

Abstract
The new iron(II)-thiolate complexes [(iPrBIP)FeII(SPh)(Cl)] (1) and [(iPrBIP)FeII(SPh)(OTf)] (2) (BIP = bis(iminopyridine)) were prepared as models for cysteine dioxygenase (CDO), which converts Cys to Cys-SO2H at a (His)3FeII center. Reaction of 1 and 2 with O2 leads to Fe-oxygenation and S-oxygenation, respectively. For 1 + O2, the spectroscopic and reactivity data, including 18O isotope studies, are consistent with an assignment of an iron(IV)-oxo complex as the product of oxygenation ([(iPrBIP)FeIV(O)(Cl)] (3)). In contrast, 2 + O2 results in direct S-oxygenation to give a sulfonato product (PhSO3−). The positioning of the thiolate ligand in 1 versus 2 appears to play a critical role in determining the outcome of O2 activation. The thiolate ligands in 1 and 2 are essential for O2 reactivity, and exhibit an important influence over the FeIII/FeII redox potential.
Determining the factors that govern the activation of dioxygen by both heme and nonheme iron metalloenzymes is of fundamental importance. Mononuclear nonheme iron oxygenases typically contain a 2-His-1-carboxylate ligand set bound to the catalytic iron center. An interesting exception is cysteine dioxygenase (CDO), which utilizes a (His)3FeII(H2O) center to activate O2 and oxidize cysteine to sulfinic acid (CysSO2H), a key metabolic process vital for human health.1 Despite the importance of CDO from a health perspective, little is known about the mechanism of this dioxygenase.2 In general, the oxidation of Cys to disulfide, sulfenic acid (Cys(O)H) and other oxidized products has been implicated in oxidative stress response.3 Thus understanding the fundamental mechanistic pathways of biologically relevant sulfur oxidations is of high current interest.4
Although many studies on iron(II) model complexes have yielded key insights into the reactivity of nonheme iron centers, relatively few have involved the use of O2 as the oxidant, in part because of the inherent difficulties with activating and controlling O2.5 In an earlier report, we described the synthesis of an N3S(thiolate)FeII model complex of CDO, which contains the 3 neutral N binding motif found in the enzyme, and reacts with O2 selectively to yield an S-oxygenated sulfonato product.6 The thiolate donor was covalently tethered to a bis(imino)pyridine (BIP) framework, in part to favor S-oxygenation as opposed to disulfide formation. To our knowledge, this reaction was the first example of an FeII-thiolate complex reacting with O2 to give S-, as opposed to Fe-oxygenation (e.g., FeIII-O-FeIII species).7
Herein we report the synthesis of two new unsymmetrical FeII-thiolate BIP complexes, [(iPrBIP)FeII(SPh)(Cl)] (1) and [(iPrBIP)FeII(SPh)(OTf)] (2) (iPrBIP = 2,6-(ArN=CMe)2C5H3N), Ar = 2,6-iPr2C6H3), in which the thiolate ligands are not covalently tethered to the BIP framework. The reactivity of these complexes toward O2 has been examined together with non-thiolate-ligated analogs. We show that coordination of the thiolate ligands is crucial for O2 activation by (BIP)FeII. We also show that S-oxygenation is possible for a terminal thiolate, and furthermore that the positioning of the thiolate donor specifies the outcome of oxygenation at either sulfur or iron.
Significant efforts have gone into the synthesis and study of (BIP)Fe complexes for their use in N2 activation and catalysis.8 However, unsymmetrical derivatives of formula [(BIP)FeII(X)(Y)] (X ≠ Y) are scarce. Careful control of stoichiometry, together with the appropriate conditions (solvent, temperature), allowed for the isolation of the monothiolato complexes 1 and 2 (Figure 1). The molecular structures of 1 and 2 reveal 5-coordinate FeII ions with the desired single terminal thiolate ligands bound to the iron. Bond distances and angles are consistent with high-spin FeII BIP complexes.8a,d,e A distinguishing feature of the structures of 1 and 2 is the positioning of the thiolate ligand. In complex 1, the PhS− group sits in a pseudo-axial position in relation to the N3Cl plane, trans to the open coordination site that subtends the obtuse N1-Fe-N3 angle (141.2°). This positioning may be aided by a π-stacking interaction between the pyridine and PhS− groups. In contrast, the PhS− ligand of 2 is bound in a pseudo-equatorial arrangement with the iPrBIP ligand, and is cis to the open coordination site.
Figure 1.
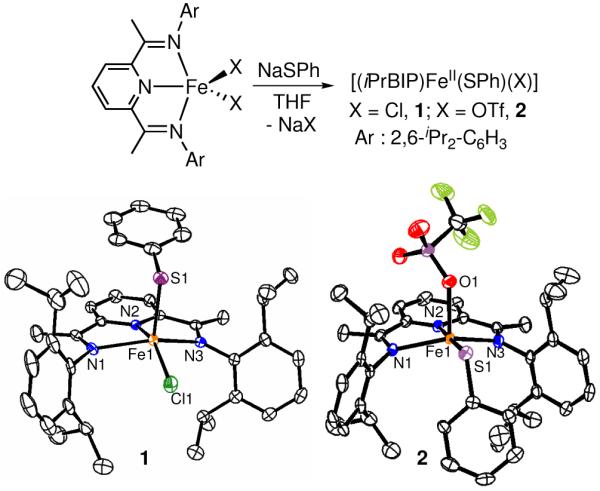
Synthetic scheme and displacement ellipsoid plots (50% probability level) for 1 and 2 at 110 K. H atoms are omitted for clarity.
Both 1 and 2 exhibit relatively sharp, paramagnetically shifted peaks in the 1H NMR spectrum (CD2Cl2) typical of high-spin (BIP)FeX2 complexes, and these spectra are consistent with their solid-state structures. Magnetic susceptibility for 1 measured by Evan’s method in CD2Cl2 gives μeff = 5.2 μB, close to the spin-only value for a high-spin FeII (S = 2) ion.
Reaction of 1 (10 – 20 mM) with a slight excess of dry O2 (5 equiv) leads to a color change from dark blue to green over the course of 1 h. A decrease of the λmax for 1 at 715 nm (ε ~ 4000 M−1 cm−1) is observed, and a new band for the green species appears at λmax 690 nm (ε ~ 1500 M−1 cm−1) (Figure 2; for time-dependence see Fig. S5). This spectrum is similar to that reported for a closely related bis(imino)pyridine iron(IV)-oxo complex (λmax 660 nm, ε ~ 1200 M−1 cm−1).9 Analysis by laser-desorption ionization mass spectrometry (LDIMS(+)) reveals a dominant isotopic cluster at m/z = 588, whose isotope and fragmentation pattern (Figs. 2, S8 and S9) are consistent with an FeIV(O) complex, [(iPrBIP)FeIV(O)(Cl)]+ (3). The thiolate ligand is oxidized to disulfide during the production of 3, as determined by 1H NMR (PhS-SPh, 85%). Introduction of 18O2 in place of 16O2 causes a shift of two mass units for the LDIMS of 3, giving m/z = 590 (80% 18O incorporation). Finally, green 3 is EPR-silent (X-band, 15 K). These data are consistent with the assignment of 3 as an FeIV(O) species.
Figure 2.
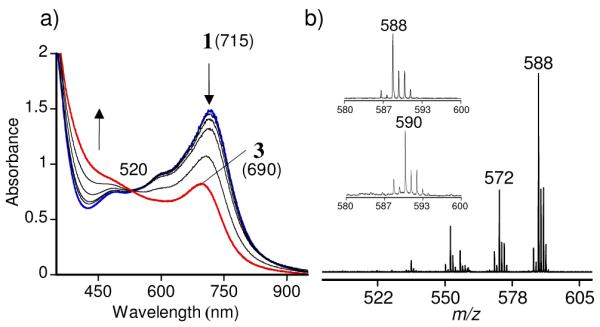
a) UV-vis spectral changes for the reaction of 1 (715 nm, 0.370 mM) with excess O2 in CH2Cl2, leading to formation of 3 (690 nm). b) LDIMS of 3 formed in the reaction of 1 + O2. Peaks at m/z of 588 and 572 correspond to [(iPrBIP)FeIV(O)(Cl)]+ and [(iPrBIP)FeII(Cl)]+, respectively. Inset: isotopic cluster for 3 prepared from 16O2 (top) and 18O2 (bottom).
If the reaction of 1 with excess O2 in CH2Cl2 is carried out in the presence of PPh3 (5 equiv), OPPh3 is produced in good yield (70%, 31P NMR). Alternatively, formation of green 3, followed by removal of O2 under vacuum and addition of PPh3 (50 – 300 equiv) under Ar, results in the smooth decay of the peak for 3 at 690 nm (Figure S6). This decay follows good pseudo-first-order behavior, and the rate constants (kobs) thus obtained were found to increase linearly with [PPh3], yielding a second-order rate constant of k2 = 3.6 ± 0.3 × 10−3 M−1 s−1 for oxygen-atom-transfer from 3 to PPh3 (Figure S7). This relatively slow reactivity may in part be due to the steric encumbrance imposed by the 2,6-iPr2-C6H3 substituents. The 18O-labeled 3 produces 18OPPh3 with modest isotopic incorporation (16O:18O 85:15). However, addition of excess H2 18O to the reaction of 3-16O and PPh3 results in a significant increase in the isotopically labeled product 18OPPh3 (50% 18O) (eq 1). These data indicate that the O atom in 3 undergoes facile exchange with exogenous H2O, as seen for other iron terminal oxo species.10 Although further spectroscopic studies are needed to definitively characterize the structure of 3, all of the spectroscopic data and reactivity presented here strongly
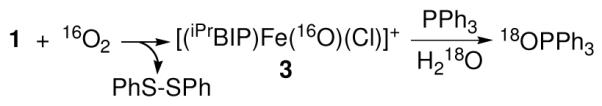 |
(1) |
support the formulation of 3 as a terminal iron-oxo complex generated from 1 + O2, with the PhS− ligand undergoing concomitant oxidation to disulfide.
The formation of nonheme FeIV(O) complexes from FeII and O2 can be induced by the addition of external co-reductants (e.g. cyclohexene or NADH).5b,c,e In the case of 1, the thiolate ligand functions as a built-in co-reductant to assist in the activation of O2. In comparison, the covalently-tethered thiolate complex [FeII(N3S(thiolate))(OTf)] (4) also serves to activate dioxygen, but in this case participation from sulfur leads to direct oxygenation of the S atom.6
To our surprise, the addition of stoichiometric amounts of O2 to the triflate complex 2 follows a very different oxidation pathway than followed by the chloro analog 1. An immediate color change from dark blue to brown is noted upon addition of O2, but LDIMS reveals a cluster at m/z 694 corresponding to S-oxygenated [FeII(iPrBIP)(PhSO3)]+. Attempts to crystallize [FeII(BIP)(PhSO3)]+ have led thus far only to the crystallization of the known FeII(iPrBIP)(OTf)2 complex, but the production of benzenesulfonic acid was readily confirmed by 1H NMR, and quantitation by reverse-phase HPLC after hydrolytic workup gave a yield of 30% for PhSO3H (based on total Fe). The use of labeled 18O2 results in ~90% incorporation of 18O into the PhSO3− ligand. Despite the fact that the thiolate donor in 2 is not part of a chelate ring, sulfur oxygenation does occur, as seen for the covalently tethered 4. In contrast, no evidence for PhSO3H was detected by LDIMS or HPLC for 1 + O2 in control experiments.
The reactivity of the related non-thiolate-ligated complexes Fe(iPrBIP)Cl2 (5) and Fe(iPrBIP)(OTf)2 (6) was next examined for comparison with 1 and 2. These complexes are completely inert toward O2 in both solution (e.g. CH2Cl2, CH3CN) and the solid-state (eq 2). Addition of PPh3 to oxygenated solutions of 5 and 6 showed no formation of OPPh3. The incorporation of a thiolate donor thus clearly plays a critical role in the activation of O2 by these nonheme iron(II) complexes.
| (2) |
The redox potentials of 1, 2, 5, and 6 are compared in Table 1. The thiolate-ligated complexes exhibit significantly lower redox potentials than the non-thiolate analogs, correlating nicely with their relative O2 reactivities. A similar correlation was made for [FeII(TMC)(OTf)2] (TMC = 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane), which exhibits a solvent-dependent redox potential, and reacts with O2 to give an FeIV(O) complex only in solvents where E1/2(FeIII/II) < −0.1 V (e.g. THF).5a Similarly, nonheme iron(II) complexes with more positive E1/2 values fail to react with O2 to give oxoiron(IV) species. From Table 1, an E1/2(FeIII/II) < −0.1 V appears to be a prerequisite for O2 activation in nonheme iron(II) complexes, and inclusion of a single thiolate donor is sufficient to lower the redox potential of (iPrBIP)FeII complexes into this range. It should be noted that the E1/2 values for 1 and 2 remain more than 1 V above the one-electron reduction potential for the O2/O2− couple in organic solvents,5h ruling out an outer-sphere mechanism of O2 activation.
Table 1.
Redox potentials for (iPrBIP)FeII and related nonheme FeII complexes.
| Compound | E1/2 (ΔEpp)a | O2 reactivity |
|---|---|---|
| [(iPrBIP)FeII(SPh)(Cl)] 1 | −0.173b (0.114) (r) | yes |
| [(iPrBIP)FeII(SPh)(OTf)] 2 | −0.372b (0.149) (r) | yes |
| [(iPrBIP)FeII(Cl)2] 5 | 0.025b (0.153) (r) | no |
| [(iPrBIP)FeII(OTf)2] 6 | 0.613b,c (ir) | no |
| [(TMC)FeII(OTf)2]d | −0.14e (qr) | yes |
| [(TMC)FeII(OTf)2]d | 0.02f (r) | no |
| [(TPA)FeII]2+, d,g | 0.36h (r) | no |
V vs Fc+/Fc; ΔEpp = peak-to-peak separation; r = reversible, ir = irreversible, qr = quasi-reversible.
In CH2Cl2, scan rate of 0.1 V/s.
Anodic peak potential.
In MeCN/THF (1:1).
In MeCN/CH2Cl2 (1:1).
TPA: tris(2-pyridylmethyl)amine.
In neat MeCN.
Given the structural and electronic similarities between the two thiolate-ligated complexes 1 and 2, why does their reactivity with O2 follow such dramatically different paths? Scrutiny of the structures of 1 and 2 appears to hold the key. The PhS− ligand in 1 is bound trans to the open site available for O2 binding, whereas it is bound cis in 2. A plausible mechanism for O2 activation in 1 thus begins with coordination of O2 to the open site trans to the thiolate donor, followed by electron-transfer from both the iron and sulfur centers to the bound O2 (Scheme 1a). In this case, intramolecular attack of an Fe-O2 intermediate on the sulfur donor would be strongly disfavored by the trans orientation of the PhS− ligand. In contrast, the analogous Fe-(O2) intermediate in 2 would be generated cis to the thiolate ligand, providing a facile pathway for intramolecular S-oxygenation (Scheme 1b). Similarly, the thiolate donor in the covalently-tethered 4 is also found cis to the open coordinate on site.
Scheme 1.
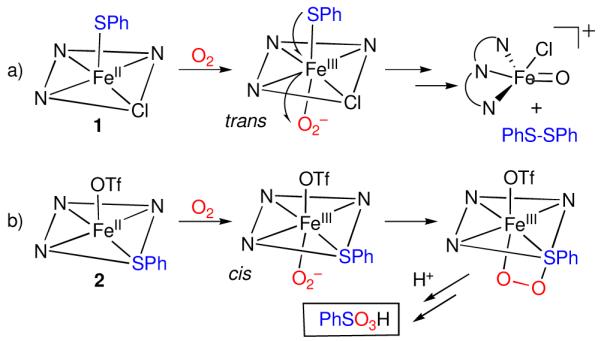
Proposed mechanisms of O2 activation by 1 and 2.
This hypothesis depends upon the feasibility of attaining a 6-coordinate structure with the sterically encumbered BIP ligand in 1 and 2. For less bulky BIP analogs, where Ar =2,6-Me2-C6H3, 6-coordinate FeII complexes are known,8d but to our knowledge there are no examples with Ar = 2,6-iPr2-C6H3. Thus we were pleased to isolate [FeII(iPrBIP)(H2O)2(NCCH3)](OTf)2 (7) as a product from reactions of 2 + O2, whose molecular structure is given in Figure 3. Despite the large steric encumbrance provided by the flanking 2,6-iPr2C6H3 substituents, a 6-coordinate geometry is clearly attainable in 7.
Figure 3.
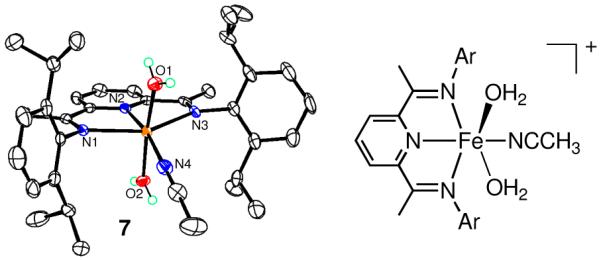
Displacement ellipsoid plots (left, 50% probability level) and molecular structure (right) of 7. H atoms, except for those attached to the water molecules, and the OTf− ions have been omitted for clarity.
In summary, we have demonstrated that a thiolate donor is essential for the activation of O2 by nonheme iron (BIP)FeII complexes, and can serve as either a co-reductant or as a site for O-capture. The relative positioning of the PhS− ligand in relation to the potential O2 binding site appears to play a critical role in determining whether oxygenation occurs at iron or sulfur.11 It is also shown that S-oxygenation can occur for terminal, iron-bound thiolates, contrary to established precedent. It has been proposed that the Cys substrate in CDO coordinates to the Fe center through a chelate ring involving sulfur and the amino group.1 The findings presented here suggest that this unusual binding mode for Cys is not required for S-oxygenation to occur.
Supplementary Material
Acknowledgment
The NIH (GM62309) is gratefully acknowledged for financial support. We thank Ms. A. McQuilken for assistance with the cyclic voltammetry of complex 6.
Footnotes
Supporting Information Available: Experimental procedures and characterization data for 1 – 6. Details of the X-ray crystallography (PDF) and crystallographic information files of 1, 2 and 7 (CIF). This material is available free of charge via the Internet at htpp://pubs.acs.org.
References
- (1)(a).Joseph CA, Maroney MJ. Chem. Commun. 2007:3338–3349. doi: 10.1039/b702158e. [DOI] [PubMed] [Google Scholar]; (b) Stipanuk MH, Ueki I, Dominy JE, Simmons CR, Hirschberger LL. Amino Acids. 2009;37:55–63. doi: 10.1007/s00726-008-0202-y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) McCoy JG, Bailey LJ, Bitto E, Bingman CA, Aceti DJ, Fox BG, Phillips GN. Proc. Natl. Acad. Sci. U.S.A. 2006;103:3084–3089. doi: 10.1073/pnas.0509262103. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Simmons CR, Krishnamoorthy K, Granett SL, Schuller DJ, Dominy JE, Begley TP, Stipanuk MH, Karplus PA. Biochemistry. 2008;47:11390–11392. doi: 10.1021/bi801546n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Siakkou E, Wilbanks SM, Jameson GNL. Anal. Biochem. 2010;405:127–131. doi: 10.1016/j.ab.2010.06.013. [DOI] [PubMed] [Google Scholar]; (f) Diebold AR, Neidig ML, Moran GR, Straganz GD, Solomon EI. Biochemistry. 2010;49:6945–6952. doi: 10.1021/bi100892w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Gardner JD, Pierce BS, Fox BG, Brunold TC. Biochemistry. 2010;49:6033–6041. doi: 10.1021/bi100189h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2)(a).Pierce BS, Gardner JD, Bailey LJ, Brunold TC, Fox BG. Biochemistry. 2007;46:8569–8578. doi: 10.1021/bi700662d. [DOI] [PubMed] [Google Scholar]; (b) Ye S, Wu X, Wei L, Tang DM, Sun P, Bartlam M, Rao ZH. J. Biol. Chem. 2007;282:3391–3402. doi: 10.1074/jbc.M609337200. [DOI] [PubMed] [Google Scholar]; (c) de Visser SP, Straganz GD. J. Phys. Chem. A. 2009;113:1835–1846. doi: 10.1021/jp809700f. [DOI] [PubMed] [Google Scholar]; (d) Aluri S, de Visser SP. J. Am. Chem. Soc. 2007;129:14846–14847. doi: 10.1021/ja0758178. [DOI] [PubMed] [Google Scholar]
- (3)(a).Paulsen CE, Carroll KS. Chem. Biol. 2009;16:217–225. doi: 10.1016/j.chembiol.2009.01.003. [DOI] [PubMed] [Google Scholar]; (b) Leonard SE, Reddie KG, Carroll KS. ACS Chem. Biol. 2009;4:783–799. doi: 10.1021/cb900105q. [DOI] [PubMed] [Google Scholar]; (c) Poole LB, Karplus PA, Claiborne A. Annu. Rev. Pharmacol. Toxicol. 2004;44:325–347. doi: 10.1146/annurev.pharmtox.44.101802.121735. [DOI] [PubMed] [Google Scholar]
- (4)(a).Heinecke J, Ford PC. J. Am. Chem. Soc. 2010;132:9240–9243. doi: 10.1021/ja102221e. [DOI] [PubMed] [Google Scholar]; (b) O’Toole MG, Kreso M, Kozlowski PM, Mashuta MS, Grapperhaus CA. J. Biol. Inorg. Chem. 2008;13:1219–1230. doi: 10.1007/s00775-008-0405-4. [DOI] [PubMed] [Google Scholar]; (c) Grapperhaus CA, Darensbourg MY. Acc. Chem. Res. 1998;31:451–459. [Google Scholar]; (d) Chohan BS, Shoner SC, Kovacs JA, Maroney MJ. Inorg. Chem. 2004;43:7726–7734. doi: 10.1021/ic049110n. [DOI] [PubMed] [Google Scholar]; (e) Galardon E, Giorgi M, Artaud I. Chem. Commun. 2004:286–287. doi: 10.1039/b312318a. [DOI] [PubMed] [Google Scholar]; (f) Noveron JC, Olmstead MM, Mascharak PK. J. Am. Chem. Soc. 2001;123:3247–59. doi: 10.1021/ja001253v. [DOI] [PubMed] [Google Scholar]
- (5)(a).Kim SO, Sastri CV, Seo MS, Kim J, Nam W. J. Am. Chem. Soc. 2005;127:4178–4179. doi: 10.1021/ja043083i. [DOI] [PubMed] [Google Scholar]; (b) Hong S, Lee YM, Shin W, Fukuzumi S, Nam W. J. Am. Chem. Soc. 2009;131:13910–13911. doi: 10.1021/ja905691f. [DOI] [PubMed] [Google Scholar]; (c) Lee YM, Hong S, Morimoto Y, Shin W, Fukuzumi S, Nam W. J. Am. Chem. Soc. 2010;132:10668–10670. doi: 10.1021/ja103903c. [DOI] [PubMed] [Google Scholar]; (d) MacBeth CE, Golombek AP, Young VG, Yang C, Kuczera K, Hendrich MP, Borovik AS. Science. 2000;289:938–941. doi: 10.1126/science.289.5481.938. [DOI] [PubMed] [Google Scholar]; (e) Thibon A, England J, Martinho M, Young VG, Frisch JR, Guillot R, Girerd JJ, Munck E, Que L, Jr., Banse F. Angew. Chem. Int. Ed. 2008;47:7064–7067. doi: 10.1002/anie.200801832. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Do LH, Lippard SJ. Inorg. Chem. 2009;48:10708–10719. doi: 10.1021/ic901711c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Korendovych IV, Kryatov SV, Rybak-Akimova EV. Acc. Chem. Res. 2007;40:510–521. doi: 10.1021/ar600041x. [DOI] [PubMed] [Google Scholar]; (h) Kryatov SV, Taktak S, Korendovych IV, Rybak-Akimova EV, Kaizer J, Torelli S, Shan XP, Mandal S, MacMurdo VL, Payeras AMI, Que L., Jr. Inorg. Chem. 2005;44:85–99. doi: 10.1021/ic0485312. [DOI] [PubMed] [Google Scholar]; (i) Martinho M, Blain G, Banse F. Dalton Trans. 2010;39:1630–1634. doi: 10.1039/b918061c. [DOI] [PubMed] [Google Scholar]
- (6).Jiang YB, Widger LR, Kasper GD, Siegler MA, Goldberg DP. J. Am. Chem. Soc. 2010;132:12214–12215. doi: 10.1021/ja105591q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7)(a).Musie G, Lai CH, Reibenspies JH, Sumner LW, Darensbourg MY. Inorg. Chem. 1998;37:4086–4093. doi: 10.1021/ic980475f. [DOI] [PubMed] [Google Scholar]; (b) Theisen RM, Shearer J, Kaminsky W, Kovacs JA. Inorg. Chem. 2004;43:7682–7690. doi: 10.1021/ic0491884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8)(a).Gibson VC, Redshaw C, Solan GA. Chem Rev. 2007;107:1745–1776. doi: 10.1021/cr068437y. [DOI] [PubMed] [Google Scholar]; (b) Manuel TD, Rohde J-U. J. Am. Chem. Soc. 2009;131:15582–15583. doi: 10.1021/ja9065943. [DOI] [PubMed] [Google Scholar]; (c) Scott J, Gambarotta S, Korobkov I, Knijnenburg Q, de Bruin B, Budzelaar PHM. J. Am. Chem. Soc. 2005;127:17204–17206. doi: 10.1021/ja056135s. [DOI] [PubMed] [Google Scholar]; (d) Britovsek GJP, England J, Spitzmesser SK, White AJP, Williams DJ. Dalton Trans. 2005:945–955. doi: 10.1039/b414813d. [DOI] [PubMed] [Google Scholar]; (e) Russell SK, Darmon JM, Lobkovsky E, Chirik PJ. Inorg. Chem. 2010;49:2782–2792. doi: 10.1021/ic902162z. [DOI] [PubMed] [Google Scholar]; (f) Bart SC, Chlopek K, Bill E, Bouwkamp MW, Lobkovsky E, Neese F, Wieghardt K, Chirik PJ. J. Am. Chem. Soc. 2006;128:13901–13912. doi: 10.1021/ja064557b. [DOI] [PubMed] [Google Scholar]; (g) Kendall AJ, Zakharov LN, Gilbertson JD. Inorg. Chem. 2010;49:8656–8658. doi: 10.1021/ic101408e. [DOI] [PubMed] [Google Scholar]; (h) Tang JK, Gamez P, Reedijk J. Dalton Trans. 2007:4644–4646. doi: 10.1039/b712849p. [DOI] [PubMed] [Google Scholar]
- (9).Annaraj J, Kim S, Seo MS, Lee YM, Kim Y, Kim SJ, Choi YS, Jang HG, Nam W. Inorg. Chim. Acta. 2009;362:1031–1034. [Google Scholar]
- (10)(a).Seo MS, In JH, Kim SO, Oh NY, Hong J, Kim J, Que L, Jr., Nam W. Angew. Chem. Int. Ed. 2004;43:2417–2420. doi: 10.1002/anie.200353497. [DOI] [PubMed] [Google Scholar]; (b) Nam W. Acc. Chem. Res. 2007;40:522–531. doi: 10.1021/ar700027f. [DOI] [PubMed] [Google Scholar]; (c) Que L., Jr. Acc. Chem. Res. 2007;40:493–500. doi: 10.1021/ar700024g. [DOI] [PubMed] [Google Scholar]
- (11).The importance of H+ in controlling the formation of FeIV(O) vs S-oxygenates in the reaction of a nonheme iron(II)-thiolate complex with O-atom donors (e.g. mCPBA) has recently appeared. McDonald AR, Bukowski MR, Farquhar ER, Jackson TA, Koehntop KD, Seo MS, De Hont RF, Stubna A, Halfen JA, Münck E, Nam W, Que L., Jr. J. Am. Chem. Soc. 2010;132:17118–17129. doi: 10.1021/ja1045428.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.