

Abstract
Objective
The mechanisms that regulate the physical interaction of pericytes and endothelial cells and the effects of these interactions on interendothelial cell junctions are not well understood. We determined the extent to which vascular pericytes could regulate pericyte-endothelial adhesion and the consequences that this disruption might have on the function of the endothelial barrier.
Methods and Results
Human retinal microvascular endothelial cells were co-cultured with pericytes, and the effect on the monolayer resistance of endothelial cells and expression of the cell junction molecules N-cadherin and VE-cadherin were measured. The molecules responsible for the effect of pericytes or pericyte conditioned media on the endothelial resistance and cell junction molecules were further analyzed. Our results indicate that pericytes increase the barrier properties of endothelial cell monolayers. This barrier function is maintained through the secretion of pericyte-derived sphingosine 1-phosphate (S1P). S1P aids in maintenance of microvascular stability by up-regulating the expression of N-cadherin and VE-cadherin, and down-regulating the expression of angiopoietin 2.
Conclusion
Under normal circumstances, the retinal vascular pericytes maintain pericyte-endothelial contacts and vascular barrier function through the secretion of S1P. Alteration of pericyte-derived S1P production may be an important mechanism in the development of diseases characterized by vascular dysfunction and increased permeability.
Keywords: Pericytes, endothelial cells, S1P, adherens junctions, permeability
The unique structural organization of the retinal microvasculature, referred to as the blood-retinal barrier (BRB), is primarily responsible for the tight regulation of vascular permeability within this tissue. The foundation of the BRB is the monolayer of endothelial cells and the complex array of interacting proteins that form the inter-endothelial cell junctions. The principal proteins found in endothelial tight junctions are occludin, and claudin-5, that create a tight seal so that water-soluble molecules cannot easily penetrate between the cells (1-2). Also contributing to the function of the BRB at the level of the endothelial junctions is the adhesive protein VE-cadherin that has been shown to interact closely with and regulate the function of the endothelial tight junctions (3).
The microvascular endothelial cells of the retina are further supported by interactions with other cell types including vascular pericytes and glial cells. It is well known that perictye-endothelial interactions are necessary for the development and maintenance of a functional microcirculation in many different tissues (4-6). Pericytes are recruited to newly formed capillaries by secretion of PDGF by the endothelium (7-9). The arrival of pericytes coincides with the production of basement membrane components by the endothelium (10), and pericyte-endothelial cell contact results in the activation of TGF-β that inhibits endothelial cell proliferation (11). Endothelial cell survival and vascular stabilization is further promoted by the production of angiopoietin-1 by perivascular cells that competes with endothelial cell produced angiopoietin-2 for binding to the Tie-2 receptor (12).
The physical interaction of pericytes with endothelial cells has been reported to be mediated in part by N-cadherin. Stimulation of the sphingosine 1-phosphate receptor (S1Pr1) on endothelial cells induces N-cadherin trafficking to the cell surface and promotes cell-cell interactions (13-14). A recent study also reports that Ephrin-B2/EphB interactions may also be important in pericyte-endothelial cell adhesion (15).
Disruption of pericyte recruitment and subsequent cell-cell interactions in genetically altered mouse models leads to an alteration of normal vascular structure and function during development (5). In the adult, selective pericyte loss is the earliest histological change in diabetic retinopathy leading to capillary dysfunction, the development of retinal edema and associated visual loss (16-19). One important consequence of pericyte loss in diabetes may be enhanced endothelial cell proliferation and angiogenesis. The alteration of pericyte/endothelial adhesion may be mediated by elevated levels of Angiopoietin-2 (Ang-2) that have been reported in the diabetic retina and vitreous (20-23). In diabetic experimental animals, pericyte loss appears to be preceded by alterations of vascular permeability suggesting that more subtle changes in endothelial-pericyte interactions might occur during the earliest stages of the disease (24-26). These changes may include the novel mechanism of pericyte detachment and migration that may be a precursor to the pericyte loss seen in the diabetic retina (27).
The mechanisms that regulate the interaction of pericytes and endothelial cells in the retinal microvasculature, and the factors involved in the local regulation of microvascular permeability in the retina have not been well characterized. Therefore the purpose of this study was to determine the extent to which vascular pericytes could regulate pericyte-endothelial adhesion and the consequences that this disruption might have on the function of the retinal endothelial barrier.
Methods
Cell Culture
Human retinal microvascular endothelial cells (HRECs) (ACBRI-181) and human retinal microvascular pericytes (ACBRI-183) were obtained from Cell Systems (Kirkland, WA). The Muller glial cell line MIO-M1 was obtained from the Institute of Ophthalmology, University College, London. Pericytes were determined to be positive for the expression of smooth muscle actin, desmin and the PDGF receptor (Supplemental Figure I). HRECs were grown on fibronectin-coated dishes in MCDB-131 supplemented with 10 % FBS, 10ng/ml EGF, 1 μg/ml Hydrocortisone, 0.2mg/ml EndoGro and 0.09mg/ml Heparin (VEC Technologies, Rensselaer, NY) and used between passages 1 and 7.
Real time PCR
Total RNA was isolated from cells, converted to cDNA and analyzed using the appropriate TaqMan assays. The relative levels of mRNA were determined by the comparative Ct method with normalization to either GAPDH or 18s RNA.
Electrical Cell-substrate Impedance Sensing
Measurements of endothelial monolayer resistance were performed with the electrical cell-substrate impedance sensing (ECIS) system Zθ (Applied Biophysics) as described previously (20).
Western blotting
Cell extracts were prepared by lysing cells in 25mM Tris, 150mM NaCl, 10mM MgCl2, 2mM EDTA, 1% Triton-X 100 and 0.05mM orthovanadate. Lysates were centrifuged at 14,000g for 10 minutes to collect the insoluble (membrane) fraction. The insoluble fractions were examined by Western blotting with antibodies to N-cadherin and VE-cadherin (Abcam, Cambridge, MA). Membranes were re-probed with antibody to β-tubulin (Abcam, Cambridge, MA) as a loading control. Blots were analyzed using an Odyssey Infrared Imaging System (LI-COR, Nebraska USA) to determine band density.
Immunofluorescence Microscopy
Isolated human retinal endothelial cells or co-cultures of endothelial cells and pericytes were fixed in 3:1 methanol/acetic acid and washed extensively with PBS. The cells were stained with anti-N-cadherin antibody and anti-α smooth muscle actin for co-cultures and examined by confocal microscopy using a Leica TCS SP5 microscope.
Statistical methods
Unless otherwise indicated, the difference among means for all quantitative analyses were tested by one-way ANOVA and corrected using the Bonferroni’s multiple comparison post test. Differences among the means of individual groups were compared using the Bonferroni’s multiple comparison post test. Values for p< 0.05 were considered significant.
See Supplemental materials available at http://atvb.Ahajournals.org for an expanded Methods section.
Results
Pericytes Upregulate N-cadherin and Increase the Barrier Properties of Endothelial Cell Monolayers
Several studies have reported on the role of pericytes as mediators of microvascular maturation and stabilization. This stabilizing effect of pericytes may require the physical interaction of pericytes with endothelial cells that has been postulated to be mediated by N-cadherin. The addition of a low density of pericytes (50-100 cells/coverslip) to a pre-established monolayer of endothelial cells resulted in the increased expression of N-cadherin as seen by immunofluorescence staining (Figures 1A and B). The upregulation of N-cadherin in endothelial cells can be seen in regions of the endothelial monolayer near where the pericytes have attached.
Figure 1.
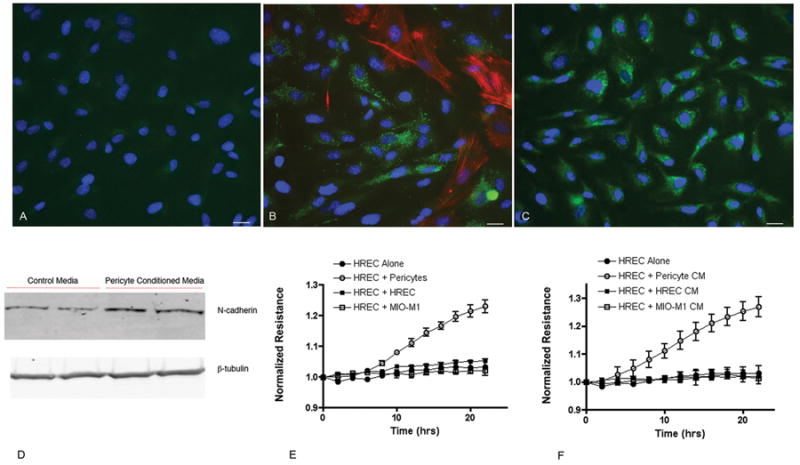
Confocal images of human retinal endothelial cells (HREC) alone (A), co-cultures of HREC and pericytes (B) and HREC after the addition of pericyte conditioned media (C). Cells are stained for smooth muscle actin (red), N-cadherin (green) and DNA (blue). The increase in N-cadherin expression seen by immunostaining is confirmed by Western blot analysis of HREC treated with pericyte conditioned media (D). The addition of pericytes (E) or pericyte conditioned media (F) to confluent monolayers of HREC increases the monolayer resistance as measured by ECIS. The addition of HREC or the Muller glial cell line MIO-M1, or their conditioned media, did not alter the monolayer resistance. Values represent the mean ± SEM from triplicate wells. For (E) values at each time point starting at 8 hours are significantly different and for (F) values at each time point starting at 6 hours are significantly different.
To determine whether pericyte-endothelial contact was required for the observed effects on endothelial cells, conditioned media from cultures of pericytes was added to endothelial cell monolayers. Increased N-cadherin expression was observed in endothelial cells by both immunostaining and Western blotting after the addition of pericyte conditioned media (Figure 1C and 1D).
Pericytes were added to monolayers of endothelial cells at approximately a 1:1 ratio and the monolayer resistance was examined using ECIS. The addition of pericytes caused a significant increase in the resistance of the pre-established endothelial cell monolayer beginning approximately 6 hours after addition (Figure 1E). This effect was specific to pericytes, as the addition of other cell types (endothelial cells or the Müller glial cell line MIO-M1) had no effect on monolayer resistance. In the presence of pericytes, the endothelial cells reached a significantly higher maximum resistance over the 24-hour period compared to endothelial cells alone (Supplemental Figure IIA). Changes in monolayer resistance could also be elicited independent of cell-cell contact by the addition of pericyte conditioned media suggesting that a secreted factor may be involved. This was again specific to the conditioned media from pericytes and resulted in a greater than 20% increase in monolayer resistance (Figure 1F and Supplemental Figure IIB).
To assess whether the expression of N-cadherin by endothelial cells is required for pericyte adhesion, a chimeric protein designed to block N-cadherin interactions (N-cadherin Fc) was added at the time of pericyte addition. In the absence of N-cadherin Fc, the pericytes attached and spread on the endothelial cell monolayer within 5 hours of plating (Figure 2A). When N-cadherin was blocked with the fusion protein the pericytes were unable to attach and spread normally as evidence by an altered morphology characterized by cells with long thin processes and a significant decrease in average cell area (Figures 2B and 2D). The effect of the blocker appeared to be transient as perictyes in treated cultures demonstrated a normal morphology after approximately 12 hours of culture (Figure 2C). The ability of pericytes to functionally increase the resistance of the endothelial cell monolayer was correlated with the adhesion and morphology of the cells (Figure 2E). Monolayer resistance remained at control levels for up to 8 hours after plating when cultures were treated with the N-cadherin Fc and pericytes exhibited altered morphology. Resistance gradually increased by 10 hours and continued to increase with time as the pericytes assumed a more normal flattened morphology.
Figure 2.
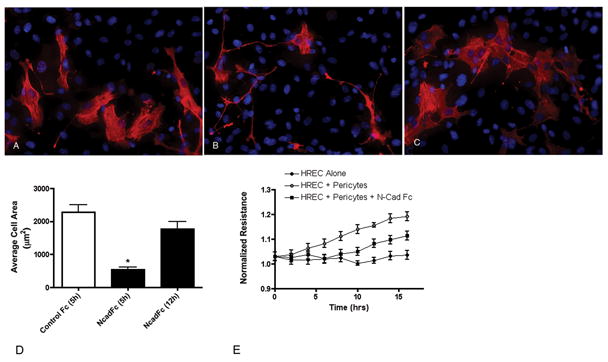
N-cad Fc disrupts pericyte-endothelial interactions. A suspension of retinal pericytes was added to a pre-established monolayer of retinal endothelial cells in the presence of control IgG Fc (0.5μg/ml) (A) or N-cadherin Fc (0.5μg/ml) (B) and incubated for 5 hours. The presence of N-cadherin Fc significantly inhibited the spreading of pericytes and the extent of contact between pericytes and endothelial cells. Cultures with N-cadherin Fc 12 hours after plating demonstrated a normal flattened morphology (C). The differences in pericyte morphology in the different conditions was confirmed by quantitation of cell area (D). Values represent the mean ± SEM from triplicate coverslips. * Significantly less than cells in the presence of control IgG Fc at 5 hours or N-cadherin Fc after 12 hours of incubation. (P<0.001). The altered morphology of pericytes in the presence of N-cadherin Fc was correlated with changes in the ability of the pericytes to increase the resistance of the endothelial monolayer (E). Values at each time point for HREC + Pericytes are significantly greater than HREC alone or HREC + Pericytes + N-cad Fc beginning at 6 hours. Values for HREC + Pericytes + N-cad Fc are significantly different from HREC alone beginning at 10 hours.
Pericyte-Derived Sphingosine 1-Phosphate Induces Adhesion Molecule Expression and Increases in Endothelial Barrier Properties
The majority of previous work has focused on the role of PDGF, TGF-β and the angiopoietins in recruiting pericytes, inhibiting endothelial cell proliferation and migration and stabilizing pericyte-endothelial interactions (28-31). Pericyte conditioned media was added to endothelial monolayers in an ECIS assay in the presence of anti-TGF-β or the angiopoietin inhibitor Tie-2 Fc. Neither of these treatments inhibited the effect of the pericyte conditioned media on monolayer resistance (Figures 3A and 3C).
Figure 3.
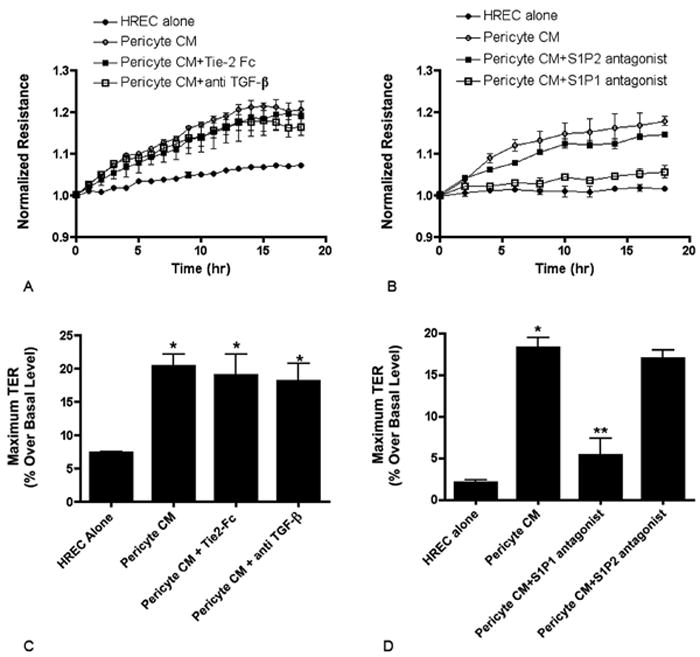
Stimulation of the S1P1 receptor mediates the effects of pericyte conditioned media. (A) ECIS normalized resistance in HREC alone, HREC plus pericyte conditioned media, HREC plus pericyte conditioned media in the presence of Tie-2 Fc chimera (200ng/ml) or HREC with pericyte conditioned media in the presence of anti TGF-β antibody (50ng/ml). The change in resistance was monitored for 18 hours after the addition of perictye conditioned media and the resistance values normalized to time 0. Values represent the mean ± SEM from triplicate wells. Values for pericyte CM, Tie-2 Fc and anti TGF-β are significantly greater than HREC alone at all time points beginning at 4 hours. (B) HREC monolayers incubated with pericyte CM alone or with the S1P1 antagonist VPC23019 (0.3μM), or the S1P2 antagonist JTE-013 (1μM). Values for pericyte CM, and S1P2 antagonist are significantly greater than HREC alone and S1P1 antagonist at all time points beginning at 4 hours. (C and D) Quantitation of percent change in resistance compared to basal level at time 0. * Significantly greater than HREC alone (P<0.001). ** Significantly less than pericyte CM and pericyte CM+S1P2 antogonist (P<0.05).
We subsequently tested the hypothesis that the bioactive lipid sphingosine 1-phosphate (S1P), that has previously been shown to mediate changes in endothelial barrier properties (32-34), may be mediating the pericyte effect. The conditioned media of pericytes depleted of lipids failed to produce an increase in monolayer resistance in an ECIS assay (Supplemental Figure III). In additional experiments, endothelial monolayers were treated with pericyte conditioned media in the presence or absence of S1P receptor antagonists. The addition of an antagonist of the S1P1 receptor (VPC23019) resulted in inhibition of the pericyte conditioned media effect with respect to monolayer resistance (Figures 3B). Monolayers exposed to pericyte conditioned media without any treatment or in the presence of an antagonist to the S1P2 receptor (JTE-013) showed normal increases in resistance. The maximum achieved resistance was quantified and found to be significantly reduced by inhibition of the S1P1 receptor (Figure 3D). In addition to inhibiting the increase in resistance, the S1P1 antagonist also blocked the pericyte conditioned media upregulation of N-cadherin expression as determined by immunostaining (Supplemental Figure IV).
A role for S1P and S1P1 in the regulation of the endothelial barrier by pericytes was further examined by the selective knockdown of sphingosine kinase-1 and S1P1 in pericytes and endothelial cells respectively. Pericytes treated with translation-blocking morpholinos demonstrated a significant reduction in the level of Sphk1 protein (Figure 4A). The conditioned media from these cells had reduced levels of S1P as determined by ELISA (Supplemental Figure V) and failed to induce the increase in endothelial monolayer resistance compared to untreated pericytes or pericytes treated with a control morpholino (Figures 4B and 4C).
Figure 4.
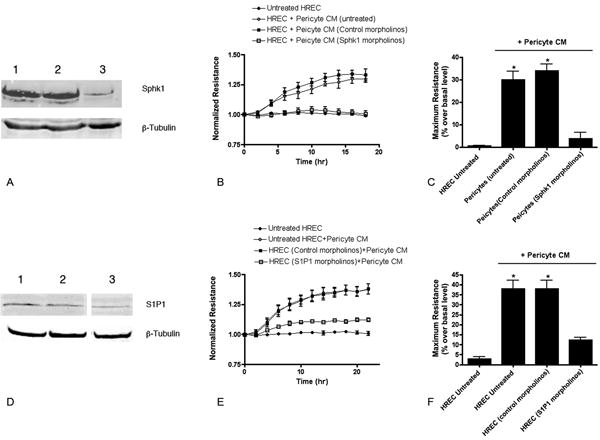
Knockdown of Sphk1 in pericytes or S1P1 in endothelial cells reduces the ability of pericytes to increase the endothelial monolayer resistance. (A) Representative Western blot for Sphk1 in untreated perictyes (lane 1), perictyes treated with control morpholinos (lane 2) or Sphk1 morpholinos (lane 3) (consistent among N=3 separate cell cultures for each treatment). (B) Conditioned media from each set of cells tested in an ECIS assay. (C) Quantitation of percent change in resistance compared to basal level at time 0. (D) Representative Western blot for S1P1 in HREC (lane 1), HREC treated with control morpholinos (lane 2) or S1P1 morpholinos (lane 3) (consistent among N=3 separate cultures for each treatment). (E) Conditioned media from untreated pericytes added to HREC with each of the treatments in an ECIS assay. (F) Quantitation of percent change in resistance compared to basal level at time 0. * Significantly greater than HREC untreated or morpholino-treated cells (P<0.0001).
Similarly we were able to confirm the role of S1P1 in mediating the pericyte effects. Endothelial cells were treated with translation-blocking morpholinos and demonstrated a reduction in the levels of S1P1 protein (Figure 4D). When these cells were treated with normal pericyte conditioned media, the cells demonstrated a significantly reduced response in terms of monolayer resistance compared to untreated endothelial cells or cells treated with the control morpholinos (Figures 4E and 4F).
The secretion of S1P by pericytes was further supported by the presence of the enzyme responsible for the production of S1P, sphingosine kinase-1 (Sphk1), in cultured cells. Sphingosine kinase-1 was detected in extracts of pericyte cells and was absent from the conditioned media (Supplemental Figure VI). The S1P in the pericyte conditioned media was measured by ELISA and found to be present at a concentration of approximately 181+/- 31 nM in 1×106 cells (Supplemental Figure V).
S1P Effects the Resistance of the Endothelial Cell Barrier Through the Regulation of Adhesion Proteins and Angiopoietin-2 Expression
Isolated endothelial cells were subsequently treated with purified S1P and found to significantly increase the expression of the adhesion molecules N-cadherin and VE-cadherin. For both of these adhesion molecules, increases were seen in mRNA levels as well as the level of protein associated with the cell membrane (Figure 5). The role of these proteins in mediating the endothelial cell barrier properties was investigated by ECIS. Endothelial cells plated onto ECIS electrodes in the presence of N-cadherin Fc or cells treated with N-cadherin morpholinos developed a level of monolayer resistance equivalent to that of cells without treatment (Figure 6). On the contrary, the monolayers in the presence of VE-cadherin Fc did not achieve the maximum level of resistance seen in untreated or N-cadherin Fc-treated cells and demonstrated a significant destabilization of the barrier over time. This result suggests a primary role for VE-cadherin in production of the endothelial barrier resistance and suggests that the function of N-cadherin may be restricted to facilitating endothelial-pericyte interactions.
Figure 5.
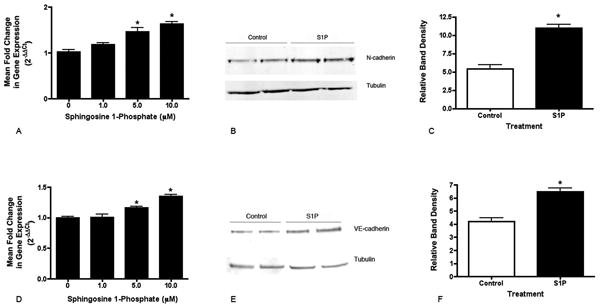
Purified S1P increases the expression of adhesion proteins in human retinal endothelial cells. Confluent HREC monolayers were incubated for 12 hours in the presence of S1P and collected for PCR and Western blotting. Both N-cadherin mRNA (A) and VE-cadherin mRNA (D) were signficantly upregulated in S1P-treated cells in a dose-dependent manner. Representative Western blots for N-cadherin (B) and VE-cadherin (E) from HREC incubated with 10μM S1P demonstrate increased protein associated with the insoluble membrane fraction of the cells. Quantitation of band density normalized to tubulin levels indicates a significant increase in both N-cadherin (C) and VE-cadherin (F) proteins. *Significantly greater than untreated and 1mM (P<0.01).
Figure 6.
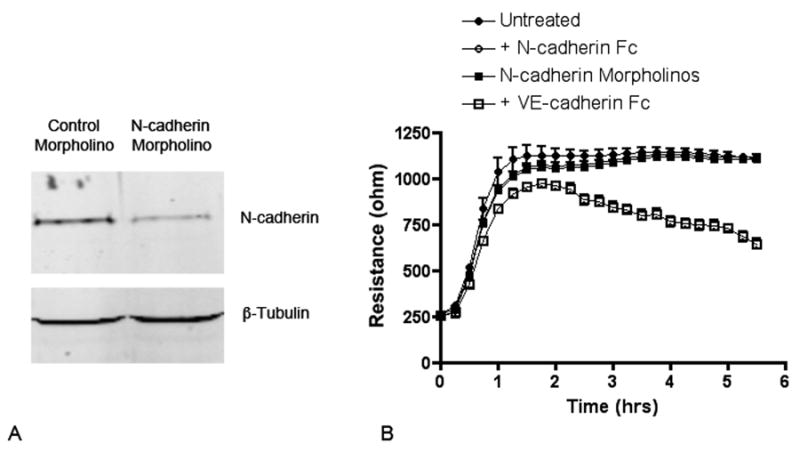
The adhesion proteins N-cadherin and VE-cadherin have different functions in establishment and maintenance of the endothelial cell barrier. (A) Western blot for N-cadherin in HREC treated with N-cadherin morpholinos or control morpholinos. (B) Morpholino-treated HREC or untreated HREC were plated onto ECIS electrodes in the absence or presence of N-cadherin Fc (0.5μg/ml) or VE-cadherin Fc (0.5μg/ml) and the developing resistance was monitored. The decreased expression of N-cadherin or the addition of N-cadherin Fc had no effect on the resistance that developed over the subsequent 6 hours compared to control cells. Cells incubated with VE-cadherin Fc demonstrated significantly less resistance at all time points beginning at approximately 20 minutes after plating and continued to decrease for more than 5 hours.
The integrity of the endothelial barrier can also be effected by various growth factors. Previous studies have demonstrated that angiopoietin-2 is a potent destabilizer of existing microvessels by antagonizing the effects of angiopoietin-1 (35-37). We have recently reported that Ang-2 effectively decreases the endothelial barrier properties in an ECIS assay (20). Low levels of angiopoietin-2 expression would therefore be expected to promote a quiescent microvasculature and the maintenance of normal barrier properties. Endothelial cells treated with S1P demonstrated a significant decrease in the level of angiopoietin-2 mRNA as compared to untreated cells (Supplemental Figure VII) further suggesting an important role for the pericyte produced S1P in the regulation of microvascular permeability.
Discussion
Pericytes play an important role in the formation, maturation and maintenance of the microvasculature. In adult tissues, pericytes are contractile cells that are postulated to regulate regional flow through the microcirculation (38, 39). The vascular beds found in the nervous system, in which vascular permeability is limited, have a ratio of pericytes to endothelial cells that is much higher than that found in tissues with normally higher levels of permeability (40-42). In addition to the number of pericytes present, the degree to which the pericytes physically interact with the endothelial cells also varies in different tissues and may impact the function of the microcirculation in that area (43). For example, the degree of pericyte coverage of the retinal capillaries is greater than that seen in other tissues (the reported endothelial to pericyte ratio is 1:1), and thus probably provides greater integrity to the retinal microcirculation (44). The physical interaction of pericytes with endothelia establishes junctional contact between the cells that includes the adhesion protein N-cadherin (45,13).
In the present study we have demonstrated that the interaction of pericytes with endothelial cells in co-culture upregulates the expression of N-cadherin in endothelial cells as determined by immunostaining. The expression of functional N-cadherin is required for the normal attachment and spreading of pericytes on the endothelial cell monolayer. A similar observation was made by Paik et al. (14) using immortalized embryonic endothelial cells treated with siRNA to knock down N-cadherin and a smooth muscle cell line. In the present study, disruption of N-cadherin using the N-cadherin Fc chimera resulted in dramatic shape changes in the retinal pericytes that were eventually normalized over time possibly due to the turnover and synthesis of new N-cadherin or loss of activity of the N-cadherin Fc. There is little knowledge about the role of N-cadherin in vivo in the retinal vasculature, except for the report by Gerhardt and collegues that describes the punctate presence of N-cadherin associated with the ablumenal endothelial membrane in the brain and retina of the chicken (13). Preliminary studies from our laboratory have detected N-cadherin by Western blotting in isolated vessels from the mouse retina (unpublished data) suggesting that it may also play an important role in endothelial-pericyte interactions in vivo.
The addition of pericytes also significantly increased the barrier properties of the endothelial cell monolayer over time. The first significant change in monolayer resistance was detected beginning approximately 5 hours after the addition of pericytes. This coincided with the timing of pericyte attachment and spreading on the endothelial monolayer. The ability of pericytes to increase the barrier was associated with functional N-cadherin interactions and the shape of the pericytes. The addition of N-cadherin Fc dramatically altered the shape of the pericytes and their ability to form extensive contact with the endothelial cells. These pericytes also lost the ability to modulate the barrier. This effect appeared to be only transient and was associated with the altered pericyte cell shape as an increase in the barrier resistance was realized over time as the pericytes attained a more normal flattened morphology typical of untreated cells.
The ability of pericytes to modulate N-cadherin expression and the barrier properties of the endothelial cells was not dependent upon cell contact as the conditioned media from pericytes alone had the same effect, suggesting the presence of a pericyte secreted factor. Subsequent experiments determined that the factor produced by the pericytes was sphingosine 1-phosphate (S1P) and that it was exerting its effects on the endothelial cells through activation of the S1P1 receptor. A recent study by Zachariah and Cyster (46) is the only study to date to demonstrate a pericyte-derived source of S1P. In this study the S1P produced by the neural crest-derived pericytes of the thymus may be acting as a chemoattractant for the SIP1 expressing thymic cells leading to their perivascular accumulation and subsequent egress. The present study is the first to demonstrate the secretion of S1P by retinal–derived pericytes supporting this unique function for pericytes and suggesting that it may be generalizable to multiple microvascular beds. We measured S1P production directly by ELISA and found it to be in the range of 180nM in cultures of approximately 1×106 cells. This together with the detection of high levels of sphingosine kinase-1, strongly suggest that retinal vascular pericytes produce S1P. Current studies are underway to correlate these findings with the in vivo situation by determining the localization of S1P in the retinal vasculature and by selectively knocking down the expression of S1P1 and Sphk1 in the retina using Cre-lox technologies.
Previous studies have demonstrated that sphingosine 1-phosphate is also present at high levels in the human plasma (300-500nM) and has been reported to be produced by platelets (47), red blood cells (48) and endothelial cells (49). Although there are high levels of S1P in the plasma, studies report that the majority of the plasma S1P may be buffered by binding to lipoproteins decreasing its ability to stimulate cell surface receptors (50-52) suggesting that the localized production of S1P by pericytes may be an important source for regulation of endothelial cell function. Such a role for pericyte-derived S1P would require the expression of S1P1 receptors on the extracellular or basal surface of the endothelial cells. A recent study has in fact confirmed that brain endothelial cells express high levels of S1P1 on the basolateral surface of the cell in culture (53).
An interesting finding in our studies was the correlation of pericyte morphology with the ability of these cells to enhance the endothelial barrier. Sphingosine 1-phosphate production may be reduced in the N-cadherin Fc-treated cells and additional studies are ongoing to determine if the expression and/or activity of sphingosine kinase in pericytes is regulated in part by cell shape. Numerous reports are present in the literature that demonstrate a correlation between gene expression and cell shape (54-56). If this is the case, one might expect that conditions which alter the stable interaction of pericytes with endothelial cells in vivo, including the turnover or downregulation of N-cadherin expression, might lead to alterations in pericyte morphology, S1P production and changes in microvascular permeability.
Previous studies have reported on the role of S1P as a regulator of endothelial barrier function. These studies demonstrated the reorganization of the actin cytoskeleton and the formation of focal adhesion-adherens junction complexes associated with changes in monolayer resistance within 5 to 60 minutes after the addition of purified S1P (57-59). This rapid cellular response, mediated by the S1P1 receptor involves several possible downstream signaling molecules including Rac, ERK and PI3 kinase (60-63). In the present study we were unable to detect changes in resistance before approximately 5 hours after the addition of pericytes or pericyte conditioned media. These differences may be due to differences in the effective S1P concentration used for stimulation. In addition we observed increased expression (mRNA and protein) of both N-cadherin and VE-cadherin in endothelial cells 6 hours after the addition of pericyte conditioned media or purified S1P suggesting that the sustained increase in monolayer resistance seen in this study may be due to the increased expression and maintenance of cell junction proteins. The contribution of these proteins to the formation of the endothelial barrier resistance was assessed using Fc fusion proteins or morpholinos. Results from these experiements suggest that the initial formation and long-term maintenance of increased endothelial resistance is only dependent upon the activity of VE-cadherin and that N-cadherin expression may function solely for the purpose of maintaining normal endothelial cell-pericyte interactions.
Pericyte-endothelial interactions also appear to be effected by the levels and activity of angiopoietin-1 (Ang1) and angiopoietin-2 (Ang2). The constitutive secretion of Ang-1 in normal quiescent vessels is thought to stabilize the vessels in part by maintaining contacts between endothelial cells and between endothelial cells and pericytes (64). Ang2 is thought to antagonize the action of Ang1 leading to vessel destabilization (12). Ang2 expression is increased in the retinas of diabetic animals, decreases the barrier properties of the endothelial monolayer and is postulated to lead to the loss of vascular pericytes and subsequent retinal vascular dysfunction (20,65,66). The effect of Ang-2 in leading to a reduction of pericyte coverage of retinal capillaries may not be solely due to the induction of pericyte apoptosis. Work by Pfister et al., (27) has reported that Ang-2 can induce more subtle changes in pericyte attachment and migration on specific regions of the capillary network that may lead to alterations in endothelial cell behavior. In the present study we have observed that S1P can downregulate the expression of Ang2 by endothelial cells. This finding suggests that in normal circumstances, the retinal vascular pericytes may play an important role in the maintainence of pericyte-endothelial contacts and therefore vascular barrier function through the secretion of S1P. Further studies are in progress to determine if one consequence of diabetes is the alteration of pericyte S1P production that might contribute to alteration of the endothelial cell-cell junctions and breakdown of the blood-retinal barrier. Results from these experiments provide a possible mechanism involved in the pathogenesis of diabetic retinopathy and may provide a novel target for the development of new therapies for this disease.
Supplementary Material
Acknowledgments
None
Sources of Funding: Funding for this study was provided by the National Institutes of Health Grant, EY12604.
Footnotes
Disclosures: None
References
- 1.Schneeberger EE, Lynch RD. The tight junctional complex. Am J Physiol Cell Physiol. 2004;286:C1213–1228. doi: 10.1152/ajpcell.00558.2003. [DOI] [PubMed] [Google Scholar]
- 2.Dejana E, Tournier-Lasserve E, Weinstein BM. The control of vascular integrity by endothelial cell junctions: Molecular basis and pathological implications. Developmental Cell. 2009;16:209–221. doi: 10.1016/j.devcel.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 3.Dejana E. Endothelial cell-cell junctions: Happy together. Natl Rev Mol Cell Biol. 2004;5:261–270. doi: 10.1038/nrm1357. [DOI] [PubMed] [Google Scholar]
- 4.Hirschi KK, D’Amore PA. Pericytes in the Microvasculature. Cardiovascular Res. 1996;32:687–698. [PubMed] [Google Scholar]
- 5.Lindahl P, Johansson BR, Leveen P, Betsholtz C. Pericyte loss and Microaneurysm formation in PDGF-B-Deficient Mice. Science. 1997;277:242–245. doi: 10.1126/science.277.5323.242. [DOI] [PubMed] [Google Scholar]
- 6.Hellstrom M, Gerhardt H, Kalen M, Li X, Eriksson U, Wolburg H, Betsholtz C. Lack of Pericytes Leads to Endothelial Hyperplasia and Abnormal Vascular Morphogenesis. J Cell Biol. 2001;153:543–554. doi: 10.1083/jcb.153.3.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stratman AN, Schwindt AE, Malotte KM, Davis GE. Endothelial-derived PDGF-BB and HB-EGF coordinately regulate pericyte recruitment during vasculogenic tube assembly and stabilization. Blood. 2010;116:4720–4730. doi: 10.1182/blood-2010-05-286872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lindblom P, Gerhardt H, Liebner S, Abramsson A, Enge M, Hellstrom M, Backstrom G, Fredriksson S, Landegren U, Nystrom HC, Bergstrom G, Dejana E, Ostman A, Lindahl P, Betsholtz C. Endothelial PDGF-B retention is required for proper investment of pericytes in the microvessel wall. Genes Dev. 2003;17:1835–1840. doi: 10.1101/gad.266803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bjarnegard M, Enge M, Norlin J, Norlin J, Gustafsdottir S, Fredriksson S, Abramsson A, Takemoto M, Gustafsson E, Fässler R, Betsholtz C. Endothelium-specific ablation of PDGFB leads to pericyte loss and glomerular, cardiac and placental abnormalities. Development. 2004;131:1847–1857. doi: 10.1242/dev.01080. [DOI] [PubMed] [Google Scholar]
- 10.Stratman AN, Malotte KM, Mahan RD, Davis MJ, Davis GE. Pericyte recruitment during vasculogenic tube assembly stimulates endothelial basement membrane matrix formation. Blood. 2009;114:5091–5101. doi: 10.1182/blood-2009-05-222364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Antonelli-Orlidge A, Saunders KB, Smith SR, D’Amore PA. An activated form of transforming growth factor beta is produced by cocultures of endothelial cells and pericytes. Proc Natl Acad Sci U S A. 1989;86:4544–4548. doi: 10.1073/pnas.86.12.4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maisonpierre PC, Suri C, Jones PF, Bartunkova S, Wiegand SJ, Radziejewski C, Compton D, McClain J, Aldrich TH, Papadopoulos N, Daly TJ, Davis S, Sato TN, Yancopoulos GD. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science. 1997;277:55–60. doi: 10.1126/science.277.5322.55. [DOI] [PubMed] [Google Scholar]
- 13.Gerhardt H, Wolburg H, Redies C. N-cadherin mediates pericyte-endothelial interaction during brain angiogenesis in the chicken. Develop Dynamics. 2000;218:472–479. doi: 10.1002/1097-0177(200007)218:3<472::AID-DVDY1008>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 14.Paik J-H, Skoura A, Chae S-S, Cowan AE, Han DK, Proia RL, Hla T. Sphinogsine 1-phosphate receptor regulation of N-cadherin mediates vascular stabilization. Genes and Development. 2004;18:2392–2403. doi: 10.1101/gad.1227804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Foo SS, Turner CJ, Adams S, Compagni A, Aubyn D, Kogata N, Lindblom P, Shani M, Zicha D, Adams RH. Ephrin-B2 controls cell motility and adhesion during blood-vessel-wall assembly. Cell. 2006;124:161–173. doi: 10.1016/j.cell.2005.10.034. [DOI] [PubMed] [Google Scholar]
- 16.Ejaz S, Chekarova I, Ejaz A, Sohail A, Lim CW. Importance of pericytes and mechanisms of pericyte loss during diabetic retinopathy. Diabetes, Obesity and Metabolism. 2008;10:53–63. doi: 10.1111/j.1463-1326.2007.00795.x. [DOI] [PubMed] [Google Scholar]
- 17.Cunha-Vaz JG, Faria de Abreu JR, Campos AJ, Figo GM. Early breakdown of the blood-retinal barrier in diabetes. Br J Ophthalmol. 1975;59:649–656. doi: 10.1136/bjo.59.11.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bhagat N, Grigorian RA, Tutela A, Zarbin MA. Diabetic macular edema: pathogenesis and treatment. Surv Ophthalmol. 2009;54:1–32. doi: 10.1016/j.survophthal.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 19.Cogan DG, Toussaint D, Kuwabara T. Retinal vascular patterns. IV. Diabetic retinopathy. Archives of Ophthalmology. 1961;66:366–378. doi: 10.1001/archopht.1961.00960010368014. [DOI] [PubMed] [Google Scholar]
- 20.Rangasamy S, Srinivasan R, Maestas J, McGuire PG, Das A. A Potential Role for Angiopoietin 2 in the Regulation of the Blood-Retinal Barrier in Diabetic Retinopathy. Invest Ophthalmol Vis Sci. 2011;52:3784–3791. doi: 10.1167/iovs.10-6386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Watanabe D, Suzuma K, Suzuma I, Ohashi H, Ojima T, Kurimoto M, Murakami T, Kimura T, Takagi H. Vitreous levels of angiopoietin 2 and vascular endothelial growth factor in patients with proliferative diabetic retinopathy. Am J Ophthalmol. 2005;139:476–481. doi: 10.1016/j.ajo.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 22.Hammes H-P, Lin J, Wagner P, Feng Y, vom Hagen F, Krzizok T, Renner O, Breier G, Brownlee M, Deutsch U. Angiopoietin-2 causes Pericyte dropout in the normal retina. Evidence for involvement in Diabetic retinopathy. Diabetes. 2004;53:1104–1110. doi: 10.2337/diabetes.53.4.1104. [DOI] [PubMed] [Google Scholar]
- 23.Ohashi H, Takagi H, Koyama S, Oh H, Watanabe D, Antonetti DA, Matsubara T, Nagai K, Arai H, Kita T, Honda Y. Alteration in expression of angiopoietins and the Tie-2 receptor in the retina of streptozotocin induced diabetic rats. Mol Vis. 2004;10:608–617. [PubMed] [Google Scholar]
- 24.Stitt AW, Li YM, Gardiner TA, Bucala R, Archer DB, Vlassara H. Advanced Glycation end products (AGEs) co-localize with AGE receptors in the retinal vasculature of diabetic and of AGE-infused rats. Am J Pathol. 1997;150:523–531. [PMC free article] [PubMed] [Google Scholar]
- 25.Qaum T, Xu Q, Joussen AM, Clemens MW, Qin W, Miyamoto K, Hassessian H, Wiegand SJ, Rudge J, Yancopoulos GD, Adamis AP. VEGF-initiated blood-retinal barrier breakdown in early diabetes. Invest Ophthalmol Vis Sci. 2001;42:2408–2413. [PubMed] [Google Scholar]
- 26.Zhang SX, Ma J, Sima J, Chen Y, Hu MS, Ottlecz A, Lambrou GN. Genetic difference in susceptibility to the blood-retina barrier breakdown in diabetes and oxygen-induced retinopathy. Am J Pathol. 2005;166:313–321. doi: 10.1016/S0002-9440(10)62255-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pfister F, Feng Y, vom Hagen F, Hoffmann S, Molema G, Hillebrands JL, Shani M, Deutsch U, Hammes HP. Pericyte migration: a novel mechanism of pericyte loss in experimental diabetic retinopathy. Diabetes. 2008;57:2495–2502. doi: 10.2337/db08-0325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hirschi KK, Rohovsky SA, D’Amore PA. PDGF, TGF-beta and heterotypic cell-cell interactions mediate endothelial cell induced recruitment of 10T1/2 cells and their differentiation to a smooth muscle fate. J Cell Biol. 1998;141:805–814. doi: 10.1083/jcb.141.3.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hellstrom M, Kalen M, Lindahl P, Abramsson A, Betsholtz C. Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development. 1999;126:3047–3055. doi: 10.1242/dev.126.14.3047. [DOI] [PubMed] [Google Scholar]
- 30.Hawighorst T, Skobe M, Streit M, Hong YK, Velasco P, Brown LF, Riccardi L, Lange-Asschenfeldt B, Detmar M. Activation of the Tie-2 receptor by angiopoietin-1 enhances tumor vessel maturation and impairs squamous cell carcinoma growth. Am J Pathol. 2002;160:1381–1392. doi: 10.1016/S0002-9440(10)62565-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Uemura A, Ogawa M, Hirashima M, Fujiwara T, Koyama S, Takagi H, Honda Y, Wiegand SJ, Yancopoulos GD, Nishikawa S. Recombinant angiopoietin-1 restores higher-order architecture of growing blood vessels in mice in the absence of mural cells. J Clin Invest. 2002;110:1619–1628. doi: 10.1172/JCI15621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Camerer E, Regard JB, Cornelissen I, Srinivasan Y, Duong DN, Palmer D, Pham TH, Wong JS, Pappu R, Coughlin SR. Sphingosine-1-phosphate in the plasma compartment regulates basal and inflammation-induced vascular leak in mice. J Clin Invest. 2009;119:1871–1879. doi: 10.1172/JCI38575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Singleton PA, Dudek SM, Ma SF, Garcia JG. Transactivation of sphingosine1-phosphate receptors is essential for vascular barrier regulation. Novel role for hyaluronan and CD44 receptor family. J Biol Chem. 2006;281:34381–34393. doi: 10.1074/jbc.M603680200. [DOI] [PubMed] [Google Scholar]
- 34.Sanchez T, Skoura A, Wu MT, Casserly B, Harrington EO, Hla T. Induction of vascular permeability by the sphingosine-1-phosphate receptor-2 (S1P2R) and its downstream effectors ROCK and PTEN. Arterioscler Thromb Vasc Biol. 2007;27:1312–1318. doi: 10.1161/ATVBAHA.107.143735. [DOI] [PubMed] [Google Scholar]
- 35.Maisonpierre PC, Suri C, Jones PF, Bartunkova S, Wiegand SJ, Radziejewski C, Compton D, McClain J, Aldrich TH, Papadopoulos N, Daly TJ, Davis S, Sato TN, Yancopoulos GD. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science. 1997;277:55–60. doi: 10.1126/science.277.5322.55. [DOI] [PubMed] [Google Scholar]
- 36.Holash J, Wiegand SJ, Yancopoulos GD. New model of tumor angiogenesis: dynamic balance between vessel regression and growth mediated by angiopoietins and VEGF. Oncogene. 1999;18:5356–5362. doi: 10.1038/sj.onc.1203035. [DOI] [PubMed] [Google Scholar]
- 37.Lobov IB, Brooks PC, Lang RA. Angiopoietin-2 displays VEGF-dependent modulation of capillary structure and endothelial cell survival in vivo. Proc Natl Acad Sci USA. 2002;99:11205–11210. doi: 10.1073/pnas.172161899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Das A, Frank RN, Weber ML, Kennedy A, Reidy CA, Mancini MA. ATP causes retinal pericytes to contract in vitro. Exp Eye Res. 1988;46:349–362. doi: 10.1016/s0014-4835(88)80025-3. [DOI] [PubMed] [Google Scholar]
- 39.Kelley C, D’Amore P, Hechtman HB, Shepro D. Microvascular pericyte contractility in vitro: comparison with other cells of the vascular wall. J Cell Biol. 1987;104:483–490. doi: 10.1083/jcb.104.3.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Armulik A, Abramsson A, Betsholtz C. Endothelial/pericyte interactions. Circ Res. 2005;97:512–523. doi: 10.1161/01.RES.0000182903.16652.d7. [DOI] [PubMed] [Google Scholar]
- 41.Ozerdem U, Grako KA, Dahlin-Huppe K, Monosov E, Stallcup WB. NG2 proteoglycan is expressed exclusively by mural cells during vascular morphogenesis. Dev Dyn. 2001;222:218–227. doi: 10.1002/dvdy.1200. [DOI] [PubMed] [Google Scholar]
- 42.Sims DE. Diversity within pericytes. Clin Exp Pharmacol Physiol. 2000;27:842–846. doi: 10.1046/j.1440-1681.2000.03343.x. [DOI] [PubMed] [Google Scholar]
- 43.Tilton RG, Kilo C, Williamson JR. Pericyte-endothelial relationships in cardiac and skeletal muscle capillaries. Microvasc Res. 1979;18:325–335. doi: 10.1016/0026-2862(79)90041-4. [DOI] [PubMed] [Google Scholar]
- 44.Frank RN, Turczyn TJ, Das A. Pericyte coverage of retinal and cerebral capillaries. Invest Ophthalmol Vis Sci. 1990;31:999–1007. [PubMed] [Google Scholar]
- 45.Gerhardt H, Betsholtz C. Endothelial-pericyte interactions in angiogenesis. Cell Tissue Res. 2003;314:15–23. doi: 10.1007/s00441-003-0745-x. [DOI] [PubMed] [Google Scholar]
- 46.Zachariah MA, Cyster JG. Neural crest-derived pericytes promote egress of mature thymocytes at the corticomedullary junction. Science. 2010;328:1129–1135. doi: 10.1126/science.1188222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Igarashi Y, Yatomi Y. Sphingosine 1-phosphate is a blood constituent released from activated platelets, possibly playing a variety of physiological and pathophysiological roles. Acta Biochim Pol. 1998;45:299–309. [PubMed] [Google Scholar]
- 48.Pappu R, Schwab SR, Cornelissen I, Pereira JP, Regard JB, Xu Y, Camerer E, Zheng YW, Huang Y, Cyster JG, Coughlin SR. Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine-1-phosphate. Science. 2007;316:295–298. doi: 10.1126/science.1139221. [DOI] [PubMed] [Google Scholar]
- 49.Venkataraman K, Lee YM, Michaud J, Thangada S, Ai Y, Bonkovsky HL, Parikh NS, Habrukowich C, Hla T. Vascular endothelium as a contributor of plasma sphingosine 1-phosphate. Circ Res. 2008;102:669–676. doi: 10.1161/CIRCRESAHA.107.165845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Murata N, Sato K, Kon J, Tomura H, Yanagita M, Kuwabara A, Ui M, Okajima F. Interaction of sphingosine 1-phosphate with plasma components, including lipoproteins, regulates the lipid receptor-mediated actions. Biochem J. 2000;352:809–815. [PMC free article] [PubMed] [Google Scholar]
- 51.Sabbadini RA. Sphingosine-1-phosphate antibodies as potential agents in the treatment of cancer and age-related macular degeneration. Br J Pharmacol. 2011;162:1225–1238. doi: 10.1111/j.1476-5381.2010.01118.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Christoffersen C, Obinata H, Kumaraswamy SB, Galvani S, Ahnström J, Sevvana M, Egerer-Sieber C, Muller YA, Hla T, Nielsen LB, Dahlbäck B. Endothelium-protective sphingosine-1-phosphate provided by HDL-associated apolipoprotein M. Proc Natl Acad Sci USA. 2011;108:9613–9618. doi: 10.1073/pnas.1103187108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhu D, Wang Y, Singh I, Bell RD, Deane R, Zhong Z, Sagare A, Winkler EA, Zlokovic BV. Protein S controls hypoxic/ischemic blood-brain barrier disruption through the TAM receptor Tyro3 and sphingosine 1-phosphate receptor. Blood. 2010;115:4963–4972. doi: 10.1182/blood-2010-01-262386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rosette C, Karin M. Cytoskeletal control of gene expression: depolymerization of microtubules activates NF-kappa B. J Cell Biol. 1995;128:1111–1119. doi: 10.1083/jcb.128.6.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang N, Butler JP, Ingber DE. Mechanotransduction across the cell surface and through the cytoskeleton. Science. 1993;260:1124–1127. doi: 10.1126/science.7684161. [DOI] [PubMed] [Google Scholar]
- 56.McBeath R, Pirone DM, Nelson CM, Bhadriraju K, Chen CS. Cell shape, cytoskeletal tension, and RhoA regulate stem cell lineage commitment. Dev Cell. 2004;6:483–495. doi: 10.1016/s1534-5807(04)00075-9. [DOI] [PubMed] [Google Scholar]
- 57.Singleton PA, Dudek SM, Chiang ET, Garcia JG. Regulation of sphingosine 1-phosphate-induced endothelial cytoskeletal rearrangement and barrier enhancement by S1P1 receptor, PI3 kinase, Tiam1/Rac1, and alpha-actinin. FASEB J. 2005;19:1646–1656. doi: 10.1096/fj.05-3928com. [DOI] [PubMed] [Google Scholar]
- 58.Sun X, Shikata Y, Wang L, Ohmori K, Watanabe N, Wada J, Shikata K, Birukov KG, Makino H, Jacobson JR, Dudek SM, Garcia JG. Enhanced interaction between focal adhesion and adherens junction proteins: involvement in sphingosine 1-phosphate-induced endothelial barrier enhancement. Microvasc Res. 2009;77:304–313. doi: 10.1016/j.mvr.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee MJ, Thangada S, Claffey KP, Ancellin N, Liu CH, Kluk M, Volpi M, Sha’afi RI, Hla T. Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phosphate. Cell. 1999;99:301–312. doi: 10.1016/s0092-8674(00)81661-x. [DOI] [PubMed] [Google Scholar]
- 60.Hla T, Lee MJ, Ancellin N, Paik JH, Kluk MJ. Lysophospholipids—receptor revelations. Science. 2001;294:1875–1878. doi: 10.1126/science.1065323. [DOI] [PubMed] [Google Scholar]
- 61.Spiegel S, Milstien S. Sphingosine 1-phosphate, a key cell signaling molecule. J Biol Chem. 2002;277:25851–25854. doi: 10.1074/jbc.R200007200. [DOI] [PubMed] [Google Scholar]
- 62.Garcia JG, Liu F, Verin AD, Birukova A, Dechert MA, Gerthoffer WT, Bamberg JR, English D. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J Clin Invest. 2001;108:689–701. doi: 10.1172/JCI12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dudek SM, Jacobson JR, Chiang ET, Birukov KG, Wang P, Zhan X, Garcia JG. Pulmonary endothelial cell barrier enhancement by sphingosine 1-phosphate: roles for cortactin and myosin light chain kinase. J Biol Chem. 2004;279:24692–24700. doi: 10.1074/jbc.M313969200. [DOI] [PubMed] [Google Scholar]
- 64.Holash J, Wiegand SJ, Yancopoulos GD. New model of tumor angiogenesis: dynamic balance between vessel regression and growth mediated by angiopoietins and VEGF. Oncogene. 1999;18:5356–5362. doi: 10.1038/sj.onc.1203035. [DOI] [PubMed] [Google Scholar]
- 65.Cai J, Kehoe O, Smith GM, Hykin P, Boulton ME. The angiopoietin/Tie-2 system regulates pericyte survival and recruitment in diabetic retinopathy. Invest Ophthalmol Vis Sci. 2008;49:2163–2171. doi: 10.1167/iovs.07-1206. [DOI] [PubMed] [Google Scholar]
- 66.Hammes HP, Lin J, Wagner P, Feng Y, Vom Hagen F, Krzizok T, Renner O, Breier G, Brownlee M, Deutsch U. Angiopoietin-2 causes pericyte dropout in the normal retina: evidence for involvement in diabetic retinopathy. Diabetes. 2004;53:1104–1110. doi: 10.2337/diabetes.53.4.1104. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.