexcitation – congestive heart failure (CHF) – is associated with a rightward shift in the distribution of burst amplitudes (Sverrisdottír et al. 1998, 2000); in other words there are more large bursts and fewer small bursts. Given that respiratory modulation of MSNA is reflected in respiratory-related changes in burst amplitude (and the temporal distribution of sympathetic firing when expressed as a cyclic histogram) it would appear that the reduced variance in burst amplitudes of itself suggests a lower modulation. It may be that in states of elevated MSNA (such as CHF, HT and COPD) the overall increase in vasoconstrictor drive limits the expression of any increase in respiratory-related vasoconstrictor drive. Alternatively, given that respiratory modulation of MSNA in human subjects appears to be largely independent of central respiratory drive at rest (it is observed even in artificially ventilated subjects in whom central inspiratory drive has been suppressed; Macefield & Wallin, 1995), it is quite possible that the mechanism proposed for the sympathoexcitation in the spontaneously hypertensive rat differs from the sympathoexcitation observed in either human essential hypertension or COPD.
Methodological considerations
As noted in our previous studies of patients with CHF, OSAS or COPD (Macefield et al. 1999; Elam et al. 2002; Ashley et al. 2010), we did not withdraw pharmacological or physiotherapeutic treatment; despite this, MSNA remained very high, so it is unlikely that the ongoing treatment affects our conclusions. Nevertheless, we should acknowledge that 5 of our 13 HT patients were on angiotensin II receptor antagonists and two were on ACE inhibitors. Given that the renin–angiotensin system has been shown to contribute to respiratory–sympathetic coupling in the hypertensive rat (Toney et al. 2010), it is possible that in these seven patients pharmacological blockade may have attenuated the respiratory modulation. Of course, this would not affect respiratory modulation of MSNA in COPD, which, because of the elevated chemical drive to breathe, would be expected to be high. There were differences in age between those patients with essential hypertension and those with COPD, but at least those with HT were matched in age to the older healthy controls. We know that MSNA increases with age (Sverrisdottír et al. 2000; Hart et al. 2009); we observed this difference between the younger and older healthy controls in the present study. Nevertheless, we do not believe age is an important parameter in this situation: what is important is that we have examined four groups of subjects with differing levels of MSNA, with or without hypertension.
It is worth noting that, unlike some studies that have addressed respiratory rhythmicity in human subjects, we specifically did not constrain the respiratory pattern by asking subjects to breathe to a metronome; subjects breathed spontaneously and there was no significant difference in respiratory period across each of the groups. However, the respiratory excursions we recorded were not calibrated with respect to volume, so we do not know whether the depth of respiration differed across groups. The times of occurrence of the inspiratory peaks were used to generate cross-correlation histograms between the times of occurrence of sympathetic spikes and respiration; the times of occurrence of the R-waves were used to generate cross-correlograms between MSNA and ECG. By using the raw nerve data, rather than the RMS-processed signal, we believe we have used a more sensitive measure of calculating respiratory rhythmicity, particularly as we generated the cross-correlation and autocorrelation histograms with a bin width of 50 ms. Moreover, by fitting low- or high-order smoothed polynomials to the histograms we could differentiate between the slower respiratory-related modulation and the faster cardiac-related modulation of MSNA, and calculate modulation from the peak–trough difference in the numbers of spikes. However, it should be pointed out that our method of analysis differed from that used by Simms et al. (2009) in the rat: their analysis was based on phrenic-nerve-triggered averaging of the integrated bursts recorded from the thoracic sympathetic chain, so the means of assessing respiratory modulation differed in the two studies. Nevertheless, despite these differences, both studies compared respiratory modulation across groups; accordingly, the conclusions reached in both studies stand on their own.
Conclusions
In conclusion, we have shown that respiratory modulation of muscle sympathetic nerve activity in human essential hypertension is not increased, unlike the clear increase observed in the neonatal and adult spontaneously hypertensive rat. Moreover, respiratory modulation of MSNA in COPD is also not increased, despite the presence of a high chemical drive to respiration. These observations argue against the utility of an increase in respiratory-related MSNA as being causal for the increase in muscle vasoconstrictor drive seen in essential hypertension.
Glossary
Abbreviations
- COPD
chronic obstructive pulmonary disease
- HT
essential hypertension
- MSNA
muscle sympathetic nerve activity
- OHC
older healthy controls
- OSAS
obstructive sleep apnoea syndrome
- SHR
spontaneous hypertensive rat
- YHC
young healthy controls
Author contributions
V.G.M. was responsible for the conception and design of the study. R.F. and V.G.M. were each involved in data acquisition. R.F. was responsible for the analysis and interpretation of the data. V.G.M. drafted the article and both authors approved the final submission. The study was conducted at the School of Medicine, University of Western Sydney.
References
- Ashley C, Burton D, Sverrisdottir YB, Sander M, McKenzie DK, Macefield VG. Firing probability and mean firing rates of human muscle vasoconstrictor neurones are elevated during chronic asphyxia. J Physiol. 2010;588:701–711. doi: 10.1113/jphysiol.2009.185348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bainton CR, Richter DW, Seller H, Ballantyne D, Klein JP. Respiratory modulation of sympathetic activity. J Autonom Nerv Syst. 1985;12:77–90. doi: 10.1016/0165-1838(85)90041-4. [DOI] [PubMed] [Google Scholar]
- Barman SM, Gebber GL. Basis for synchronization of sympathetic and phrenic nerve discharges. Am J Physiol. 1976;231:1601–1607. doi: 10.1152/ajplegacy.1976.231.5.1601. [DOI] [PubMed] [Google Scholar]
- Bent LR, Bolton PS, Macefield VG. Modulation of muscle sympathetic bursts by sinusoidal galvanic vestibular stimulation in human subjects. Exp Brain Res. 2006;174:701–711. doi: 10.1007/s00221-006-0515-6. [DOI] [PubMed] [Google Scholar]
- Boczek-Funcke A, Häbler H-J, Jänig W, Michaelis M. Respiratory modulation of the activity in sympathetic neurones supplying muscle, skin and pelvic organs in the cat. J Physiol. 1992;449:333–361. doi: 10.1113/jphysiol.1992.sp019089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson JT, Hedner J, Elam M, Ejnell H, Sellgren J, Wallin BG. Augmented resting sympathetic activity in awake patients with obstructive sleep apnea. Chest. 1993;103:1763–1768. doi: 10.1378/chest.103.6.1763. [DOI] [PubMed] [Google Scholar]
- Cohen MI, Gootman PM. Periodicities in efferent discharge of splanchnic nerve of the cat. Am J Physiol. 1970;218:1092–1101. doi: 10.1152/ajplegacy.1970.218.4.1092. [DOI] [PubMed] [Google Scholar]
- Czyzyk-Krzeska MF, Trzebski A. Respiratory-related discharge pattern of sympathetic nerve activity in the spontaneously hypertensive rat. J Physiol. 1990;426:355–368. doi: 10.1113/jphysiol.1990.sp018142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Troyer A, Leeper JB, McKenzie DK, Gandevia SC. Neural drive to the diaphragm in patients with severe COPD. Am J Respir Crit Care Med. 1997;155:1335–1340. doi: 10.1164/ajrccm.155.4.9105076. [DOI] [PubMed] [Google Scholar]
- Eckberg DL, Nerhed C, Wallin BG. Respiratory modulation of muscle sympathetic and vagal cardiac outflow in man. J Physiol. 1985;365:181–196. doi: 10.1113/jphysiol.1985.sp015766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckberg DL, Rea RF, Andersson OK, Hedner T, Pernow J, Lundberg JM, Wallin BG. Baroreflex modulation of sympathetic activity and sympathetic neurotransmitters in humans. Acta Physiol Scand. 1988;133:221–231. doi: 10.1111/j.1748-1716.1988.tb08401.x. [DOI] [PubMed] [Google Scholar]
- Elam M, Macefield VG, McKenzie D. The firing pattern of single muscle vasoconstrictor neurons in awake patients with the obstructive sleep apnoea syndrome. J Appl Physiol. 2002;93:297–303. doi: 10.1152/japplphysiol.00899.2001. [DOI] [PubMed] [Google Scholar]
- Fischer T, Schobel HP, Frank H, Andreae M, Schneider KY, Heusser K. Pregnancy-induced sympathetic hyperactivity: a precursor of preeclampsia. Eur J Clin Invest. 2004;34:443–448. doi: 10.1111/j.1365-2362.2004.01350.x. [DOI] [PubMed] [Google Scholar]
- Gandevia SC, Leeper JB, McKenzie DK, De Troyer A. Discharge frequencies of parasternal intercostal and scalene motor units during breathing in normal and COPD subjects. Am J Respir Crit Care Med. 1996;153:622–628. doi: 10.1164/ajrccm.153.2.8564108. [DOI] [PubMed] [Google Scholar]
- Grassi G, Colombo M, Seravalle G, Spaziani D, Mancia G. Dissociation between muscle and skin sympathetic nerve activity in essential hypertension, obesity, and congestive heart failure. Hypertension. 1998;31:64–67. doi: 10.1161/01.hyp.31.1.64. [DOI] [PubMed] [Google Scholar]
- Grassi G, Seravalle G, Cattaneo BM, Lanfranchi A, Vailati S, Giannattasio C, Del Bo A, Sala C, Bolla GB, Pozzi M. Sympathetic activation and loss of reflex sympathetic control in mild congestive heart failure. Circulation. 1995;92:3206–3211. doi: 10.1161/01.cir.92.11.3206. [DOI] [PubMed] [Google Scholar]
- Hagbarth KE, Vallbo ÅB. Pulse and respiratory grouping of sympathetic impulses in human peripheral nerves. Acta Physiol Scand. 1968;74:96–108. doi: 10.1111/j.1748-1716.1968.tb04218.x. [DOI] [PubMed] [Google Scholar]
- Hart EC, Joyner MJ, Wallin BG, Johnson CP, Curry TB, Eisenach JH, Charkoudian N. Age-related changes in the sympathetic-hemodynamic balance in men. Hypertension. 2009;54:127–133. doi: 10.1161/HYPERTENSIONAHA.109.131417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausberg M, Kosch M, Harmelink P, Barenbrock M, Hohage H, Kisters K, Dietl KH, Rahn KH. Sympathetic nerve activity in end-stage renal disease. Circulation. 2002;106:1974–1979. doi: 10.1161/01.cir.0000034043.16664.96. [DOI] [PubMed] [Google Scholar]
- Hedner J, Ejnell H, Sellgren J, Hedner T, Wallin BG. Is high and fluctuating muscle nerve sympathetic activity in the sleep apnoea syndrome of pathogenetic importance for the development of hypertension? J Hypertens Suppl. 1988;6:S529–S531. doi: 10.1097/00004872-198812040-00166. [DOI] [PubMed] [Google Scholar]
- Heindl S, Lehnert M, Criée CP, Hasenfuss G, Andreas S. Marked sympathetic activation in patients with chronic respiratory failure. Am J Respir Crit Care Med. 2001;164:597–601. doi: 10.1164/ajrccm.164.4.2007085. [DOI] [PubMed] [Google Scholar]
- Johansson M, Elam M, Rundqvist B, Eisenhofer G, Herlitz H, Lambert G, Friberg P. Increased sympathetic nerve activity in renovascular hypertension. Circulation. 1999;18:2537–2542. doi: 10.1161/01.cir.99.19.2537. [DOI] [PubMed] [Google Scholar]
- Koizumi K, Seller H, Kaufman A, Brooks CM. Pattern of sympathetic discharges and their relation to baroreceptor and respiratory activities. Brain Res. 1971;27:281–294. doi: 10.1016/0006-8993(71)90254-x. [DOI] [PubMed] [Google Scholar]
- Lambert E, Straznicky N, Schlaich M, Esler M, Dawood T, Hotchkin E, Lambert G. Differing pattern of sympathoexcitation in normal-weight and obesity-related hypertension. Hypertension. 2007;50:862–868. doi: 10.1161/HYPERTENSIONAHA.107.094649. [DOI] [PubMed] [Google Scholar]
- Leimbach WN, Wallin BG, Victor RG, Aylward PE. Direct evidence from intraneural recordings for increased central sympathetic outflow in patients with heart failure. Circulation. 1986;73:913–919. doi: 10.1161/01.cir.73.5.913. [DOI] [PubMed] [Google Scholar]
- Macefield VG, Elam M, Wallin BG. Firing properties of single postganglionic sympathetic neurones recorded in awake human subjects. Autonom Neurosci. 2002;95:146–159. doi: 10.1016/s1566-0702(01)00389-7. [DOI] [PubMed] [Google Scholar]
- Macefield VG, Rundqvist B, Sverrisdottir YB, Wallin BG, Elam M. Firing properties of single muscle vasoconstrictor neurons in the sympathoexcitation associated with congestive heart failure. Circulation. 1999;100:1708–1713. doi: 10.1161/01.cir.100.16.1708. [DOI] [PubMed] [Google Scholar]
- Macefield VG, Wallin BG. Modulation of muscle sympathetic activity during spontaneous and artificial ventilation and apnoea in humans. J Autonom Nervous Syst. 1995;53:137–147. doi: 10.1016/0165-1838(94)00173-h. [DOI] [PubMed] [Google Scholar]
- Miyajima E, Yamada Y, Yoshida Y, Matsukawa T, Shionoiri H, Tochikubo O, Ishii M. Muscle sympathetic nerve activity in renovascular hypertension and primary aldosteronism. Hypertension. 2001;17:1057–1062. doi: 10.1161/01.hyp.17.6.1057. [DOI] [PubMed] [Google Scholar]
- Narkiewicz K, van de Borne PJH, Pesek CA, Dyken ME, Montano N, Somers VK. Selective potentiation of peripheral chemoreflex sensitivity in obstructive sleep apnea. Circulation. 1999;99:1183–1189. doi: 10.1161/01.cir.99.9.1183. [DOI] [PubMed] [Google Scholar]
- Paton JF. A working heart–brainstem preparation of the mouse. J Neurosci Methods. 1996;65:63–68. doi: 10.1016/0165-0270(95)00147-6. [DOI] [PubMed] [Google Scholar]
- Raupach T, Bahr F, Herrmann P, Luethje L, Heusser K, Hasenfuss G, Bernardi L, Andreas S. Slow breathing reduces sympathoexcitation in COPD. Eur Respir J. 2008;32:387–392. doi: 10.1183/09031936.00109607. [DOI] [PubMed] [Google Scholar]
- Schlaich MP, Lambert E, Kaye DM, Krozowski, Campbell DJ, Lambert G, Hastings J, Aggarwal A, Esler MD. Sympathetic augmentation in hypertension: role of nerve firing, norepinephrine reuptake, and angiotensin neuromodulation. Hypertension. 2004;43:169–175. doi: 10.1161/01.HYP.0000103160.35395.9E. [DOI] [PubMed] [Google Scholar]
- Schlaich MP, Socratous F, Hennebry S, Eikelis N, Lambert EA, Straznicky N, Esler MD, Lambert GW. Sympathetic activation in chronic renal failure. J Am Soc Nephrol. 2009;20:933–939. doi: 10.1681/ASN.2008040402. [DOI] [PubMed] [Google Scholar]
- Schobel HP, Fischer T, Heuszer K, Geier H, Schmeider RE. Preeclampsia – a state of sympathetic hyperactivity. N Engl J Med. 1996;335:1480–1485. doi: 10.1056/NEJM199611143352002. [DOI] [PubMed] [Google Scholar]
- Seals DR, Suwarno NO, Dempsey JA. Influence of lung volume on sympathetic nerve discharge in normal humans. Circ Res. 1990;67:130–141. doi: 10.1161/01.res.67.1.130. [DOI] [PubMed] [Google Scholar]
- Seals DR, Suwarno NO, Joyner MJ, Iber C, Copeland JG, Dempsey JA. Respiratory modulation of muscle sympathetic nerve activity in intact and lung-denervated humans. Circ Res. 1993;72:440–454. doi: 10.1161/01.res.72.2.440. [DOI] [PubMed] [Google Scholar]
- Simms AE, Paton JF, Pickering AE, Allen AM. Amplified respiratory–sympathetic coupling in the spontaneously hypertensive rat: does it contribute to hypertension? J Physiol. 2009;587:597–610. doi: 10.1113/jphysiol.2008.165902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somers VK, Dyken ME, Clary MP, Abboud FM. Sympathetic neural mechanisms in obstructive sleep apnea. J Clin Invest. 1995;96:1897–1904. doi: 10.1172/JCI118235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundlöf G, Wallin BG. Human muscle nerve sympathetic activity at rest. Relationship to blood pressure and age. J Physiol. 1978;274:621–637. doi: 10.1113/jphysiol.1978.sp012170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sverrisdóttir YB, Rundqvist B, Elam M. Relative burst amplitude in human muscle sympathetic nerve activity: a sensitive indicator of altered sympathetic traffic. Clin Auton Res. 1998;8:95–100. doi: 10.1007/BF02267819. [DOI] [PubMed] [Google Scholar]
- Sverrisdóttir YB, Rundqvist B, Johannsson G, Elam M. Sympathetic neural burst amplitude distribution: A more specific indicator of sympathoexcitation in human heart failure. Circulation. 2000;102:2076–2081. doi: 10.1161/01.cir.102.17.2076. [DOI] [PubMed] [Google Scholar]
- Toney GM, Pedrino GR, Fink GD, Osborn JW. Does enhanced respiratory–sympathetic coupling contribute to peripheral neural mechanisms of angiotensin II–salt hypertension? Exp Physiol. 2010;95:587–594. doi: 10.1113/expphysiol.2009.047399. [DOI] [PMC free article] [PubMed] [Google Scholar]