

Abstract
As part of an ongoing effort to explore the effect of major groove electrostatics on the thermodynamic stability and structure of DNA, a 7-deaza-2′-deoxyadenosine:dT (7-deaza-dA:dT) base pair in the Dickerson–Drew dodecamer (DDD) was studied. The removal of the electronegative N7 atom on dA and the replacement with an electropositive C–H in the major groove was expected to have a significant effect on major groove electrostatics. The structure of the 7-deaza-dA:dT base pair was determined at 1.1 Å resolution in the presence of Mg2+. The 7-deaza-dA, which is isosteric for dA, had minimal effect on the base pairing geometry and the conformation of the DDD in the crystalline state. There was no major groove cation association with the 7-deaza-dA heterocycle. In solution, circular dichroism showed a positive Cotton effect centered at 280 nm and a negative Cotton effect centered at 250 nm that were characteristic of a right-handed helix in the B-conformation. However, temperature-dependent NMR studies showed increased exchange between the thymine N3 imino proton of the 7-deaza-dA:dT base pair and water, suggesting reduced stacking interactions and an increased rate of base pair opening. This correlated with the observed thermodynamic destabilization of the 7-deaza-dA modified duplex relative to the DDD. A combination of UV melting and differential scanning calorimetry experiments were conducted to evaluate the relative contributions of enthalpy and entropy in the thermodynamic destabilization of the DDD. The most significant contribution arose from an unfavorable enthalpy term, which probably results from less favorable stacking interactions in the modified duplex, which was accompanied by a significant reduction in the release of water and cations from the 7-deaza-dA modified DNA.
Introduction
Nucleoside analogs containing pyrrolopyrimidine bases,1,2 or 7-deazapurines, are used as isosteric analogs of adenine and guanine in biochemical and biophysical studies.3−7 The 7-deazapurines are also used to study the effects of site-specific alteration of the electrostatic potential of the DNA major groove, where it has been shown for 7-deazaguanine that there is a significant alteration in DNA hydration and cation binding.7,8 The 7-deazaadenosine base was identified in the antibiotic tubercidin, a ribonucleoside isolated from various species of Streptomyces.2,9−11 The incorporation of 7-deaza-dA into DNA hinders the processing of the double helix by proteins, e.g., restriction endonucleases.(12) It slightly reduces the bending of DNA in oligodeoxynucleotides containing d(GGCA6C)·d(CCGT6G) tracts.13,14 The preparation of phosphoramidites containing 7-deaza-dA has been described by Seela et al.15,16
There remains a paucity of quantitative data as to how substitution of adenine with 7-deaza-dA alters the structure and thermodynamics of the DNA double helix. Thermal denaturation of (7-deaza-dA)11A·T12 as compared to dA12·dT12 led to the conclusion that destabilization induced by 7-deaza-dA was associated with an unfavorable entropy change.(17) Pope et al.(18) conducted a high-angle X-ray fiber diffraction study of poly[d(7-deaza-dA-T)]·poly[d(7-deaza-dA-T)]. They suggested that replacement of dA by 7-deaza-dA caused slight alterations to the structure of A-DNA, but greater perturbations to B-DNA. When 7-deaza-dG was incorporated into the Dickerson–Drew dodecamer (DDD)19,20 it had minimal effect on the overall conformation determined by NMR or crystallography.7,21 However, duplex stability was reduced adjacent to the modification site due to a loss of enthalpic stabilization. Moreover, 7-deaza-dG caused a reduction in hydration and cation binding. This was attributed to the elimination of a high affinity major groove cation binding site.(21) Clearly, while 7-deaza-dG was an isostere of dG, it altered the ensemble of DNA, water and salts, and thermodynamic stability of the DDD.(7)
In studies presented herein, an adenine at position A6 in the DDD19,20 has been replaced by 7-deaza-dA15,16 to form the DDD-1 duplex [5′-d(C1G2C3G4A5Y6T7T8C9G10C11G12)-3′]2 (Y=7-deaza-dA) (Chart 1). Crystallography has been used to determine the structure of the DDD-1 duplex. A combination of thermal melting studies monitored by UV absorbance, differential scanning calorimetry (DSC), and NMR studies have been performed. The corresponding decamer DD-1, [5′-d(G1C2G3A4Y5T6T7C8G9C10)-3′]2, which does not form an intramolecular hairpin at low salt concentrations, was also used in thermodynamic studies. We demonstrate that 7-deaza-dA has minimal effect upon base pairing geometry and conformation of the DDD. However, the 7-deaza-dA:dT base pair is thermodynamically destabilized, which is primarily attributed to unfavorable enthalpy terms dominated by less favorable stacking interactions, resulting from changes in the base electrostatics and electronic dipole–dipole interactions. There is also a net release of electrostricted waters from the duplex.
Chart 1. (a) Structure of 7-deaza-dA and (b) Sequences and numbering of the nucleotides for unmodified DD, 7-deaza-dA DD, unmodified DDD, 7-deaza-dA DDD (NMR), and 7-deaza-dA DDD (X-ray) duplexes.

a In solution, the two strands exhibit pseudo-dyad symmetry. In the crystal structure, the two strands were not symmetry related and the nucleotides were individually numbered.
Materials and Methods
Sample Preparation
The oligodeoxynucleotides 5′-CGCGAYTTCGCG-3′ (DDD-1) and 5′-GCGAYTTCGC-3′, (DD-1), Y = 7-deaza-dA, were synthesized by the University of Nebraska Medical Center Eppley Institute Molecular Biology Shared Resource. The 7-deaza-dA phosphoramidite was obtained commercially (Glen Research, Sterling, VA, U.S.A.). The oligodeoxynucleotides were purified using semipreparative reverse-phase HPLC (Phenomenex, Phenyl-Hexyl, 5 μm, 250 mm × 10.0 mm) equilibrated with 0.1 M triethylammonium acetate (pH 7.0). The unmodified oligodeoxynucleotides, 5′-CGCGAATTCGCG-3′ (DDD) and 5′-GCGAATTCGC-3′ (DD), were synthesized by the Midland Reagent Company (Midland, TX) and purified by anion-exchange HPLC. The oligodeoxynucleotides were desalted using Sephadex G-25, lyophilized, and characterized by MALDI-TOF-MS. The oligodeoxynucleotides were dissolved in the appropriate buffers. The concentrations of single-stranded oligodeoxynucleotides were determined by UV absorbance at 260 nm using extinction coefficients of 1.11 × 105 M–1 cm–1 (dodecamers) and 9.5 × 104 M–1 cm–1 (decamers)(22) and assuming similar extinction coefficients for 7-deaza-dA and dA. The oligodeoxynucleotides were annealed by heating to 80 °C for 15 min and then cooling to room temperature.
Temperature–Unfolding Profiles (Melting Curves)
The thermodynamic parameters for the temperature-induced unfolding reactions of the duplexes were measured using a VP-DSC differential scanning calorimeter (Microcal, Inc., Northampton, MA, U.S.A.). The heat capacity profile for each DNA solution was measured against a buffer solution. The experimental curves were normalized for the heating rate, and a buffer vs buffer scan was subtracted using the program Origin (v. 5.0; Microcal, Inc.). The resulting monophasic or biphasic curves were analyzed by deconvolution with the Microcal software; their integration (∫ΔCp dT) yielded the molar unfolding enthalpy (ΔHcal), which was independent of the nature of the transition.23,24 The molar entropy (ΔScal) was obtained similarly, using ∫(ΔCp/T) dT. The free energy change at any temperature T was obtained with the Gibbs equation: ΔG°(T) = ΔHcal – TΔScal.
Absorption versus temperature profiles (UV melts) for each duplex were measured at either 260 or 275 nm using a thermoelectrically controlled UV–vis Aviv 14DS (Aviv Biomedical, Inc., Lakewood, NJ) or Lambda 40-Perkin-Elmer (Perkin-Elmer, Inc., Waltham, MA) spectrophotometers. The temperature was scanned at heating rates of 0.75–1.00 °C/min. Melting curves as a function of strand concentration (7–70 μM) were obtained to check the molecularity of each oligodeoxynucleotide (i.e., hairpin vs duplex). Additional melting curves were obtained as a function of salt(25) and osmolyte concentrations26−28 to determine the differential binding of counterions (ΔnNa+) and waters (Δnw), which accompanied the helix-to-coil transitions.29,30 For duplexes that melted via biphasic transitions only the TM of the duplex → random coil transition was used for the calculations.
In the determination of ΔnNa+, UV melts were measured in the salt range of 10–200 mM NaCl at pH 7.0, whereas in the determination of Δnw, UV melts were measured in the ethylene glycol concentration range of 0.5–4.0 m at pH 7.0 and 10 mM NaCl. The osmolalites of the solutions were obtained with a UIC vapor pressure osmometer, Model 830 (Jolliet, IL, U.S.A.). These osmolalities were then converted into water activities, aw, using the relationship ln aw = −(Osm/Mw), where Osm is the solution osmolality and Mw is the molality of H2O, 55.5 mol/kg.(31)
Circular Dichroism
Circular dichroism (CD) measurements were conducted on an Aviv model 202SF CD spectropolarimeter (Aviv Biomedical, Inc., Lakewood, NJ). To approach 100% duplex formation the spectrum of each sample was obtained using a strain-free 1 cm quartz cell at low temperatures. Typically, 1 OD of a duplex DNA was dissolved in 1 mL of 10 mM sodium phosphate buffer (pH 7.0). The reported spectra correspond to an average of three scans from 220 to 350 nm at a wavelength step of 1 nm.
NMR Spectroscopy
Modified and unmodified duplexes were prepared at 0.3 mM and 1.8 mM concentrations, respectively. The samples were prepared in 10 mM NaH2PO4, 0.1 M NaCl, and 50 μM Na2EDTA (pH 7.0). The samples were exchanged with D2O and dissolved in 0.5 mL of 99.99% D2O to observe nonexchangeable protons. For the observation of exchangeable protons, the samples were dissolved in 0.5 mL of 9:1 H2O/D2O. 1H NMR spectra for unmodified and modified oligodeoxynucleotides were recorded at 600 and 800 MHz. Chemical shifts were referenced to water. Data were processed using TOPSPIN software (Bruker Biospin Inc., Billerica, MA). The NOESY32,33 and DQF-COSY(34) spectra of samples in D2O were collected at 15 °C at 800 MHz; NOESY experiments were conducted at a mixing time of 250 ms. The NOESY spectra of the modified and unmodified sample in H2O were collected at 5 °C at 600 MHz, with a 250 ms mixing time. These experiments were performed with a relaxation delay of 2.0 s. Water suppression was performed using the WATERGATE pulse sequence.(35)
Crystallizations and Data Collection
Crystallization trials were performed with the Nucleic Acid Mini-screen (Hampton Research, Aliso Viejo, CA).(36) The hanging drop vapor diffusion technique was used. Droplets, with a volume of 2 μL, of a 1:1 mixture of sample and mini-screen buffer were equilibrated against 0.75 mL of 35% 2-methyl-2,4-pentanediol (MPD) at 18 °C. The crystal used for data collection was grown in 10% MPD, 40 mM sodium cacodylate (pH 6.0), 12 mM spermine tetra-HCl, and 80 mM NaCl. The single crystal was mounted in a nylon loop and frozen in liquid nitrogen. Diffraction data were collected at low temperature in a cold nitrogen stream on beamline 21-ID-F at LS-CAT, APS (Argonne National Laboratory, Argonne, IL). Separate data sets for high and low resolution reflections were collected. All data were processed with the program HKL2000.(37)
Crystal Structure Determination and Refinement
The diffraction data were processed in space group P212121 (orthorhombic). Phasing was carried out by the molecular replacement method using the program MOLREP in the CCP4 suite.(38) The DDD sequence with PDB entry 355D(39) was used as the starting model. Initial refinements of the model were performed with the CNS program,(40) setting aside 5% randomly selected reflections for calculating the Rfree. Rigid body refinement and simulated annealing were performed. Multiple rounds of coordinate refinements and simulated annealing led to an improved model for which sum (2Fo-Fc) and difference (Fo-Fc) Fourier electron density maps were generated. At a later stage solvent water molecules were added on the basis of Fourier 2Fo-Fc sum and Fo-Fc difference electron density maps. Water molecules were accepted based on the standard distances and B-factor criteria. Further structure refinement was performed using the program SHELX,(41) and REFMAC in CCP4.(38) A Mg2+ ion and four Na+ ions were identified in the electron density maps based on their low B-factors and the characteristic Mg2+ octahedral and Na+ tetrahedral coordination geometries. Geometry and topology files were generated for the 7-deaza-dA modified bases and anisotropic temperature factor refinement was performed afterward. The program TURBO-FRODO(42) was used to display electron density maps. The helicoidal parameters of the 7-deaza-dA-modified DDD were analyzed using the program CURVES (version 5.3).(43)
Data Deposition
Complete structure factor and final coordinates were deposited in the Protein Data Bank (www.rcsb.org): PDB ID code 3OPI.
Results
Crystallography
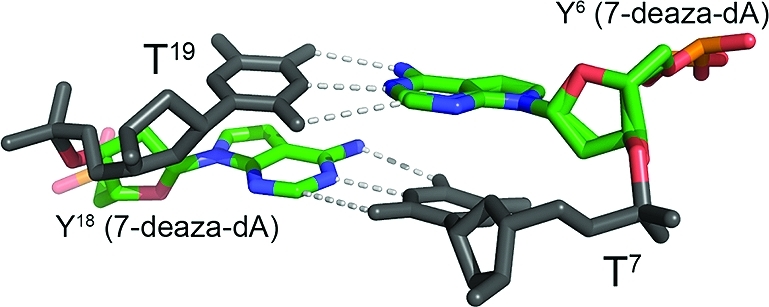
The 7-deaza-dA-modified DDD-1 diffracted at a resolution of 1.1 Å. The two strands of the DDD-1 duplex were not symmetry-related in the crystal. Therefore, each of the nucleotides was uniquely numbered (Chart 1). Minimal perturbation of the DNA duplex was observed at the 7-deaza-dA site (Figure 1).(44) The 7-deaza-dA bases were in the anti conformation about the glycosyl bonds and Watson–Crick base pairing was maintained at base pairs Y6·T19 and Y18·T7 (Figure 2). Waters formed the anticipated minor groove inner spine of hydration (Figure S1 of the Supporting Information), similar to the situation in the DDD.19,20,44 The replacement of N7-dA with a carbon atom in 7-deaza-dA6 did not alter Mg2+ binding in the crystal, e.g., as indicated by a comparison to the high resolution structure of the DDD obtained by Tereshko and Egli.(44) One Mg2+ ion was present per asymmetric unit, but two Mg2+ ions interacted with each DNA molecule as a consequence of the crystallographic 21 symmetry. This Mg2+ interacted via six coordinated waters with the G2 and G22 nucleotides in the major groove (Figure 1). It also interacted via coordinated waters with the Y6 and T7 phosphate oxygens from an adjacent DNA molecule. It did not interact directly with the Y6 7-deaza-dA base (Figure 1; Figure S2 of the Supporting Information). Instead, it stabilized a contact between DNA molecules. The sum electron density contoured at the 1.0 σ level for the G4, A5 and Y6 nucleotides suggested two conformations of the phosphate backbone (Figure S2 of the Supporting Information). These were each refined with occupancy 0.5. It is likely that these were due to this Mg2+-mediated lattice contact between DNA molecules. Helicoidal analyses indicated that the rise, roll, and twist parameters of the DDD-1 duplex were unaffected by these two backbone conformations (Figure S3 of the Supporting Information). The difference between the two conformations primarily involved torsion angle α (5′-P-O-C5-C4-3′) (Figure S4 of the Supporting Information). Smaller variations were observed in other torsion and glycosyl angles of the G4, A5 and Y6 nucleotides (Figure S5 of the Supporting Information). In all, 133 waters and four Na+ ions, of which one was observed at the 5′-ApT-3′ step,(44) were assigned per asymmetric unit. A summary of crystal data and data collection statistics is given in Table 1.
Figure 1.

Sum electron density contoured at the 1.0 σ level (green meshwork) surrounding the DDD-1 duplex in the region of the G4, A5 ,and Y6 nucleotides, where the phosphate groups display two alternative conformations. Bases G4 and A5 are shown in gray (one phosphate conformation) and black (second phosphate conformation). Modified base Y6 is in blue (one phosphate conformation) and navy (second phosphate group conformation). The Mg2+ ion (white sphere) is coordinated by six water molecules (red spheres). The Mg2+ ion interacts via coordinated waters with phosphate oxygens of one conformer of Y6 only (second conformation of the phosphate backbone is shown in navy) and T7 residue. Similar interactions are observed in the unmodified DDD duplex (PDB entry 355D). This interaction does not involve the N7 atom of Y6 and is maintained for the 7-deaza-dA base.
Figure 2.

Sum electron density contoured at the 1.0 σ level (green meshwork) around the modified Y6·T19 and Y18·T7 along the normal to the base pairs, viewed (a) from the side and (b) from the top approximately the named to base pairs, revealing stacking interactions. (c) Watson–Crick base pairing of 7-deaza-dA·dT. Y6 and Y18 bases are shown in blue.
Table 1. Crystal Data, Data Collection, and Refinement Statistics.
| space group | orthorhombic P212121 |
| cell parameters (Å) | a = 25.64, b = 40.31, c = 65.93 |
| temperature of data collection (° C) | –170 |
| wavelength (Å) | 0.9785 |
| max resolution (Å) | 1.1 |
| unique reflections | 27920 |
| completeness all/1.14–1.10 Å (%) | 97.8/95.8 |
| redundancy all/1.14–1.1 Å | 10.6/6.9 |
| I/σ (I) all/1.14–1.1 Å | 61.26/4.8 |
| Rmerge all/1.14–1.10 Å | 0.048/0.394 |
| Rwork | 0.161 |
| Rfree | 0.195 |
| number of DNA atoms | 486 |
| number of water molecules | 133 |
| number of ions | 1 Mg2+ |
| 4 Na+ | |
| rms distances (Å) | 0.024 |
| rms angles (°) | 1.95 |
Circular Dichroism
The CD spectra of the DDD and DDD-1 dodecamers are shown in Figure 3. These experiments were performed at 16 mM [Na+]. In both instances, a positive Cotton effect was observed, centered near 280 nm. In both instances, a negative Cotton effect was centered at 250 nm. These were characteristic of a right-handed helix in the B-DNA family. There was an 18% decrease in the intensity of the 250 nm band for DDD-1 relative to DDD. CD experiments with the decamers DD and DD-1 revealed a similar trend. The decreased intensity of the 250 nm band for DD-1 relative to DD was 10% (Figure 3).
Figure 3.

CD spectra of duplexes in 10 mM sodium phosphate buffer (pH 7.0) at 4 °C, ∼10 μM strand concentration: (a) DDD (●) and DDD-1 (O) and (b) DD (●) and DD-1 (O). The spectra without symbols are the spectra of the unmodified DDD and DD at 90 °C.
NMR Spectroscopy
In solution, the pseudodyad symmetry of the DNA duplex results in the symmetry-related resonances from the two strands being isochronous;45,46 thus, the NMR resonances are labeled for nucleotides 1–12. The 7-deaza-dA H7 and H8 protons were assigned from a combination of COSY and NOESY spectra, which established the presence of the 7-deaza-dA base at position Y6 in the DDD-1 duplex (Figure S6 in the Supporting Information). The upfield chemical shift of 1.07 ppm observed for Y6 H8 relative to A6 H8 in the DDD was attributed primarily to different electron distributions in the pyrrolopyrimidine vs purine bases, not to a conformational change in the DDD-1 duplex. The nonexchangeable DNA protons were assigned using standard methods.47,48 All sequential NOEs between the aromatic and anomeric protons of the DDD-1 duplex were observed (Figure S6 in the Supporting Information). The imino proton region of the NOESY spectrum of the DDD-1 duplex is shown in Figure 4. The sequential connectivity of the base imino protons was obtained from base pairs G2·C11 → C3·G10 → G4·C9 → A5·T8 → Y6·T7.(49) Cross peaks from A5 H2 to T8 N3H and Y6 H2 to T7 N3H were observed. For the imino protons, the greatest downfield shift of 0.49 ppm was observed for the T7 imino proton. The imino resonances of the terminal base pairs C1·G12 were missing. This was attributed to rapid exchange with water.
Figure 4.

(a) NOE connectivity for the imino protons for the base pairs G2•C11 to Y6•T7.The experiments were carried out at a mixing time of 250 ms and 600 MHz at 5 °C. (b) Interstrand NOE cross peaks between opposite bases: a1, T7N3H → Y6 H2; b1, T8 N3H → A5 H2; b2, T8 N3H → Y6 H2; c1, G2 N1H → C11N2H2; c2, G2 N1H → C11N2H1; d1, G10 N1H → C3N2H2; d2, G10 N1H → C3N2H1; e1, G4 N1H → C9N2H2; e2, G4 N1H → A5 H2; e3, G4 N1H → C9N2H1.
Unfolding Studies
(a). NMR
Spectra of the DDD-1 and DDD duplexes were collected as a function of temperature, over the range 5–65 °C (Figure 5). At 15 °C, for the 7-deaza-dA-modified duplex, the T7 imino resonance began to broaden, compared with the other peaks and with the unmodified DDD. At 45 °C, the T7 peak completely broadened. These observations indicated that the T7 imino proton was in enhanced exchange with the solvent and indicated a destabilization of the Y6·T7 base pair.
Figure 5.

1H NMR of imino proton resonances as a function of temperature. (A) 7-deaza-dA DDD-1 duplex. (B) Unmodified DDD duplex. Modified and unmodified duplexes were prepared at 0.3 mM and 1.8 mM concentration respectively. The samples were prepared in 10 mM NaH2PO4, 0.1 M NaCl, and 50 μM Na2EDTA at pH 7.0.
(b). UV Melting Studies
The unfolding of duplexes was studied by temperature-dependent UV spectroscopy. Absorption spectra at low and high temperatures revealed a greater hyperchromic effect at 260 nm for DDD and DD and at 275 nm for DDD-1 and DD-1. These were chosen as optimum wavelengths used for all UV melting studies. Typical melting curves of dodecamer and decamer duplexes are shown in Figure 6. In 10 mM NaCl, dodecamers (DDD and DDD-1) unfolded in broad biphasic transitions, whereas decamers (DD and DD-1) unfolded via monophasic transitions. The overall sequential melting behavior corresponded to duplex → hairpin and hairpin → random coil transitions, while the corresponding decamers, which formed less stable hairpins, melted through a single duplex random coil transition. The TM values were determined by taking the first derivative of the melting curves, and shape analysis of these curves are reported in Table 2. Incorporation of 7-deaza-dA was destabilizing for both dodecamer and decamer. The TM of the first transition for the dodecamer DDD-1 relative to DDD was unchanged in 16 mM Na+ (low salt) and 8.2 °C lower in 116 mM Na+ (high salt) concentrations. At higher salt concentration both melting transitions overlapped and only one transition was observed. The TM of the modified DD-1 was lower than that for DD by 3.4 °C in low salt and by 5.5 °C in high salt.
Figure 6.

UV melting curves in 10 mM sodium phosphate buffer (pH 7.0) ∼40 μM total strand concentration for (a) DDD (●) at 260 nm and DDD-1 (O) at 275 nm and (b) DD (●) at 260 nm and DD-1 (O) at 275 nm.
Table 2. Thermodynamic Profiles for the Formation of Duplexes at 20 °C.a.
| oligodeoxynucleotide | NaClb | TMc | ΔG° d,e | ΔHe | TΔSe | ΔnNa+f | Δnwf |
|---|---|---|---|---|---|---|---|
| DDD | 10 | 33.3 | –6.9 | –116.0 | –109.1 | –2.3 ± 0.2 | –38.0 ± 2.0 |
| 100 | 57.7 | –15.5 | –109.5 | –94.0 | –1.8 ± 0.1 | –30.0 ± 2.0 | |
| DDD-1 | 10 | 34.5 | –4.6 | –76.0 | –71.4 | –1.4 ± 0.1 | –19.0 ± 2.0 |
| 100 | 49.5 | –10.4 | –74.0 | –63.6 | –1.1 ± 0.1 | –15.0 ± 2.0 | |
| DD | 10 | 29.5 | –5.6 | –80.1 | –74.5 | –2.2 ± 0.2 | –30.0 ± 4.0 |
| 100 | 53.0 | –8.2 | –72.3 | –64.1 | –1.7 ± 0.1 | –22.0 ± 3.0 | |
| DD-1 | 10 | 26.1 | –3.8 | –56.4 | –52.6 | –1.5 ± 0.2 | –17.0 ± 2.0 |
| 100 | 47.5 | –5.7 | –54.1 | –48.4 | –1.3 ± 0.1 | –14.0 ± 2.0 |
Parameters are measured from UV (TM) and DSC melting curves in 10 mM sodium phosphate buffer (pH 7.0). The observed standard deviations are TM (±0.5), ΔHcal (±3%), ΔG°20 (±5%), and TΔScal (±3%).
Salt concentration in mM.
°C.
Determined at 20 °C.
kcal/mol.
Per mol DNA. ΔnNa+ was determined experimentally, using the linking number: ΔnNa+ = ∂ ln K/∂ ln [Na+], where K corresponds to two single strands in equilibrium with a duplex.
DSC of the 7-Deaza-dA-Modified Duplexes
The DSC melting curves for the DDD and DDD-1 dodecamers and the DD and DD-1 decamers are shown in Figure 7, and the thermodynamic profiles are listed in Table 2. At the lower salt concentration (16 mM Na+), the helix–coil transition was biphasic for the dodecamers. The DDD unfolded via a broad first transition and a sharper second transition. The biphasic DSC thermogram of DDD-1 revealed a broad peak with a shoulder for the first transition at lower temperature that could not be resolved. At increased salt concentration, the dodecamers unfolded via monophasic transitions. This was attributed to higher screening by salt on the duplex phosphates, relative to the phosphates of the hairpin. This shifts the duplex transition to higher temperatures, confirming the helix → hairpin → random coil transitions of each dodecamer duplex, which was observed in the UV melting studies. For the decamers, the helix–coil transitions were monophasic, confirming their unfolding through a duplex to random coil transition as seen in the UV studies. Enthalpies were determined by deconvolution of the DSC graphs; however, only the model-independent enthalpies of the duplex → random coil transitions are reported in Table 2. The dA to 7-deaza-dA substitution was destabilizing at both low and high salt concentrations.
Figure 7.

DSC curves in 10 mM sodium phosphate buffer (pH 7.0): (a) DDD (●) and DDD-1 (O) at ∼200 μM and (b) DD (●) and DD-1 (O) at ∼300 μM.
Analysis of thermograms of dodecamers revealed decreased endothermic enthalpies of 40.0 and 35.5 kcal/mol for DDD-1 relative to DDD in 10 and 100 mM NaCl, respectively (Table 3). For decamers, endothermic enthalpies of 80.1 kcal/mol for DD and a reduced unfolding enthalpy of 56.4 kcal/mol for DD-1 (Table 3) were obtained at low salt. At the higher salt concentration, the ΔΔH was 18.2 kcal/mol for DD vs DD-1.
Table 3. Differential Thermodynamic Profiles for Pairs of Dodecamer and Decamer Duplexes.
| NaCla | ΔΔHc | ΔΔG°b,c | Δ(TΔS)c | ΔΔnNa+d | ΔΔnwd |
|---|---|---|---|---|---|
| Substitution of dA6 with 7-Deaza-dA in DDD (DDD-1 Minus DDD) | |||||
| 10 | 40.0 | 2.3 | 37.7 | 0.9 | 19.0 |
| 100 | 35.5 | 5.1 | 30.4 | 0.7 | 15.0 |
| Substitution of dA5 with 7-Deaza-dA in DD (DD-1 Minus DD) | |||||
| 10 | 23.7 | 1.8 | 21.9 | 0.7 | 13.0 |
| 100 | 18.2 | 2.5 | 15.7 | 0.4 | 8.0 |
Salt concentration in mM.
Determined at 20 °C.
kcal/mol.
Per mol DNA.
Thermodynamic Profiles for the Formation of Each Duplex
The thermodynamic data is provided in Table 2. The favorable Gibbs free energies, indicating spontaneous formation of each duplex, resulted from compensation of favorable enthalpy and unfavorable entropy contributions. The favorable enthalpies arose from the formation of base pairs and base pair stacks, uptake of electrostricted waters, and release of structural waters, whereas the unfavorable entropy terms included the ordering of two strands to form a duplex, condensation of counterions, and immobilization of waters.
Relative to the unmodified oligodeoxynucleotides, the 7-deaza-dA modified oligodeoxynucleotides were destabilized at low and high salt concentrations. The inclusion of two 7-deaza-dA modifications in DDD-1 yielded a decrease in ΔG of 2.3 and 5.1 kcal/mol in 10 and 100 mM NaCl, respectively, whereas in decamers ΔG decreases of 1.8 and 2.5 kcal/mol in low and high salt, respectively, were observed following two 7-deaza-dA substitutions.
Differential Association of Water Molecules
TM dependencies on water activity were studied to determine the thermodynamic association of water molecules to DNA duplexes. By increasing concentrations of the osmolyte ethylene glycol from 0.5 to 4.0 m the activity of water was decreased. The UV melting curves showed that the TMs of the dodecamers (DDD and DDD-1) and decamers (DD and DD-1) decreased linearly with increasing osmolyte concentrations (i.e., decreasing activity of water). The TM dependence on water activity of dodecamers and decamers are shown in Figure 8. The slopes of these lines, ∂ TM/∂ log aw, in conjunction with the ΔH/RTM2 term, were used to obtain the differential association of water molecules. The Δnw values for the formation of each duplex in 10 mM NaCl are shown in Table 2. Water uptake values, expressed as mol H2O per mol duplex, measured in low salt, were 38 (DDD) and 19 (DDD-1) for dodecamers, and 30 (DD) and 17 (DD-1) for decamers. At the higher salt concentration (116 mM Na+), Δnw values followed a similar trend. Lower Δnw values at this salt concentration (Table 2) were due to increased screening of the water dipoles at higher salt concentration. The overall effect, and assuming that the random coil states of all the duplexes behave similarly at higher temperature, was that the substitution of 7-deaza-dA into duplex DNA caused a decreased association of water molecules. For instance, there was a ΔΔnw of 19 and 15 between DDD and DDD-1 at 10 mM and 100 mM NaCl, respectively, and ΔΔnw of 13 and 8 between the pair of decamer duplexes at low and high salt, respectively (Table 3). Parameters used to calculate differential water binding for dodecamers are shown in Table S1 in the Supporting Information.
Figure 8.

TM dependence on osmolyte concentration (as a function of ethylene glycol) for duplexes in 10 mM sodium phosphate buffer (pH 7.0), ∼5 μM strand concentration for (a) DDD (●) and DDD-1 (○) and ∼7 μM strand concentration for (b) DD (●) and DD-1 (○).
Differential Association of Counterions
UV melting curves at salt concentrations ranging from 16 to 216 mM [Na+] were measured to examine the thermodynamic association of counterions with the DNA duplexes. The TM values of the DDD and DDD-1 dodecamers, and DD and DD-1 decamers increased linearly with salt concentration (Figure 9), consistent with the expectation that the duplex states should have higher charge density parameters. The TM dependence on salt concentration for dodecamers and decamers are shown in Figure 9, panels a and b, respectively. The slopes of these lines, ∂ TM/∂ log[Na+], in conjunction with the experimentally determined ΔH/RTM2 terms, allowed measurement of differential counterion binding. The ΔnNa+ values for the formation of each duplex, from the association of two complementary strands, in low and high salt are shown in Table 2. In low salt, the Na+ uptake as measured in mol Na+ per mol duplex was 2.3 for the DDD dodecamer and 1.4 for the DDD-1 dodecamer, and 2.2 for the DD dodecamer and 1.5 for the DD-1 decamer. The ΔnNa+ values at the higher salt concentration of 116 mM showed a similar trend; however, the values were lower due to the higher screening of the phosphates by salt (Table 2). The average differential Na+ uptake as measured in mol Na+ per mol phosphate was estimated as 0.094 (DDD and DD) in this range of salt concentration, which was consistent with the fact that these oligodeoxynucleotides were not behaving electrostatically as long polyelectrolytes.(50) However, the main effect, assuming that the random coil states of the different single strand oligodeoxynucleotides were thermodynamically similar at higher temperatures, was that the introduction of 7-deaza-dA into the duplex DNA caused a slightly decreased association of counterions. For instance, there was a ΔΔnNa+ of 0.9 and 0.7 between DDD and DDD-1 at 10 and 100 mM NaCl, respectively, and ΔΔnNa+ of 0.7 and 0.4 between the pair of decamer duplexes at low and high salt, respectively (Table 3). Parameters used to calculate differential counterion binding for dodecamers are presented in Table S2 in the Supporting Information.
Figure 9.

TM dependencies on salt concentration for duplexes in 10 mM sodium phosphate buffer (pH 7.0), ∼5 μM strand concentration for (a) DDD (●) and DDD-1 (○) and ∼7 μM strand concentration for (b) DD (●) and DD-1 (○).
Discussion
It has been assumed that 7-deaza-dA, an isostere for dA in duplex DNA, does not substantially perturb the duplex, and thus provides a good model for dA. However, in light of suggestions that 7-deaza-dA introduces a large structural perturbation to the B-form of poly(dA-dT)·poly(dA-dT),(18) it was of interest to provide a comprehensive characterization of B-DNA with a 7-deaza-dA modification. The Dickerson–Drew dodecamer19,20 provides a well-characterized system suitable for detailed crystallographic analysis,(44) as well as NMR analysis.46,51,52 The present studies provide the first high-resolution crystallographic data for the substitution of adenine with 7-deaza-dA in duplex DNA.
Structure of the 7-Deaza-dA:dT Base Pair
The structure of the 7-deaza-dA:dT base pair in the DDD duplex reveals that 7-deaza-dA has minimal effect on duplex conformation (Figure 1) and base pair geometry (Figure 2) as compared to a canonical dA:dT base pair. Substitution of 7-deaza-dA changes the electronegative N7-dA atom to a carbon atom, which alters the electrostatics of the nucleobase. Consistent with this expectation, the downfield shift of the T7 imino resonance (Figure 5) is attributed to stronger hydrogen bonding with the more electronegative 7-deaza-dA N1 nitrogen. Thus, the observed destabilization of 7-deaza-dA does not result from a decrease in H-bonding but must be due to other changes induced by the perturbation of the electrostatic potential in the major groove. Other NMR chemical shift perturbations are minimal, which indicates that the modification does not affect the structure at the flanking nucleotides. Our results differ from those of Pope et al.,(18) who suggested that replacement of dA by 7-deaza-dA caused perturbations to B-DNA for the poly[d(7-deaza-dA-T)]·poly[d(7-deaza-dA-T)] duplex. The physical properties of poly(dA-dT) differ from the DDD, and it may be of interest to look for structural perturbations induced by 7-deaza-dA in other sequences.
7-Deaza-dA Enthalpically Destabilizes the DDD
The 7-deaza-dA substitution thermodynamically destabilizes the DDD-1 and DD-1 duplexes, compared to the unmodified DDD and DD duplexes. This is evidenced by the ΔΔG values (computed as the average of 10 and 100 mM [Na+], Table 3). At 20 °C, ΔΔG is decreased by 3.7 kcal/mol for DDD-1 and by 2.2 kcal/mol for DD-1. In both cases, the major contributor to the reduced ΔΔG values is the enthalpy term, which drops 37.8 kcal/mol for DDD-1 and 20.9 kcal/mol for DD-1 (Table 3). The differential ΔΔH values at different salt concentrations suggest the presence of heat capacity effects. The heat capacity values were 0.8 kcal/K mol (DDD) and −0.08 kcal/K mol (DDD-1), and −0.5 kcal/K mol (DD) and −0.2 kcal/K mol (DD-1). These may be due to exposures of nonpolar groups to solvent and/or to changes in structural hydration between the random coil and duplex states of DDD-1 and DD-1.(53) The present data lead to a different conclusion than did studies of (7-deaza-dA)11A·T12 as compared to dA12·dT12, conducted by Seela and Thomas.(17) They concluded that destabilization induced by 7-deaza-dA was minimal and was associated with an unfavorable entropy change.(17) It should be noted, however, that the DDD presents a different sequence context than does the A-tract dA12·dT12 sequence.(54)
Base Stacking Effects
The most significant contribution to the unfavorable ΔΔH term (Table 3) of 32.7 kcal/mol for DDD-1 (17.6 kcal/mol for DD-1) results from a reduction of stacking enthalpy in the modified duplexes, attributed to less favorable π–π interactions involving the pyrrolopyrimidine ring of 7-deaza-dA and the neighboring base pairs vs adenine. In the CD spectra, the intensities of the negative bands near 250 nm are thought to track base stacking contributions. The band intensities at 250 nm are consistent with reduced base stacking in DDD-1 and DD-1 at low temperature (Figure 3). There is an 18% decrease in the intensity of the 250 nm band for DDD-1 relative to DDD. The decreased intensity of the 250 nm band for DD-1 relative to DD is 10%. However, changes in the electronic structure of 7-deaza-dA may modulate the relative optical dipole orientations responsible for the CD bands. Exchange-mediated line broadening of DNA imino protons is often associated with the rate-limiting formation of an open state of the base pair in which the imino proton is freed from its hydrogen bond and is accessible to the base that catalyzes the proton exchange.55−59 The increased broadening of the Y6·T7 base pair thymine N3 imino resonance (Figure 5) is consistent with this model, which correlates with reduced stacking enthalpy of the DDD-1 duplex relative to the DDD duplex. However, the possibility that base pair opening is not rate-limiting cannot be ruled out, with the line broadening reflecting a more rapid hydrogen exchange catalysis for the substituted duplex.(60) In this regard, the C7–H on the 7-deaza-dA (as compared to the: N7 on the natural dA) would be anticipated to exhibit a reduced electrostatic repulsion with hydroxide or phosphate base catalyst.
Duplex Hydration
The unfavorable ΔΔH term observed upon incorporation of 7-deaza-dA is partially attributed to reduced hydration of the modified duplexes. This may, in part, be due to the more hydrophobic major groove edge of 7-deaza-dA as compared to dA. Thus, 7-deaza-dA substitution results in a ΔΔnW of 17 H2O per mol DNA for DDD-1 and 11 H2O per mol DNA for DD-1 (obtained by averaging the data obtained in 10 and 100 mM NaCl, Table 3). This “translates” into a reduction of approximately 9 H2O per mol DNA per 7-deaza-dA nucleotide for the DDD-1 duplex and 6 H2O per mol DNA per 7-deaza-dA nucleotide for the DD-1 duplex, assuming localized effects. A release of 17 water molecules from the DDD-1 duplex (11 water molecules from the DD-1) accounts for an unfavorable enthalpy term ΔΔH of 5.1 kcal/mol (3.3 kcal/mol for the DD-1).(61) The release of waters indicates increases in the volumes of the modified systems, i.e, positive ΔΔV terms. Since ΔΔG is also positive, this indicates release of electrostricted waters from DDD-1 and DD-1.(62) There may also be a compensating increase of structural water due to the more hydrophobic major groove edge of 7-deaza-dA. Another way to interpret the data is that the displacement of water by ethylene glycol, used in the osmotic stress experiments, near 7-deaza-dA will be more facile than at dA because of the reduced electrostatic interaction with solvent. In any case, similar reductions in hydration were observed for DNA modified with 7-deaza-dG nucleotides.(7)
Cation Binding
The introduction of the 7-deaza-dA:dT pair into the DDD causes a decrease in the differential association of cations. This is reflected in the ΔΔnNa+ of 0.9 and 0.7 between DDD and DDD-1 at 10 and 100 mM NaCl, respectively, and ΔΔnNa+ of 0.7 and 0.4 between the pair of decamer duplexes at low and high salt, respectively. The reduced uptake of Na+ is not attributed to the loss of a major groove high affinity cation binding site near the 7-deaza-dA nucleotide. High-resolution crystallographic structures of the DDD19,20 provide insight into the sequence-dependent distribution of waters and counterions in B-DNA.39,44,63−72 When the DDD was crystallized in the presence of Tl+, no high-occupancy cation binding sites were observed in the major groove near A6. Likewise, Tereshko and Egli(44) did not observe a high affinity cation site near A6. In the present crystallographic unit cell two Mg2+ ions interact with the DNA, but they are not associated with the major groove edge of either Y6 or Y18 (Figure 1; Figure S2 in the Supporting Information). This is consistent with the notion that cation binding in A-T tracts occurs in the minor groove.(68) It seems possible that the thermodynamically measured decrease in the association of cations could be due to the disruption of nonspecific cation binding, particularly in the minor groove. In any case, the contribution to the large ΔΔH term for the release of counterions is anticipated to be negligible since counterion release contributes predominantly to the Δ(TΔS) term.(73) In contrast, the major groove high-affinity cation sites in the DDD were associated with the major groove edge of dG nucleotides.(69) Indeed, the incorporation of 7-deaza-dG into the DDD was accompanied by changes in hydration and major groove cation organization.(7)
Summary
Introduction of the 7-deaza-dA:T base pair into the DDD has minimal effect upon base pairing geometry and DNA conformation, as evidenced by a combination of crystallographic and NMR studies. The 7-deaza-dA retains Watson–Crick hydrogen bonding, but the 7-deaza-dA:dT base pair is thermodynamically destabilized. A detailed analysis reveals that this is due to primarily to unfavorable enthalpy terms, which are dominated by less favorable stacking interactions, resulting from changes in the base electrostatics and electronic dipole–dipole interactions. There is also a net release of electrostricted waters from the duplex. The introduction of the 7-deaza-dA:dT pair into the DDD causes a decreased association of cations, which is reflected in the TΔS term.
Acknowledgments
This work was supported by NIH Grant R01 GM68430 (L.A.M., M.P.S., and B.G.) and R01 GM055237 (M.E.). Funding for the NMR spectrometers was supplied by Vanderbilt University and by NIH Grant RR-05805. Vanderbilt University and the Vanderbilt Center for Structural Biology assisted with the purchase of in-house crystallographic instrumentation. Crystallographic data were collected on the 21-ID-F beamline of the Life Sciences Collaborative Access Team (LS-CAT) at the Advanced Photon Source (Argonne National Laboratory, Argonne, IL). Supporting institutions may be found at http://ls-cat.org/members.html. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Science, under Contract W-31109-Eng-38.
Supporting Information Available
The Supporting Information includes Tables S1, parameters used to calculate differential counterion binding for the dodecamers; S2, parameters used to calculate differential water binding for dodecamers; and Figures S1, stick model and electron density of the crystal structure of 7-deaza-dA modified DDD-1; S2, interactions between Mg2+ ion and DDD-1 duplex; S3, interbase pair parameters (helical rise, roll, twist); S4, comparison of backbone torsion angles (alpha and beta) of DDD and DDD-1; S5, comparison of gamma, delta, epsilon, chi and zeta angles in the crystal structures of the DDD-1; S6, expanded plots of NOESY spectrum of the DDD-1 duplex showing sequential NOEs between the aromatic and anomeric protons and COSY spectrum. This material is available free charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Suhadolnik R. J.Nucleoside Antibiotics; Wiley-Interscience: New York, 1970. [Google Scholar]
- Smulson M. E.; Suhadolnik R. J. J. Biol. Chem. 1967, 242, 2872–2876. [PubMed] [Google Scholar]
- Mizusawa S.; Nishimura S.; Seela F. Nucleic Acids Res. 1986, 14, 1319–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malygin E. G.; Zinoviev V. V.; Petrov N. A.; Evdokimov A. A.; Jen-Jacobson L.; Kossykh V. G.; Hattman S. Nucleic Acids Res. 1999, 27, 1135–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shchyolkina A. K.; Kaluzhny D. N.; Arndt-Jovin D. J.; Jovin T. M.; Zhurkin V. B. Nucleic Acids Res. 2006, 34, 3239–3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramzaeva N.; Michalek E.; Kazimierczuk Z.; Seela F.; Rosemeyer H. Chem. Biodivers. 2007, 4, 2725–2744. [DOI] [PubMed] [Google Scholar]
- Ganguly M.; Wang F.; Kaushik M.; Stone M. P.; Marky L. A.; Gold B. Nucleic Acids Res. 2007, 35, 6181–6195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold B.; Marky L. M.; Stone M. P.; Williams L. D. Chem. Res. Toxicol. 2006, 19, 1402–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anzai K.; Nakamura G.; Suzuki S. J. Antibiot. (Tokyo) 1957, 10, 201–204. [PubMed] [Google Scholar]
- McCarty R. M.; Bandarian V. Chem. Biol. 2008, 15, 790–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarty R. M.; Somogyi A.; Lin G.; Jacobsen N. E.; Bandarian V. Biochemistry 2009, 48, 3847–3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono A.; Ohtani Y.; Sato M.; Ueda T. Nucleic Acids Symp. Ser. 1983, 67–70. [PubMed] [Google Scholar]
- Seela F.; Berg H.; Rosemeyer H. Biochemistry 1989, 28, 6193–6198. [DOI] [PubMed] [Google Scholar]
- Seela F.; Grein T. Nucleic Acids Res. 1992, 20, 2297–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seela F.; Kehne A. Tetrahedron 1985, 41, 5387–5392. [Google Scholar]
- Seela F.; Ramzaeva N.; Leonard P.; Chen Y.; Debelak H.; Feiling E.; Kroschel R.; Zulauf M.; Wenzel T.; Frohlich T.; et al. Nucleosides Nucleotides Nucleic Acids 2001, 20, 1421–1424. [DOI] [PubMed] [Google Scholar]
- Seela F.; Thomas H. Helv. Chim. Acta 1995, 78, 94–108. [Google Scholar]
- Pope L. H.; Shotton M. W.; Forsyth T.; Hughes D. J.; Denny R. C.; Fuller W. Biophys. Chem. 1998, 70, 161–172. [DOI] [PubMed] [Google Scholar]
- Wing R.; Drew H.; Takano T.; Broka C.; Tanaka S.; Itakura K.; Dickerson R. E. Nature 1980, 287, 755–758. [DOI] [PubMed] [Google Scholar]
- Drew H. R.; Wing R. M.; Takano T.; Broka C.; Tanaka S.; Itakura K.; Dickerson R. E. Proc. Natl. Acad. Sci. U.S.A. 1981, 78, 2179–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F.; Li F.; Ganguly M.; Marky L. A.; Gold B.; Egli M.; Stone M. P. Biochemistry 2008, 27, 7147–7157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavaluzzi M. J.; Borer P. N. Nucleic Acids Res. 2004, 32, e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marky L. A.; Breslauer K. J. Biopolymers 1987, 26, 1601–1620. [DOI] [PubMed] [Google Scholar]
- Rentzeperis D.; Marky L. A.; Dwyer T. J.; Geierstanger B. H.; Pelton J. G.; Wemmer D. E. Biochemistry 1995, 34, 2937–2945. [DOI] [PubMed] [Google Scholar]
- Chaires J. B. Biopolymers 1985, 24, 403–419. [DOI] [PubMed] [Google Scholar]
- Spink C. H.; Chaires J. B. Biochemistry 1999, 38, 496–508. [DOI] [PubMed] [Google Scholar]
- Qu X.; Chaires J. B. J. Am. Chem. Soc. 2001, 123, 1–7. [DOI] [PubMed] [Google Scholar]
- Yu H.; Ren J.; Chaires J. B.; Qu X. J. Med. Chem. 2008, 51, 5909–5911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantor C. R.; Schimmel P. R.. Biophysical Chemistry; Freeman: San Francisco, 1980. [Google Scholar]
- Kaushik M.; Suehl N.; Marky L. A. Biophys. Chem. 2007, 126, 154–164. [DOI] [PubMed] [Google Scholar]
- Courtenay E. S.; Capp M. W.; Anderson C. F.; Record M. T. Jr. Biochemistry 2000, 39, 4455–4471. [DOI] [PubMed] [Google Scholar]
- Jeener J.; Meier B. H.; Bachmann P.; Ernst R. R. J. Chem. Phys. 1979, 71, 4546–4553. [Google Scholar]
- Wagner R.; Berger S. J. Magn. Res. A 1996, 123, 119–121. [DOI] [PubMed] [Google Scholar]
- Piantini U.; Sorensen O. W.; Ernst R. R. J. Am. Chem. Soc. 1982, 104, 6800–6801. [Google Scholar]
- Piotto M.; Saudek V.; Sklenar V. J. Biomol. NMR 1992, 6, 661–665. [DOI] [PubMed] [Google Scholar]
- Berger I.; Kang C. H.; Sinha N.; Wolters M.; Rich A. Acta Crystallogr. D 1996, 52, 465–468. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z.; Minor W. Acta Crystallogr. A 1997, 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Acta Crystallogr. D 1994, 50, 760–763. [DOI] [PubMed] [Google Scholar]
- Shui X.; McFail-Isom L.; Hu G. G.; Williams L. D. Biochemistry 1998, 37, 8341–8355. [DOI] [PubMed] [Google Scholar]
- Brunger A. T.; Adams P. D.; Clore G. M.; DeLano W. L.; Gros P.; Grosse-Kunstleve R. W.; Jiang J. S.; Kuszewski J.; Nilges M.; Pannu N. S.; et al. Acta Crystallogr. D 1998, 54, 905–921. [DOI] [PubMed] [Google Scholar]
- Sheldrick G. M.; Schneider T. R. Methods Enzymol. 1997, 277, 319–343. [PubMed] [Google Scholar]
- Cambillau C.; Roussel A.. 1997, Université Aix-Marseille II, Marseille, France. [Google Scholar]
- Ravishankar G.; Swaminathan S.; Beveridge D. L.; Lavery R.; Sklenar H. J. Biomol. Struct. Dyn. 1989, 6, 669–699. [DOI] [PubMed] [Google Scholar]
- Tereshko V.; Minasov G.; Egli M. J. Am. Chem. Soc. 1999, 121, 470–471. [Google Scholar]
- Pardi A.; Tinoco I. Jr. Biochemistry 1982, 21, 4686–4693. [DOI] [PubMed] [Google Scholar]
- Hare D. R.; Wemmer D. E.; Chou S. H.; Drobny G.; Reid B. R. J. Mol. Biol. 1983, 171, 319–336. [DOI] [PubMed] [Google Scholar]
- Reid B. R. Q. Rev. Biophys. 1987, 20, 2–28. [DOI] [PubMed] [Google Scholar]
- Patel D. J.; Shapiro L.; Hare D. Q. Rev. Biophys. 1987, 20, 35–112. [DOI] [PubMed] [Google Scholar]
- Boelens R.; Scheek R. M.; Dijkstra K.; Kaptein R. J. Magn. Reson. 1985, 62, 378–386. [Google Scholar]
- Hud N. V.2009, In Nucleic Acid-Metal Ion Interactions; Hud N. V., Ed.; RSC Publishing: Cambridge, UK. [Google Scholar]
- Pardi A.; Morden K. M.; Patel D. J.; Tinoco I. Jr. Biochemistry 1982, 21, 6567–6574. [DOI] [PubMed] [Google Scholar]
- Tjandra N.; Tate S.; Ono A.; Kainosho M.; Bax A. J. Am. Chem. Soc. 2000, 26, 6190–6200. [Google Scholar]
- Makhatadze G. I.; Privalov P. L. J. Mol. Biol. 1990, 213, 375–384. [DOI] [PubMed] [Google Scholar]
- Hagerman P. J. Nature 1986, 321, 449–450. [DOI] [PubMed] [Google Scholar]
- Teitelbaum H.; Englander S. W. J. Mol. Biol. 1975, 92, 79–92. [DOI] [PubMed] [Google Scholar]
- Mandal C.; Kallenbach N. R.; Englander S. W. J. Mol. Biol. 1979, 135, 391–411. [DOI] [PubMed] [Google Scholar]
- Englander S. W.; Kallenbach N. R. Q. Rev. Biophys. 1984, 16, 521–655. [DOI] [PubMed] [Google Scholar]
- Leroy J. L.; Kochoyan M.; Huynh-Dinh T.; Gueron M. J. Mol. Biol. 1988, 200, 223–238. [DOI] [PubMed] [Google Scholar]
- Folta-Stogniew E.; Russu I. M. Biochemistry 1996, 35, 8439–8449. [DOI] [PubMed] [Google Scholar]
- Benight A. S.; Schurr J. M.; Flynn P. F.; Reid B. R.; Wemmer D. E. J. Mol. Biol. 1988, 200, 377–399. [DOI] [PubMed] [Google Scholar]
- Gasan A. I.; Maleev V. Y.; Semenov M. A. Stud Biophys 1990, 136, 171–178. [Google Scholar]
- Marky L. A.; Kupke D. W. Methods Enzymol. 2000, 323, 419–441. [DOI] [PubMed] [Google Scholar]
- Egli M.; Tereshko V.; Teplova M.; Minasov G.; Joachimiak A.; Sanishvili R.; Weeks C. M.; Miller R.; Maier M. A.; An H.; Dan Cook P.; Manoharan M. Biopolymers 1998, 48, 234–252. [DOI] [PubMed] [Google Scholar]
- Shui X.; Sines C. C.; McFail-Isom L.; VanDerveer D.; Williams L. D. Biochemistry 1998, 37, 16877–16887. [DOI] [PubMed] [Google Scholar]
- Minasov G.; Tereshko V.; Egli M. J. Mol. Biol. 1999, 291, 83–99. [DOI] [PubMed] [Google Scholar]
- McFail-Isom L.; Sines C. C.; Williams L. D. Curr. Opin. Struct. Biol. 1999, 9, 298–304. [DOI] [PubMed] [Google Scholar]
- Williams L. D.; Maher L. J. III. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 497–521. [DOI] [PubMed] [Google Scholar]
- Woods K. K.; McFail-Isom L.; Sines C. C.; Howerton S. B.; Stephens R. K.; Williams L. D. J. Am. Chem. Soc. 2000, 122, 1546–1547. [Google Scholar]
- Howerton S. B.; Sines C. C.; VanDerveer D.; Williams L. D. Biochemistry 2001, 40, 10023–10031. [DOI] [PubMed] [Google Scholar]
- Egli M. Chem. Biol. 2002, 9, 277–286. [DOI] [PubMed] [Google Scholar]
- Woods K. K.; Lan T.; McLaughlin L. W.; Williams L. D. Nucleic Acids Res. 2003, 31, 1536–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egli M.; Tereshko V. ACS Symp. Ser. 2004, 884, 87–109. [Google Scholar]
- Krakauer H. Biopolymers 1972, 11, 811–828. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.