

Abstract
A patient with an intermittent movement disorder has been found to have an inherited defect in pyruvate decarboxylase ((2-oxo-acid carboxy-lyase, E.C. 4.1.1.1.). The patient is a 9 yr old boy who since infancy has had repeated episodes of a combined cerebellar and choreoathetoid movement disorder. He has an elevated level of pyruvic acid in his blood, an elevated urinary alanine content, and less marked elevations in blood alanine and lactate.
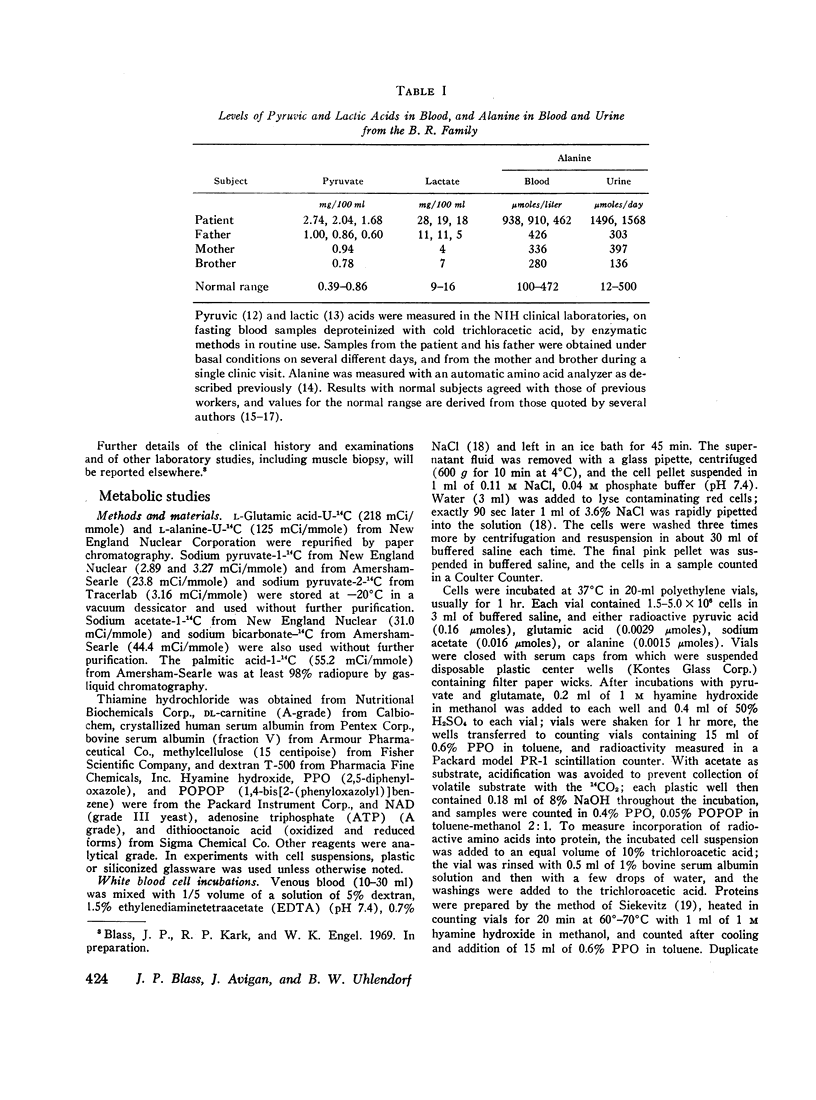
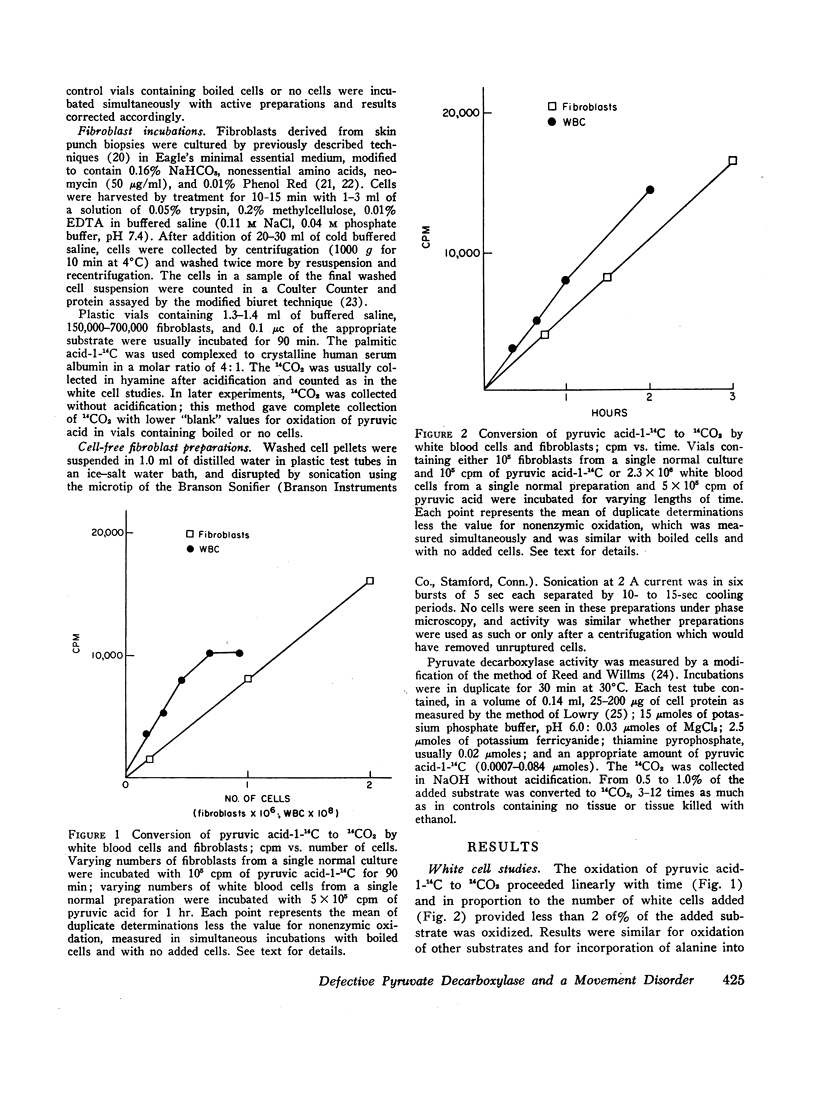
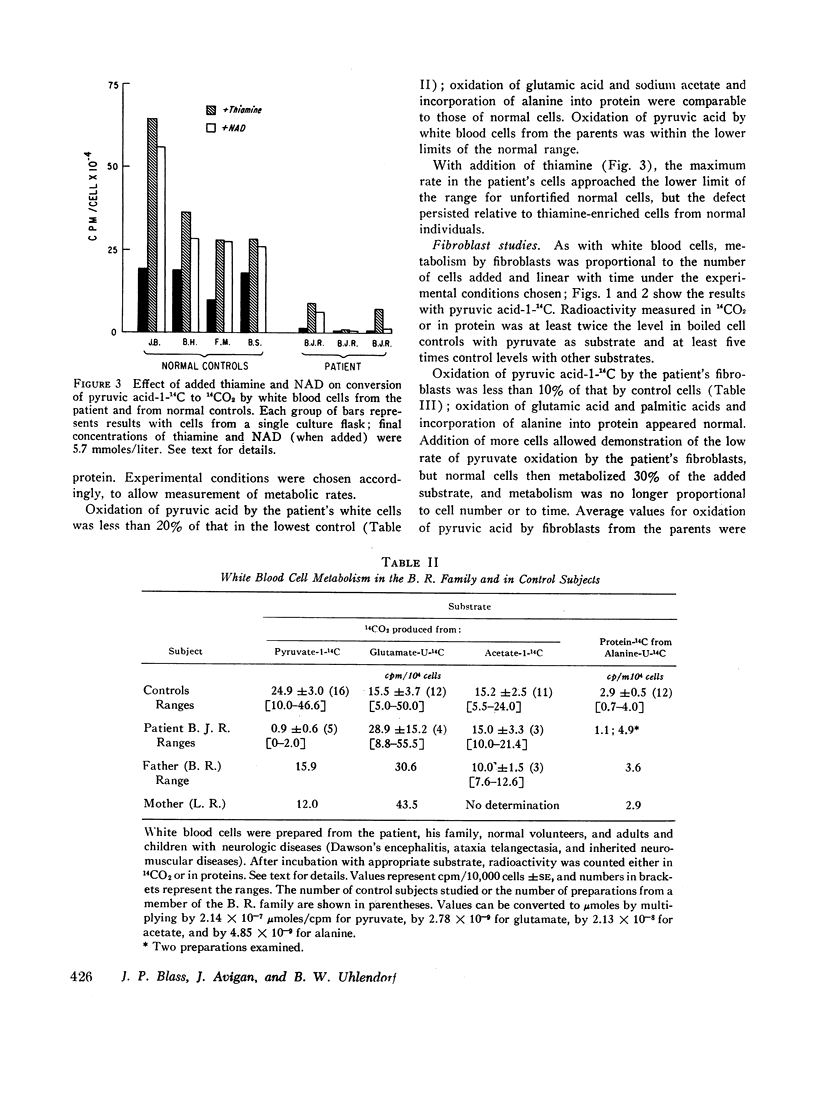
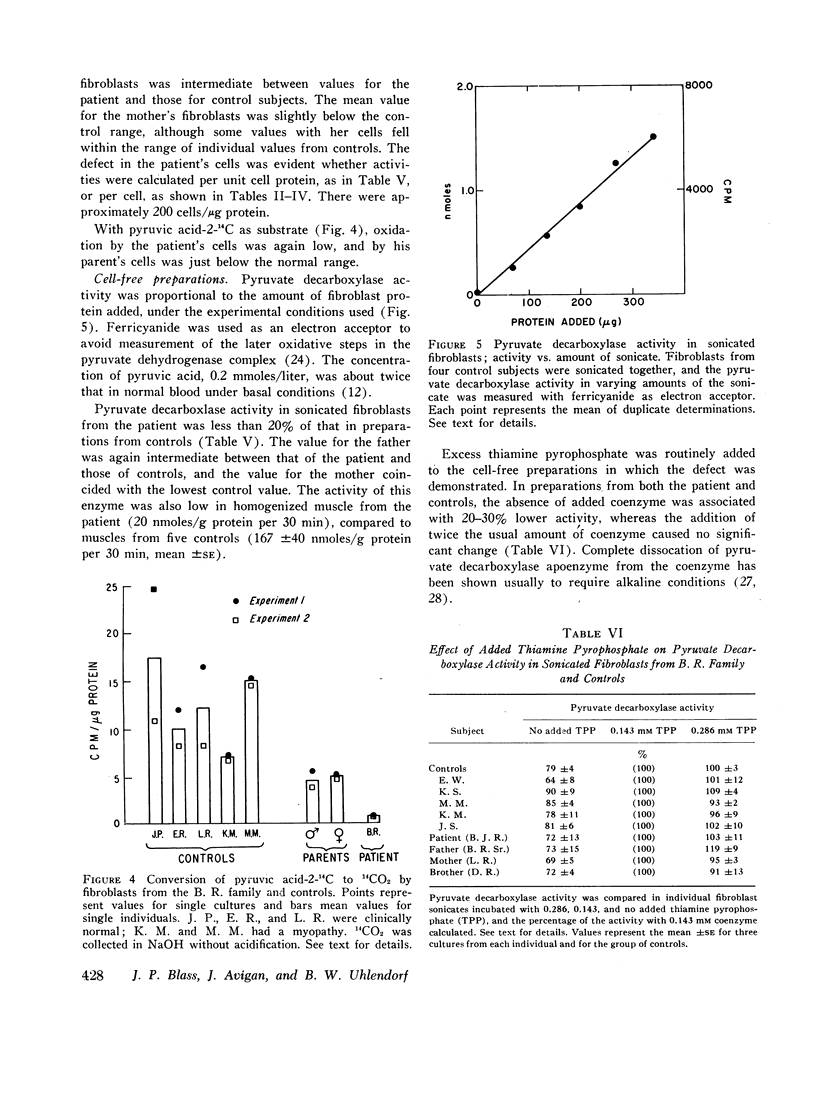
Methods were developed to study his metabolic abnormality in dilute suspensions of white blood cells and cultured skin fibroblasts, as well as in cell-free sonicates of fibroblasts. Oxidation of pyruvic acid-1-14C and pyruvic acid-2-14C by his cells and pyruvate decarboxylase activity in sonicates of his cells were less than 20% of those in cells from control subjects. Oxidation of glutamic acid-U-14C, acetate-1-14C, and palmitate-1-14C was normal, as was incorporation of alanine-U-14C into protein.
The rate of oxidation of pyruvic acid by the father's cells and the activity of pyruvate decarboxylase in the father's sonicated fibroblasts were intermediate between those of the patient and those of controls. Values for the mother were at or just below the lower limits of the ranges in controls. Kinetic data suggested the posibility of several forms of pyruvate decarboxylase in this family.
Possible mechanisms relating the chemical abnormality and the clinical symptoms in this patient are discussed.
Full text
PDF






Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- ALTSCHULE M. D., COUNTY A., GONCZ R. M., HOLLIDAY P. D., VICTOR M. Carbohydrate metabolism in brain disease. VIII. Carbohydrate metabolism in Wernicke's encephalopathy associated with alcoholism. AMA Arch Intern Med. 1957 Jan;99(1):28–39. doi: 10.1001/archinte.1957.00260010030005. [DOI] [PubMed] [Google Scholar]
- Beutler E. Glutathione reductase: stimulation in normal subjects by riboflavin supplementation. Science. 1969 Aug 8;165(3893):613–615. doi: 10.1126/science.165.3893.613. [DOI] [PubMed] [Google Scholar]
- Bressler R., Brendel K. The role of carnitine and carnitine acyltransferase in biological acetylations and fatty acid synthesis. J Biol Chem. 1966 Sep 10;241(17):4092–4097. [PubMed] [Google Scholar]
- Brues A. M. Genetic load and its varieties. Science. 1969 Jun 6;164(3884):1130–1136. doi: 10.1126/science.164.3884.1130. [DOI] [PubMed] [Google Scholar]
- COTTOM D. G. Acute cerebellar ataxia. Arch Dis Child. 1957 Jun;32(163):181–188. doi: 10.1136/adc.32.163.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke C. A., Evans D. A., Harris R., McConnell R. B., Woodrow J. C. Genetics in medicine: a review. Q J Med. 1968 Jan;37(145):1–63. [PubMed] [Google Scholar]
- Clayton B. E., Dobbs R. H., Patrick A. D. Leigh's subacute necrotizing encephalopathy: clinical and biochemical study, with special reference to therapy with lipoate. Arch Dis Child. 1967 Oct;42(225):467–478. doi: 10.1136/adc.42.225.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coronato A., Cohen A. B. Lactic acidosis secondary to pernicious anemia. Ann Intern Med. 1969 Jan;70(1):77–80. doi: 10.7326/0003-4819-70-1-77. [DOI] [PubMed] [Google Scholar]
- DENT C. E. Argininosuccinic aciduria. A new form of mental deficiency due to metabolic causes. Proc R Soc Med. 1959 Oct;52:885–885. doi: 10.1177/003591575905201025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DREYFUS P. M. Clinical application of blood transketolase determinations. N Engl J Med. 1962 Sep 20;267:596–598. doi: 10.1056/NEJM196209202671204. [DOI] [PubMed] [Google Scholar]
- Dancis J., Hutzler J., Rokkones T. Intermittent branched-chain ketonuria. Variant of maple-syrup-urine disease. N Engl J Med. 1967 Jan 12;276(2):84–89. doi: 10.1056/NEJM196701122760204. [DOI] [PubMed] [Google Scholar]
- Dunn H. G., Perry T. L., Dolman C. L. A syndrome resembling Friedreich's ataxia with relapsing polyneuropathy and hyperalaninemia. Neurology. 1968 Mar;18(3):301–301. [PubMed] [Google Scholar]
- EAGLE H. Amino acid metabolism in mammalian cell cultures. Science. 1959 Aug 21;130(3373):432–437. doi: 10.1126/science.130.3373.432. [DOI] [PubMed] [Google Scholar]
- ERICKSON R. J. FAMILIAL INFANTILE LACTIC ACIDOSIS. J Pediatr. 1965 Jun;66:1004–1016. doi: 10.1016/s0022-3476(65)80085-3. [DOI] [PubMed] [Google Scholar]
- FALLON H. J., FREI E., 3rd, DAVIDSON J. D., TRIER J. S., BURK D. Leukocyte preparations from human blood: evaluation of their morphologic and metabolic state. J Lab Clin Med. 1962 May;59:779–791. [PubMed] [Google Scholar]
- FARMER T. W., MUSTIAN V. M. Vestibulocerebellar ataxia. A newly defined hereditary syndrome with periodic manifestations. Arch Neurol. 1963 May;8:471–480. doi: 10.1001/archneur.1963.00460050021002. [DOI] [PubMed] [Google Scholar]
- GLOSTER J. A., HARRIS P. Observations on an enzymic method for the estimation of pyruvate in blood. Clin Chim Acta. 1962 Mar;7:206–211. doi: 10.1016/0009-8981(62)90011-6. [DOI] [PubMed] [Google Scholar]
- Gilbert V. E. Blood pyruvate and lactate during febrile human infections. Metabolism. 1968 Oct;17(10):943–951. doi: 10.1016/0026-0495(68)90162-5. [DOI] [PubMed] [Google Scholar]
- HARTMANN A. F., Sr, WOHLTMANN H. J., PURKERSON M. L., WESLEY M. E. Lactate metabolism. Studies of a child with a serious congenital deviation. J Pediatr. 1962 Aug;61:165–180. doi: 10.1016/s0022-3476(62)80251-0. [DOI] [PubMed] [Google Scholar]
- HUCKABEE W. E. Abnormal resting blood lactate. I. The significance of hyperlactatemia in hospitalized patients. Am J Med. 1961 Jun;30:840–848. doi: 10.1016/0002-9343(61)90172-3. [DOI] [PubMed] [Google Scholar]
- Harris H. Enzyme and protein polymorphism in human populations. Br Med Bull. 1969 Jan;25(1):5–13. doi: 10.1093/oxfordjournals.bmb.a070670. [DOI] [PubMed] [Google Scholar]
- Haworth J. C., Ford J. D., Younoszai M. K. Familial chronic acidosis due to an error in lactate and pyruvate metabolism. Can Med Assoc J. 1967 Sep 23;97(13):773–779. [PMC free article] [PubMed] [Google Scholar]
- Herndon J. H., Jr, Steinberg D., Uhlendorf B. W., Fales H. M. Refsum's disease: characterization of the enzyme defect in cell culture. J Clin Invest. 1969 Jun;48(6):1017–1032. doi: 10.1172/JCI106058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill W., Sherman H. Acute intermittent familial cerebellar ataxia. Arch Neurol. 1968 Apr;18(4):350–357. doi: 10.1001/archneur.1968.00470340036002. [DOI] [PubMed] [Google Scholar]
- Hommes F. A., Polman H. A., Reerink J. D. Leigh's encephalomyelopathy: an inborn error of gluconeogenesis. Arch Dis Child. 1968 Aug;43(230):423–426. doi: 10.1136/adc.43.230.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ISRAELS S., HAWORTH J. C., GOURLEY B., FORD J. D. CHRONIC ACIDOSIS DUE TO AN ERROR IN LACTATE AND PYRUVATE METABOLISM. REPORT OF TWO CASES. Pediatrics. 1964 Sep;34:346–356. [PubMed] [Google Scholar]
- JOINER C. L., McARDLE B., THOMPSON R. H. S. Blood pyruvate estimations in the diagnosis and treatment of polyneuritis. Brain. 1950 Dec;73(4):431–452. doi: 10.1093/brain/73.4.431. [DOI] [PubMed] [Google Scholar]
- Kamoshita S., Aguilar M. J., Landing B. H. Infantile subacute necrotizing encephalomyelopathy. Am J Dis Child. 1968 Aug;116(2):120–129. doi: 10.1001/archpedi.1968.02100020122002. [DOI] [PubMed] [Google Scholar]
- King J. L., Jukes T. H. Non-Darwinian evolution. Science. 1969 May 16;164(3881):788–798. doi: 10.1126/science.164.3881.788. [DOI] [PubMed] [Google Scholar]
- LEIGH D. Subacute necrotizing encephalomyelopathy in an infant. J Neurol Neurosurg Psychiatry. 1951 Aug;14(3):216–221. doi: 10.1136/jnnp.14.3.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonsdale D. Encephalopathy and pyruvate metabolism. N Engl J Med. 1968 Aug 8;279(6):325–325. [PubMed] [Google Scholar]
- Lonsdale D., Faulkner W. R., Price J. W., Smeby R. R. Intermittent cerebellar ataxia associated with hyperpyruvic acidemia, hyperalaninemia, and hyperalaninuria. Pediatrics. 1969 Jun;43(6):1025–1034. [PubMed] [Google Scholar]
- Lonsdale D. Hyperalaninemia with pyruvicemia. N Engl J Med. 1968 May 30;278(22):1235–1235. doi: 10.1056/nejm196805302782222. [DOI] [PubMed] [Google Scholar]
- McCandless D. W., Schenker S., Cook M. Encephalopathy of thiamine deficieny: studies of intracerebral mechanisms. J Clin Invest. 1968 Oct;47(10):2268–2280. doi: 10.1172/JCI105912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris M. D., Fisher D. A., Fiser R. Late-onset branched-chain ketoaciduria: (maple syrup urine disease). J Lancet. 1966 Mar;86(3):149–152. [PubMed] [Google Scholar]
- SIEKEVITZ P. Uptake of radioactive alanine in vitro into the proteins of rat liver fractions. J Biol Chem. 1952 Apr;195(2):549–565. [PubMed] [Google Scholar]
- Steinberg D., Vroom F. Q., Engel W. K., Cammermeyer J., Mize C. E., Avigan J. Refsum's disease--a recently characterized lipidosis involving the nervous system. Combined clinical staff conference at the National Institutes of Health. Ann Intern Med. 1967 Feb;66(2):365–395. doi: 10.7326/0003-4819-66-2-365. [DOI] [PubMed] [Google Scholar]
- Tada K., Yoshida T., Konno T., Wada Y., Yokoyama Y. Hyperalaninemia with pyruvicemia (preliminary report). Tohoku J Exp Med. 1969 Jan;97(1):99–100. doi: 10.1620/tjem.97.99. [DOI] [PubMed] [Google Scholar]
- UTTER M. F., KEECH D. B. PYRUVATE CARBOXYLASE. I. NATURE OF THE REACTION. J Biol Chem. 1963 Aug;238:2603–2608. [PubMed] [Google Scholar]
- WROBLEWSKI F., LADUE J. S. Serum glutamic pyruvic transaminase in cardiac with hepatic disease. Proc Soc Exp Biol Med. 1956 Apr;91(4):569–571. doi: 10.3181/00379727-91-22330. [DOI] [PubMed] [Google Scholar]
- Wieland O., Von Jagow-Westermann B., Stukowski B. Kinetic and regulatory properties of heart muscle pyruvate dehydrogenase. Hoppe Seylers Z Physiol Chem. 1969 Mar;350(3):329–334. doi: 10.1515/bchm2.1969.350.1.329. [DOI] [PubMed] [Google Scholar]
- Worsley H. E., Brookfield R. W., Elwood J. S., Noble R. L., Taylor W. H. Lactic acidosis with necrotizing encephalopathy in two sibs. Arch Dis Child. 1965 Oct;40(213):492–501. doi: 10.1136/adc.40.213.492. [DOI] [PMC free article] [PubMed] [Google Scholar]