

Abstract
The c-MET (mesenchymal–epithelial transition factor) receptor tyrosine kinase is an exciting novel drug target in view of its key role in oncogenesis, as well as its association with disease prognosis in a number of malignancies. Several drugs targeting c-MET are currently showing promise in clinical trials and will hopefully validate positive observations from preclinical studies. The potential efficacy of these different therapeutic agents is expected to be influenced by the mechanism of aberrant hepatocyte growth factor (HGF)/c-MET signaling pathway activation in a particular cancer, but presents a promising strategy for cancer treatment either as a single agent or as part of a combination therapeutic approach. However, there is an ongoing need to improve and accelerate the transition of preclinical research into improved therapeutic strategies for patients with cancer. The main challenges facing the development of HGF/c-MET-targeted agents for cancer treatment include the discovery of rationally designed anticancer drugs and combination strategies, as well as the validation of predictive biomarkers. This paper discusses these issues, with a particular focus on future directions in the evaluation of c-MET-driven malignancies.
Keywords: c-MET, drug development, targeted therapy, treatment resistance, patient selection, biomarkers
Introduction
Recent research has demonstrated that the c-MET (mesenchymal–epithelial transition factor) receptor tyrosine kinase and its ligand hepatocyte growth factor (HGF) (also known as the ‘HGF/MET axis’) regulate a range of cellular functions [Yap and de Bono, 2010; Ma et al. 2003; Trusolino and Comoglio, 2002; Bladt et al. 1995; Schmidt et al. 1995].
Under normal physiological conditions, HGF-induced c-MET tyrosine kinase activation is tightly regulated by paracrine ligand delivery, ligand activation at the target cell surface, and ligand-activated receptor internalization and degradation [Cecchi et al. 2010]. The importance of the HGF/c-MET pathway in the control of tissue homeostasis is supported by the well established protective activity of HGF in several degenerative diseases, including progressive nephropathies [Okada and Kalluri, 2005; Liu and Yang, 2006], liver cirrhosis [Ueki et al. 1999] and lung fibrosis [Watanbe et al. 2005]. However, activated c-MET signaling caused by deregulation of normal cellular functions is clearly implicated in oncogenesis, leading to cell growth, proliferation, angiogenesis, invasion, survival, and metastasis [Liu et al. 2008; Ma et al. 2008; Birchmeier et al. 2003]. Activation of the c-MET signaling pathway can occur via activating mutations, overexpression of the kinase itself or its ligand HGF, or by autocrine, paracrine, or endocrine loop regulation [Cecchi et al. 2010].
c-MET as a key target in oncological drug development
Clinically, c-MET has gained considerable interest through its apparent deregulation by overexpression or mutation in various cancers, including non-small cell lung cancer (NSCLC) [Ma et al. 2003, 2005; Kong-Beltran et al. 2006]. Overexpression of c-MET, along with HGF, also appears indicative of an increased aggressiveness of tumors [Boccaccio and Comoglio, 2006]. The deregulation of c-MET identifies it as an important therapeutic target in the development of future anticancer therapies. There is an increasing body of evidence that supports c-MET as a key target in oncology, for example through the development of small molecules or biological inhibitors. In addition, inhibition of c-MET affects downstream signal transduction with resulting biological consequences in tumor cells [Christensen et al. 2005]. The mutation or gene amplification of MET in selected clinical populations also suggests that certain patients may be exquisitely sensitive to targeted therapies that inhibit the HGF/MET axis [Christensen et al. 2005].
c-MET also has prognostic implications in patients with cancer [Beau-Faller et al. 2008; Boccaccio and Comoglio, 2006; Cheng et al. 2005]. Firstly, overexpression of circulating c-MET in patients with NSCLC has been significantly associated with early tumor recurrence [Cheng et al. 2005] and patients with adenocarcinoma and MET amplification have also demonstrated a trend for poor prognosis [Beau-Faller et al. 2008]. Cappuzzo and colleagues have provided clear evidence that increased MET gene copy number is a negative prognostic factor, further supporting anti-c-MET therapeutic strategies in this disease [Cappuzzo et al. 2009]. Of note, data from the same study indicated that epidermal growth factor receptor (EGFR) gene gain has no prognostic function in NSCLC, supporting its role as a predictive factor for improved survival in patients with NSCLC exposed to EGFR tyrosine kinase inhibitors (TKIs) [Cappuzzo et al. 2009].
Resistance to established agents
c-MET is involved in resistance to established agents, such as vascular endothelial growth factor receptor (VEGFR) and EGFR inhibitors. For example, the c-MET receptor and VEGFR have been found to cooperate to promote tumor survival [Eder et al. 2009]. Furthermore, c-MET has additional roles in tumor angiogenesis; firstly, as an independent angiogenic factor and also one that may interact with angiogenic proliferation and survival signals promoted through VEGF and other angiogenic proteins [Eder et al. 2009]. Combined VEGF and HGF/c-MET signaling has also been reported to have a greater effect on the prevention of endothelial cell apoptosis, formation of capillaries in vivo, and the increase of microvessel density within tumors [Boccaccio and Comoglio, 2006]. For EGFR, c-MET has been implicated in cooperating as a mediator of EGFR tyrosine phosphorylation and cell growth in the presence of EGFR inhibitors [Mueller et al. 2008]. MET amplification is responsible for EGFR-TKI acquired resistance in approximately 20% of patients [Bean et al. 2007; Engelman et al. 2007]. Recent findings from Pillay and colleagues suggest that inhibition of a dominant oncogene by targeted therapy can also alter the hierarchy of receptor tyrosine kinases, resulting in rapid therapeutic resistance [Pillay et al. 2009].
Such findings appear to suggest that c-MET inhibition, either alone or in combination with an EGFR inhibitor, may confer clinical benefit in the setting of EGFR inhibitor resistance. Indeed, available data imply that c-MET may be a clinically relevant therapeutic target for some patients with acquired resistance to gefitinib or erlotinib, particularly given that MET gene amplification occurs independently of EGFRT790M mutations [Bean et al. 2007]. The presence of MET gene amplification in combination with gain-of-function drug-sensitive EGFR mutations could together lead to cellular changes that confer enhanced fitness to cells bearing both alterations [Bean et al. 2007]. However, other mechanisms could contribute to disease progression in such patients. As the mechanism of interaction between HGF/c-MET and resistance remains unclear, further research into crosstalk and balance between these two signal pathways remains critical and necessary for the development of novel anticancer therapies.
Plasticity in cancer cell ‘addiction’
When considering the rational identification of responsive tumors, previous experience with EGFR TKIs has demonstrated that they are only efficacious in a small subset of tumors that exhibit genetic alterations of the receptor itself [Sharma et al. 2007]. However, research has also shown that cultured cell lines containing the same EGFR genetic lesions present in human tumors can undergo cell cycle arrest or apoptosis when subjected to EGFR inhibition, even under otherwise optimal conditions [Sharma et al. 2007]. This phenomenon, termed ‘oncogene addiction’, applies to all clinical scenarios in which cancer cells appear to depend on a single overactive oncogene for their proliferation and survival [Sharma et al. 2007; Sharma and Settleman, 2007]. For c-MET, further consideration needs to be given to the fact that genetic alterations of the kinase can induce oncogene addiction and therefore possibly aid prediction of therapeutic responsiveness. Importantly, research from Comoglio and colleagues has highlighted that preclinical investigations of developmental c-MET inhibitors appear to utilize a vast array of differing cell lines, most of which tend not to be genetically characterized [Comoglio et al. 2008]. Clearly, to enable identification and recruitment of potentially responsive patients in future studies, the rational selection of genetically defined cell lines will need to become mandatory, in order to lead to the development of reliable in vitro models for the testing of c-MET inhibition. Future models will need to be able to clearly display signaling abnormalities of c-MET and also to respond to c-MET inactivation with a distinct and measurable phenotypic readout.
In addition to oncogene addiction, available data suggest that c-MET can act as an ‘oncogene expedient’ even in the absence of genetic alterations [Comoglio et al. 2008]. Such findings indicate that c-MET might potentiate the effect of other oncogenes, promote malignant progression and participate in tumor angiogenesis [Comoglio et al. 2008]. In order to identity potentially responsive tumors, the different roles that c-MET can play in malignant transformation and progression warrant further research.
Ongoing development of c-MET inhibitors
The prevalence of HGF/c-MET pathway activation in human malignancies has driven a rapid growth in cancer drug development programs, with several new drugs targeting c-MET showing great promise. Several c-MET inhibitors are now under evaluation in clinical trials (Table 1), and the interest around these compounds has consistently increased since an interaction between EGFR and c-MET was observed [Bonine-Summers et al. 2007]. Clinical trials with these agents will hopefully validate positive observations from preclinical studies. c-MET inhibitor agents under development include compounds that directly inhibit HGF and/or its binding to c-MET, antibodies targeted at c-MET, and small-molecule c-MET TKIs. The potential efficacy of each of these different therapeutic agents is likely to be influenced by the mechanism of aberrant HGF/c-MET signaling pathway activation in a particular cancer but will also hopefully offer a promising new strategy for cancer treatment, either alone or as part of a combination therapeutic approach.
Table 1.
c-MET inhibitors under current development.
| Agent | Company | MOA | Phase | Reference |
|---|---|---|---|---|
| AMG102 | Amgen, CA, USA | Anti-HGF antibody | II | [Gordon et al. 2010] |
| Tivantinib (ARQ 197) | ArQule, MA, USA; Daiichi Sankyo, Tokyo, Japan | Selective c-MET TKI | III | [Yap et al. 2011; Schiller et al. 2010] |
| Cabozantinib (XL184) | Exelixis, CA, USA; Bristol- Myers Squibb, NY, USA | Nonselective c-MET, VEGFR2 and RET TKI | II | [Kurzrock et al. 2011] |
| MetMAb | Genentech, CA, USA | Anti-c-MET antibody | II | [Spigel et al. 2011] |
c-MET, mesenchymal–epithelial transition factor; HGF, hepatocyte growth factor; MOA, mechanism of action; RET, rearranged during transfection; TKI, tyrosine kinase inhibitor; VEGFR2, vascular endothelial growth factor (VEGF) receptor-2.
Future challenges
There remains an urgent need to improve and accelerate the transition of preclinical research into improved therapeutic strategies for patients with cancer [de Bono and Ashworth, 2010]. The main challenges facing the effective use of HGF/c-MET targeted antagonists for cancer treatment include optimal patient selection, diagnostic and pharmacodynamic biomarker development, and the identification and testing of rationally designed anticancer drugs and combination strategies. If the ongoing development of c-MET inhibitors is to result in a clinically useful therapeutic approach, an absolute requirement is the definition of a target patient population and a practical but analytically validated method to identify them in a clinical context [de Bono and Ashworth, 2010].
Although traditional drug development has involved a ‘compound-to-trial’ process, there is increasing evidence that this should now change to a ‘biology-to-trial’ approach, starting with unraveling of the fundamental mechanisms of cancer targets, which may then drive initial drug discovery and subsequent clinical studies [Yap et al. 2010]. The ‘one-size-fits-all’ approach currently in use does not take into account the now well established patient-to-patient variation that exists in the molecular drivers of both cancer and drug sensitivity [Janne et al. 2009; McDermott and Settleman, 2009]. A new paradigm is now emerging that involves the use of customized, adaptive, hypothesis-testing early trial designs, which incorporate analytically validated and clinically qualified biomarkers from the earliest possible stage (Figure 1) [Yap et al. 2010]. This preferred scenario recognizes that the new generation of molecularly targeted drugs has the potential for personalized medicine and the possibility of more efficacious and less toxic antitumor therapies in patients who have defined molecular aberrations. In this scenario, there is an initial need to focus on the biology of the disease, identify a possible therapeutic target, and then understand how a molecularly targeted approach could offer therapeutic benefit.
Figure 1.

The shifting focus of old versus new phase I clinical trial designs. PD, pharmacodynamics; PK, pharmacokinetics; MTD, maximum tolerated dose. Reprinted by permission from Macmillan Publishers Ltd: NATURE REVIEWS CANCER, Timothy A. Yap, et al. 2010a;10:514–523, © 2010.
Key molecular targets or pathways which are vital to certain cancers, or that present opportunities for synthetic lethality, should be actively pursued and dissected to improve our understanding of these essential pathways and to identify predictive biomarkers that could be integrated early in the drug discovery process. A strong biological basis clearly already exists for c-MET as a therapeutic target. However, there is an ongoing need to identify an altered molecular target which will provide a therapeutic window and therefore a clear basis for selective tumor cell cytotoxicity with absolute or relative sparing of normal cells [de Bono et al. 2003]. Although MET amplification or mutations have been demonstrated in a range of cancers in preclinical studies, these have, to date, not been shown to strongly predict which patients will respond to c-MET inhibitors in the clinic [Yap and de Bono, 2010; Comoglio et al. 2008].
Translating results from cancer genome mapping into clinical use will necessitate the development of analytically validated biomarker assays that can be clinically validated as potential predictors of benefit from anticancer therapies [de Bono and Ashworth, 2010]. These biomarkers will support a personalized approach as they could be used to examine intra- and inter-patient tumor molecular heterogeneity and assist selection of an optimal anticancer therapy for each individual patient. Moreover, these biomarkers could be increasingly used as intermediate endpoints of response. The upfront use and testing of putative predictive biomarkers in early clinical trial programs could minimize any possible need for retrospective subgroup dredging for predictive biomarkers in later phase trials carried out in unselected populations [Yap et al. 2010].
Selecting patients based on molecular predictors may help minimize the risk of late and costly drug attrition due to disease heterogeneity, accelerate patient benefit, and could also accelerate the drug approval process, which currently remains slow and inefficient. However, care should be taken when using predictive biomarkers to select patients since the potential beneficial effects of the targeted therapy in a more broadly defined patient population may be missed.
c-MET inhibitors in combination with other agents
Several different therapeutic strategies, aimed at inhibiting HGF/c-MET signaling, are currently in development, but it is still unclear if these agents will be most effective as distinct monotherapies or in combination with other agents. The combination of anti-c-MET therapeutic agents with either signal transduction inhibitors [ErbB family or mammalian target of rapamycin (mTOR) inhibitors] or with cytotoxic chemotherapies has been evaluated in preclinical studies which have provided insight into the rational development of combined therapeutic strategies for future clinical trial evaluation. Several studies have focused on the combination of c-MET inhibitors and agents targeting ErbB family members, with the rationale for this approach based on evidence of crosstalk between c-MET and other EGFR family members [Stommel et al. 2007; Jo et al. 2000]. In addition, cancers codependent on both c-MET and EGFR signaling have also been identified [Engelman et al. 2007], with MET amplification detected in patients with NSCLC who have clinically developed resistance to the EGFR inhibitors gefitinib or erlotinib [Bean et al. 2007; Engelman et al. 2007]. Several clinical trials are currently under way, which aim to determine if the combination of c-MET TKIs with EGFR, VEGF, or chemotherapy is a clinically effective therapeutic approach (Table 2).
Table 2.
Combination studies: c-MET inhibitor plus other pathways.
| Combination | Phase | Reference |
|---|---|---|
| EGFR | ||
| Tivantinib ± erlotinib (ArQule, MA, USA; Daiichi Sankyo, Tokyo, Japan) | III | [Sandler et al. 2011] |
| MetMAb ± erlotinib (OAM4558g) (Genentech, South San Francisco, CA, USA) | II | [Spigel et al. 2011] |
| Ficlatuzumab (AV-299) ± gefitinib (Aveo Pharmaceuticals, MA, USA) | II | [Tan et al. 2011] |
| VEGF | ||
| Rilotumumab (AMG 102) + bevacizumab or motesanib (Amgen, CA, USA) | Ib | [Rosen et al. 2010] |
| Tivantinib + sorafenib (ArQule, MA, USA; Daiichi Sankyo, Tokyo, Japan) | I | [Adjei et al. 2011] |
| Chemotherapy | ||
| Crizotinib + pemetrexed/docetaxel (Pfizer, NY, USA) | III | [clinicaltrials.gov (trial no. NCT00932893)] |
| Tivantinib + gemcitabine (ArQule, MA, USA; Daiichi Sankyo, Tokyo, Japan) | I | [clinicaltrials.gov (trial no. NCT00874042)] |
| Tivantinib + irinotecan and cetuximab (ArQule, MA, USA; Daiichi Sankyo, Tokyo, Japan) | I/II | [Bessudo et al. 2011] |
c-MET, mesenchymal–epithelial transition factor; EGFR, epidermal growth factor receptor; VEGF, vascular endothelial growth factor.
Because c-MET activation leads to increased downstream signaling through a variety of different pathways, a combined approach that inhibits c-MET and its known downstream signaling intermediates could possibly enhance therapeutic efficacy. This approach may also be effective in cancers in which multiple receptors are concurrently activated – such as by EGFR – because these receptors typically activate the same downstream signaling proteins [Toschi and Janne, 2008]. Preclinical studies exploring a combination of anti-c-MET therapeutic agents with mTOR inhibitors have also demonstrated increased growth suppression compared with mTOR inhibitors alone [Ma et al. 2005].
Chemotherapy remains the mainstay of treatment for several malignancies, even though advances in the molecular knowledge of cancer continue to support the development of selective targeted compounds. However, the use of conventional chemotherapy is often limited by de novo or acquired resistance, typically resulting from increased growth factor receptor signaling [Dai et al. 2005; Knuefermann et al. 2003]. These observations have prompted growth factor receptor inhibitors to be evaluated in combination with chemotherapy. Successful clinically validated examples of this approach include cetuximab, an anti-EGFR antibody, in colorectal cancer [Cunningham et al. 2004] and trastuzumab in patients with ERBB2-amplified breast cancer [Slamon et al. 2001]. Emerging preclinical data suggest that inhibitors of the HGF/c-MET signaling pathway may also be effective in combination with chemotherapy [Lasagna et al. 2006; Bowers et al. 2000].
The Pharmacologic Audit Trail
Pharmacodynamic and pharmacokinetic data together allow the construction of a framework, known as the ‘pharmacologic audit trail’ (PhAT), for rational decision making in clinical trials [Sarker and Workman, 2007; Workman, 2003, 2002]. The PhAT allows all the key stages in drug development to be linked and interpreted in relation to measured parameters (such as pharmacodynamic and pharmacokinetic parameters) and provides a stepwise ‘audit’ to assess the risk of failure during the development of a novel compound at any particular stage. An updated PhAT has recently been developed to reflect the evolving drug discovery and development landscape, implementing the evaluation of potential predictive assays earlier in the drug development process and strategies to reverse resistance mechanisms (Figure 2) [Yap et al. 2010]. This updated version recommends inclusion of the identification and initial clinical qualification of robust predictive biomarker assays for patient selection early in the drug development process. The inclusion of intermediate endpoint biomarkers, which should be identified and studied in the audit trail as early predictors of antitumor activity, is also recommended.
Figure 2.
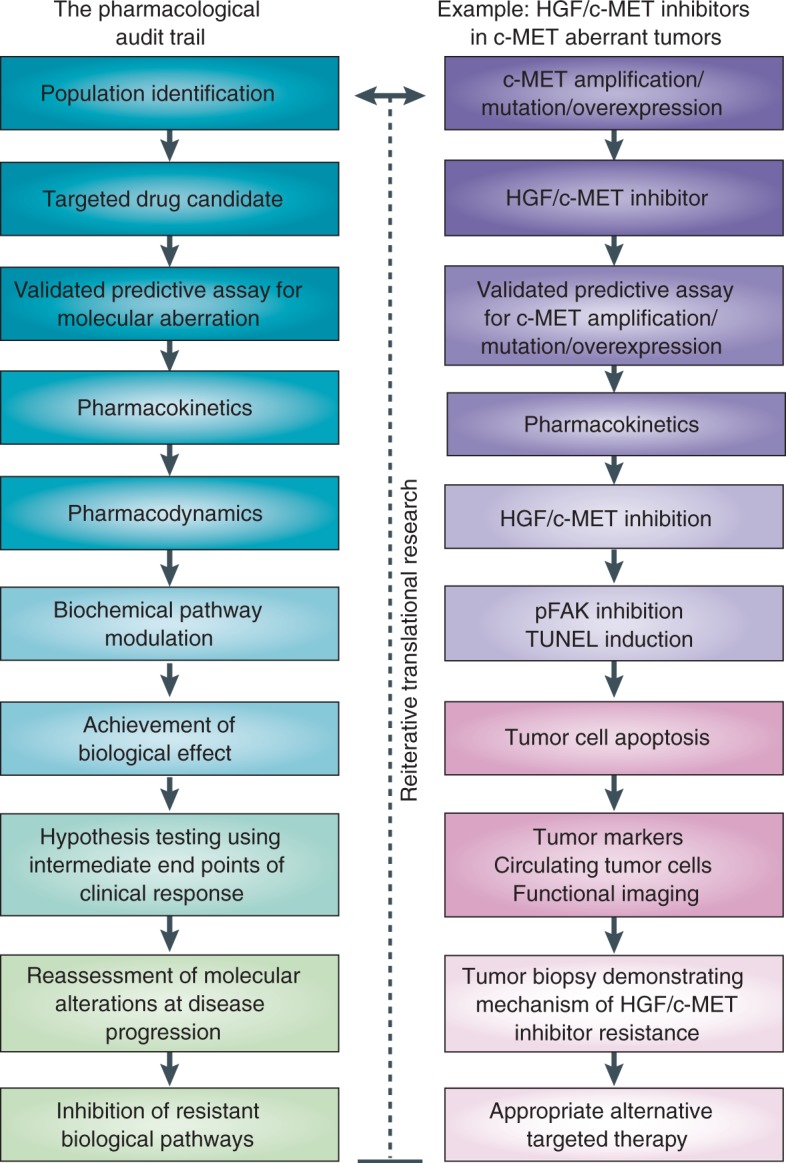
Pharmacological audit trail. PARP, poly(ADP)-ribose polymerase; PBMC, peripheral blood mononuclear cell; PhAT, pharmacological audit trail. Adapted by permission from Macmillan Publishers Ltd: NATURE REVIEWS CANCER, Timothy A. Yap, et al. 2010a;10:514–523. © 2010.
Because there is an ongoing need to acquire more data from preclinical models on the relationship of anticancer drug antitumor activity and the required degree and duration of target blockade, careful assessment is warranted as to whether this is safely achievable in clinical trials and the PhAT should be seen as a useful tool.
Conclusions
Optimal methods for the assessment of HGF/c-MET overexpression or MET amplification have yet to be determined. Traditional histopathological diagnosis remains important when evaluating the extent of phenotypic aggressiveness, but personalized molecular diagnosis is needed to understand whether a tumor in one specific patient carries a particular genetic alteration that could be targeted by a particular therapy. In the case of c-MET, the current challenge is to identify the genetically defined responsive patient subsets that could benefit from c-MET inhibition and therefore enable appropriate patient selection strategies to be implemented in future clinical studies. This calls for a vast preclinical strategy of tumor categorization based on genetic makeup, responsiveness to c-MET inhibition and follow-up validation of surrogate indicators of c-MET activity. Treatment selection should be driven by a detailed understanding of the genetics and biology of the patient and their cancer. There is also increasing evidence for the traditional route of drug development and registration to be adapted for the development of molecularly targeted agents. Several different c-MET inhibitors are currently in development, each focusing on one or more of the steps that regulate c-MET activation. Finally, understanding the other key activated signaling pathways that occur concurrently with HGF/c-MET activation will be critical in the rational development of combination therapeutic strategies.
Acknowledgements
Matthew Joynson, a medical writer, assisted with the styling of this manuscript. The authors wrote and revised the main draft of the article.
Funding
Editorial assistance was funded by Daiichi Sankyo Europe GmbH.
Conflict of interest statement
Professor Johann S. de Bono has received honoraria from Daiichi Sankyo Europe GmbH for speaking at scientific symposia. Dr Timothy A. Yap and Professor Johann S. de Bono have been involved in conducting trials of c-MET inhibitors sponsored by ArQule, Inc. and Johnson and Johnson. Dr Timothy A. Yap has served as a consultant for Merck. Professor Johann S. de Bono has served as a consultant for ArQule, Inc., Johnson and Johnson, Genentech, Merck, Pfizer Oncology, AstraZeneca, Exelixis, and GlaxoSmithKline.
References
- Adjei A.A., Sosman J.A., Martell R.E., Dy G.K., Goff L.W., Ma W.W., et al. (2011) Efficacy in selected tumor types in a phase I study of the c-MET inhibitor ARQ 197 in combination with sorafenib. J Clin Oncol 29(Suppl): abstract 3034 [Google Scholar]
- Bean J., Brennan C., Shih J.-Y., Riely G., Viale A., Wang L., et al. (2007) MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A 104: 20932–20937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beau-Faller M., Ruppert A.M., Voegeli A.C., Neuville A., Meyer N., Guerin E., et al. (2008) MET gene copy number in non-small cell lung cancer: Molecular analysis in a targeted tyrosine kinase inhibitor naïve cohort. J Thorac Oncol 3: 331–339 [DOI] [PubMed] [Google Scholar]
- Bessudo A., Bendell J.C., Gabrail N.Y., Kopp M.V., Mueller L., Hart L.L., et al. (2011) Phase I results of the randomized, placebo-controlled, phase I/II study of the novel oral c-MET inhibitor, tivantinib (ARQ 197), irinotecan (CPT-11), and cetuximab in patients with wild-type KRAS metastatic colorectal cancer who have received front-line systemic therapy. J Clin Oncol 29(Suppl): abstract 3582 [Google Scholar]
- Birchmeier C., Birchmeier W., Gherardi E., VandeWoude G.F. (2003) Met, metastasis, motility and more. Nat Rev Mol Cell Biol 4: 915–925 [DOI] [PubMed] [Google Scholar]
- Bladt F., Riethmacher D., Isenmann S., Aguzzi A., Birchmeier C. (1995) Essential role for the c-MET receptor in the migration of myogenic precursor cells into the limb bud. Nature 376: 768–771 [DOI] [PubMed] [Google Scholar]
- Boccaccio C., Comoglio P.M. (2006) Invasive growth: a MET-driven genetic programme for cancer and stem cells. Nat Rev Cancer 6: 637–645 [DOI] [PubMed] [Google Scholar]
- Bonine-Summers A.R., Aakre M.E., Brown K.A., Arteaga C.L., Pietenpol J.A., Moses H.L., et al. (2007) Epidermal growth factor receptor plays a significant role in hepatocyte growth factor mediated biological responses in mammary epithelial cells. Cancer Biol Ther 6: 561–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers D.C., Fan S., Walter K.A., Abounader R., Williams J.A., Rosen E.M., et al. (2000) Scatter factor/hepatocyte growth factor protects against cytotoxic death in human glioblastoma via phosphatidylinositol 3-kinase- and AKT-dependent pathways. Cancer Res 60: 4277–4283 [PubMed] [Google Scholar]
- Cappuzzo F., Marchetti A., Skokan M., Rossi E., Gajapathy S., Felicioni L., et al. (2009) Increased MET gene copy number negatively affects survival of surgically resected non-small-cell lung. J Clin Oncol 27: 1667–1674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cecchi F., Rabe D.C., Bottaro D.P. (2010) Targeting the HGF/Met signalling pathway in cancer. Eur J Cancer 46: 1260–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng T.L., Chang M.Y., Huang S.Y., Sheu C.C., Kao E.L., Cheng Y.J., et al. (2005) Overexpression of circulating c-met messenger RNA is significantly correlated with nodal stage and early recurrence in non-small cell lung cancer. Chest 128: 1453–1460 [DOI] [PubMed] [Google Scholar]
- Christensen J.G., Burrows J., Salgia R. (2005) c-Met as a target for human cancer and characterization of inhibitors for therapeutic intervention. Cancer Lett 225: 1–26 [DOI] [PubMed] [Google Scholar]
- Clinicaltrials.gov (trial no. NCT00874042). Dose escalation study of ARQ 197 in combination with gemcitabine in adult patients with advanced solid tumors. http://clinicaltrials.gov/ct2/show/NCT00874042?term=NCT00874042&rank=1 (accessed 19 July 2011)
- Clinicaltrials.gov (trial no. NCT00932893). An investigational drug, PF-02341066 is being studied versus standard of care in patients with advanced non-small cell lung cancer with a specific gene profile involving the anaplastic lymphoma kinase (ALK) gene. http://clinicaltrials.gov/ct2/show/NCT00932893?term=NCT00932893&rank=1 (accessed 19 July 2011)
- Comoglio P.M., Giordano S., Trusolino L. (2008) Drug development of MET inhibitors: targeting oncogene addiction and expedience. Nature Rev Drug Discov 7: 504–516 [DOI] [PubMed] [Google Scholar]
- Cunningham D., Humblet Y., Siena S., Khayat D., Bleiberg H., Santoro A., et al. (2004) Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med 351: 337–345 [DOI] [PubMed] [Google Scholar]
- Dai Q., Ling Y.H., Lia M., Zou Y.Y., Kroog G., Iwata K.K., et al. (2005) Enhanced sensitivity to the HER1/epidermal growth factor receptor tyrosine kinase inhibitor erlotinib hydrochloride in chemotherapy-resistant tumor cell lines. Clin Cancer Res 11: 1572–1578 [DOI] [PubMed] [Google Scholar]
- de Bono J.S., Ashworth A. (2010) Translating cancer research into targeted therapeutics. Nature 467: 543–549 [DOI] [PubMed] [Google Scholar]
- de Bono J.S., Tolcher A.W., Rowinsky E.K. (2003) The future of cytotoxic therapy: selective cytotoxicity based on biology is the key. Breast Cancer Res 5: 154–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eder J.P., Vande Woude G.F., Boerner S.A., LoRusso P.M. (2009) Novel therapeutic inhibitors of the c-Met signaling pathway in cancer. Clin Cancer Res 15: 2207–2214 [DOI] [PubMed] [Google Scholar]
- Engelman J.A., Zejnullahu K., Mitsudomi T., Song Y., Hyland C., Park J.O., et al. (2007) MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 316: 1039–1043 [DOI] [PubMed] [Google Scholar]
- Gordon M.S., Sweeney C.S., Mendelson D.S., Eckhardt S.G., Anderson A., Beaupre D.M., et al. (2010) Safety, pharmacokinetics, and pharmacodynamics of AMG 102, a fully human hepatocyte growth factor-neutralizing monoclonal antibody, in a first-in-human study of patients with advanced solid tumors. Clin Cancer Res 16: 699–710 [DOI] [PubMed] [Google Scholar]
- Janne P.A., Gray N., Settleman J. (2009) Factors underlying sensitivity of cancers to small-molecule kinase inhibitors. Nature Rev Drug Discov 8: 709–723 [DOI] [PubMed] [Google Scholar]
- Jo M., Stolz D.B., Esplen J.E., Dorko K., Michalopoulos G.K., Strom G.C. (2000) Cross-talk between epidermal growth factor receptor and c-Met signal pathways in transformed cells. J Biol Chem 275: 8806–8811 [DOI] [PubMed] [Google Scholar]
- Knuefermann C., Lu Y., Liu B., Jin W., Liang K., Wu L., et al. (2003) HER2/PI-3K/Akt activation leads to a multidrug resistance in human breast adenocarcinoma cells. Oncogene 22: 3205–3212 [DOI] [PubMed] [Google Scholar]
- Kong-Beltran M., Seshagiri S., Zha J., Zhu W., Bhawe K., Mendoza N., et al. (2006) Somatic mutations lead to an oncogenic deletion of met in lung cancer. Cancer Res 66: 283–289 [DOI] [PubMed] [Google Scholar]
- Kurzrock R., Sherman S.I., Ball D.W. (2011) Activity of XL184 (cabozantinib), an oral tyrosine kinase inhibitor, in patients with medullary thyroid cancer. J Clin Oncol 29: 2660–2666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasagna N., Fantappie O., Solazzo M., Morbidelli L., Marchetti S., Cipriani G., et al. (2006) Hepatocyte growth factor and inducible nitric oxide synthase are involved in multidrug resistance-induced angiogenesis in hepatocellular carcinoma cell lines. Cancer Res 66: 2673–2682 [DOI] [PubMed] [Google Scholar]
- Liu X., Yao W., Newton R.C., Scherle P.A. (2008) Targeting the c-MET signaling pathway for cancer therapy. Expert Opin Investig Drugs 17: 997–1011 [DOI] [PubMed] [Google Scholar]
- Liu Y., Yang J. (2006) Hepatocyte growth factor: new arsenal in the fights against renal fibrosis?. Kidney Int 70: 238–240 [DOI] [PubMed] [Google Scholar]
- Ma P.C., Jagadeeswaran R., Jagadeesh S., Tretiakova M.S., Nallasura V., Fox E.A., et al. (2005) Functional expression and mutations of c-Met and its therapeutic inhibition with SU11274 and small interfering RNA in non-small cell lung cancer. Cancer Res 65: 1479–1488 [DOI] [PubMed] [Google Scholar]
- Ma P.C., Kijima T., Maulik G., Fox E.A., Sattler M., Griffin J.D., et al. (2003) c-MET mutational analysis in small cell lung cancer: novel juxtamembrane domain mutations regulating cytoskeletal functions. Cancer Res 63: 6272–6281 [PubMed] [Google Scholar]
- Ma P.C., Schaefer E., Christensen J.G., Salgia R. (2005) A selective small molecule c-MET inhibitor, PHA665752, cooperates with rapamycin. Clin Cancer Res 11: 2312–2319 [DOI] [PubMed] [Google Scholar]
- Ma P.C., Tretiakova M.S., MacKinnon A.C., Ramnath N., Johnson C., Dietrich S., et al. (2008) Expression and mutational analysis of MET in human solid cancers. Genes Chromosomes Cancer 47: 1025–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott U., Settleman J. (2009) Personalized cancer therapy with selective kinase inhibitors: an emerging paradigm in medical oncology. J Clin Oncol 27: 5650–5659 [DOI] [PubMed] [Google Scholar]
- Mueller K.L., Hunter L.A., Ethier S.P., Boerner J.L. (2008) Met and c-Src cooperate to compensate for loss of epidermal growth factor receptor kinase activity in breast cancer cells. Cancer Res 68: 3314–3322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada H., Kalluri R. (2005) Cellular and molecular pathways that lead to progression and regression of renal fibrogenesis. Curr Mol Med 5: 467–474 [DOI] [PubMed] [Google Scholar]
- Pillay V., Allaf L., Wilding A.L., Donoghue J.F., Court N.W., Greenall S.A., et al. (2009) The plasticity of oncogene addiction: implications for targeted therapies directed to receptor tyrosine kinases. Neoplasia 11: 448–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen P.J., Sweeney C.J., Park D.J., Beaupre D.M., Deng H., Leitch I.M., et al. (2010) A phase Ib study of AMG 102 in combination with bevacizumab or motesanib in patients with advanced solid tumors. Clin Cancer Res 16: 2677–2687 [DOI] [PubMed] [Google Scholar]
- Sandler A., Schiller J.H., Hirsh V., Sequist L.V., Soria J., Von Pawel J., et al. (2011) A phase III, randomized, double-blind, placebo-controlled study of erlotinib plus ARQ 197 versus erlotinib plus placebo in previously treated subjects with locally advanced or metastatic, nonsquamous, non-small cell lung cancer (NSCLC). J Clin Oncol 29(Suppl): abstract TPS217 [DOI] [PubMed] [Google Scholar]
- Sarker D., Workman P. (2007) Pharmacodynamic biomarkers for molecular cancer therapeutics. Adv Cancer Res 96: 213–268 [DOI] [PubMed] [Google Scholar]
- Schiller J.H., Akerley W.L., Brugger W., Ferrari D., Garmey E.G., Gerberet D.E., et al. (2010) Results from ARQ 197-209: a global randomized placebo-controlled phase II clinical trial of erlotinib plus ARQ 197 versus erlotinib plus placebo in previously treated EGFR inhibitor-naive patients with locally advanced or metastatic non-small cell lung cancer (NSCLC). J Clin Oncol 28(Suppl): abstract LBA7502 [Google Scholar]
- Schmidt C., Bladt F., Goedecke S., Brinkmann V., Zschiesche W., Sharpe M., et al. (1995) Scatter factor/hepatocyte growth factor is essential for liver development. Nature 373: 699–702 [DOI] [PubMed] [Google Scholar]
- Sharma S., Bell D., Settleman J., Haber D. (2007) Epidermal growth factor receptor mutations in lung cancer. Nature Rev Cancer 7: 169–181 [DOI] [PubMed] [Google Scholar]
- Sharma S., Settleman J. (2007) Oncogene addiction: setting the stage for molecularly targeted cancer therapy. Genes Dev 21: 3214–3231 [DOI] [PubMed] [Google Scholar]
- Slamon D.J., Leyland-Jones B., Shak S., Fuchs H., Paton V., Bajamonde A., et al. (2001) Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 344: 783–792 [DOI] [PubMed] [Google Scholar]
- Spigel D.R., Ervin T.J., Ramlau R., Daniel D.B., Goldschmidt J.H., Blumenschein G.R., et al. (2011) Final efficacy results from OAM4558g, a randomized phase II study evaluating MetMAb or placebo in combination with erlotinib in advanced NSCLC. J Clin Oncol 29(Suppl): abstract 7505 [Google Scholar]
- Stommel J.M., Kimmelman A.C., Ying H., Nabioullin R., Ponugoti A.H., Wiedemeyer R., et al. (2007) Coactivation of receptor tyrosine kinases affects the response of tumour cells to targeted therapies. Science 318: 287–290 [DOI] [PubMed] [Google Scholar]
- Tan E., Park K., Lim W.T., Ahn M., Ng Q.S., Ahn J.S., et al. (2011) Phase Ib study of ficlatuzumab (formerly AV-299), an anti-hepatocyte growth factor (HGF) monoclonal antibody (MAb) in combination with gefitinib (G) in Asian patients (pts) with NSCLC. J Clin Oncol 29(Suppl): abstract 7571 [Google Scholar]
- Toschi L., Janne P.A. (2008) Single-agent and combination therapeutic strategies to inhibit hepatocyte growth factor/MET signaling in cancer. Clin Cancer Res 14: 5941–5946 [DOI] [PubMed] [Google Scholar]
- Trusolino L., Comoglio P.M. (2002) Scatter-factor and semaphorin receptors: cell signalling for invasive growth. Nature Rev Cancer 2: 289–300 [DOI] [PubMed] [Google Scholar]
- Ueki T., Kaneda Y., Tsutsui H., Nakanishi K., Sawa Y., Morishita R., et al. (1999) Hepatocyte growth factor gene therapy of liver cirrhosis in rats. Nature Med 5: 226–230 [DOI] [PubMed] [Google Scholar]
- Watanabe M., Ebina M., Orson F.M., Nakamura A., Kubota K., Koinuma D., et al. (2005) Hepatocyte growth factor gene transfer to alveolar septa for effective suppression of lung fibrosis. Mol Ther 12: 58–67 [DOI] [PubMed] [Google Scholar]
- Workman P. (2002) Challenges of PK/PD measurements in modern drug development. Eur J Cancer 38: 2189–2193 [DOI] [PubMed] [Google Scholar]
- Workman P. (2003) How much gets there and what does it do? The need for better pharmacokinetic and pharmacodynamic endpoints in contemporary drug discovery and development. Curr Pharm Des 9: 891–902 [DOI] [PubMed] [Google Scholar]
- Yap T.A., de Bono J.S. (2010) Targeting the HGF/c-Met axis: state of play. Mol Cancer Ther 9: 1077–1079 [DOI] [PubMed] [Google Scholar]
- Yap T.A., Olmos D., Brunetto A.T., Tunariu N., Barriuso J., Riisnaes R., et al. (2011) Phase I trial of a selective c-MET inhibitor ARQ 197 incorporating proof of mechanism pharmacodynamic studies. J Clin Oncol 29: 1271–1279 [DOI] [PubMed] [Google Scholar]
- Yap T.A., Sandhu S.K., Workman P., de Bono J.S. (2010) Envisioning the future of early anti-cancer drug development. Nature 10: 514–523 [DOI] [PubMed] [Google Scholar]