

Abstract
One key adaptation that Mycobacterium tuberculosis established to survive long term in vivo is a reliance on lipids as an energy source. M. tuberculosis H37Rv has 36 fadD genes annotated as putative fatty acyl-CoA synthetase genes, which encode enzymes that activate fatty acids for metabolism. One such gene, fadD5 (Rv0166), is located within the mce1 operon, a cluster of genes associated with M. tuberculosis persistence. We disrupted the putative fatty acid binding site of fadD5 in H37Rv M. tuberculosis. No significant differences were found in the growth of the mutant and wild-type strains in vitro in nutrient-rich broth or in activated RAW264.7 cells. However, the fadD5 mutant was diminished in growth in minimal medium containing mycolic acid, but not other long-chain fatty acids. C57BL/6 mice infected with the fadD5 mutant survived significantly longer than those infected with wild-type, and the mutant never attained the plateau phase of infection in the mouse lungs. The steady-state infection phase was maintained for up to 168 days at a level one to two logs less than that shown by wild-type. These observations raise a rather intriguing possibility that FadD5 may serve to recycle mycolic acids for the long-term survival of the tubercle bacilli.
Keywords: Mycobacterium tuberculosis, fatty acyl-CoA synthetase, fadD, mce1 operon, fatty acid metabolism
INTRODUCTION
Tuberculosis (TB) remains as one of the most common human diseases today, causing nearly two million deaths per year [1, 2]. The success of the pathogen responsible for causing TB depends upon its ability to asymptomatically persist in its host for years. One-third of the world’s population is latently infected with Mycobacterium tuberculosis [3]. The organism in such hosts faces harsh external pressures, including nutrient deprivation [4]. Details regarding the bacterium’s strategies to acquire nutrients during infection remain unclear.
M. tuberculosis stands out among the prokaryotes for genetic dedication to lipid metabolism; only 50 genes in E. coli are annotated for lipid metabolic roles compared to roughly 250 genes in M. tuberculosis [5]. The abundance of lipid metabolic genes suggests that M. tuberculosis may utilize a variety of lipid metabolic processes for its long-term survival inside its host. Evidence suggests that M. tuberculosis processes lipids as a primary carbon source during infection [5–7].
The fadD5 (Rv0166) gene is one in a family of 36 annotated fadD genes scattered throughout the M. tuberculosis genome. The large number of FadD homologues in M. tuberculosis is notable compared to the single fadD gene present in, for example, E. coli [8]. The fadD5 gene belongs to a 13-gene operon in M. tuberculosis called mce1 [5]. Previously, a mutant disrupted in this operon was shown to cause accelerated death in BALB/c and C57BL/6 mice. The mce1 operon mutant is unable to establish a steadystate infection in mice, and therefore it was suggested to play a role in M. tuberculosis latent infection [7, 9, 10].
The M. tuberculosis genome contains four homologous copies of the operons mce1–4 [5]. Each operon encodes two putative integral membrane proteins (YrbEA and YrbEB) at the 5’ end followed by six putative cell membrane-associated proteins (MceA–F) resembling components of ATP-binding cassette (ABC) transporters [5, 6, 9, 11]. Absent in the other mce operons (mce2–4) is the fadD5 gene (Rv0166), which is annotated in the H37Rv genome as a putative fatty acyl-CoA synthetase (FACS). The fadD5 gene has been shown to be co-transcribed with the mce1 operon genes and negatively regulated by Mce1R [12]. The fadD5 gene is transcribed in ex vivo-infected macrophage-like cells [12]. Because of its association with an operon associated with virulence and latency, we aimed to explore the importance of fadD5 during infection and its possible role in fatty acid catabolism.
MATERIALS AND METHODS
Bacterial strains and growth conditions
Wild-type (WT) M. tuberculosis H37Rv and its derivative strains (fadD5 mutant, fadD5 complemented, and mce1 operon mutant strains) were grown in Middlebrook 7H9 broth containing 10% ADC, 0.2% glycerol, and 0.05% Tween-80, or on Middlebrook 7H11 agar containing OADC, 0.5% glycerol, and cycloheximide (100 µg mL−1) (Sigma Aldrich). Bacteria were grown in kanamycin-containing medium (20 µg mL−1) when required. Bacteria were washed and prepared as single cell suspensions prior to mouse infections and growth curve measurements. To test for growth in a specific carbon source, we cultured the M. tuberculosis strains in complete Sauton’s medium with glycerol or modified Sauton’s medium with oleate (100 µg mL−1), palmitate (100 µg mL−1), caprylate (100 µg mL−1), or mycolic acid derived from a human strain (25 µg mL−1) (Sigma Aldrich) as the sole carbon substrate. Bacterial growth curves were measured by O.D.580 from three independent cultures at the indicated time points.
Disruption and complementation of the fadD5 gene
The fatty acyl-CoA ligase (FACL) enzymatic family share a 25-amino acid FACS signature motif [13]. FadD5 carries a stretch of 25 amino acids 56% identical to the consensus E. coli FACS motif. We therefore identified this stretch in FadD5 as the FACS motif and targeted it for functional disruption (Fig. 1B).
Fig. 1. The fadD5 gene in the mce1 operon and its comparison to FACS consensus sequence.
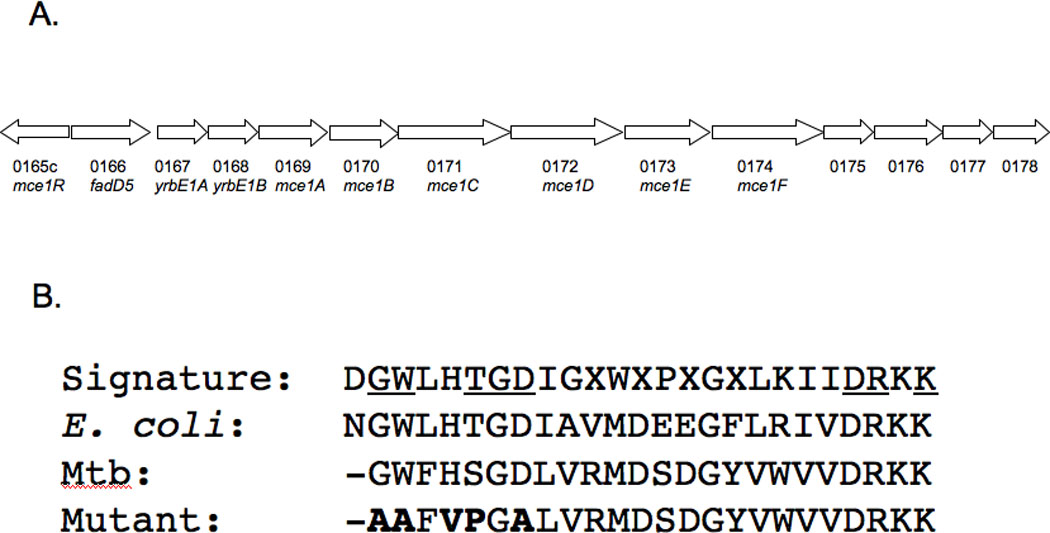
(A) Position of the Rv0166 fadD5 gene within the mce1 operon of the M. tuberculosis H37Rv genome. (B) Alignment of the FACS signature motif (as defined by Black, P.N., et al., 1997) of E. coli and M. tuberculosis FadD enzymes. Among the 25 amino acid residues characterizing the signature motif, 13 are highly conserved and 8 are invariant (underlined). Dashes indicate the absence of a homologous residue, and residues in bold reflect the in-frame substitutions created in the M. tuberculosis fadD5 mutant strain.
We replaced five amino acids within the FACS motif region (Fig. 1B). Substitutions of any of these five amino acids in E. coli resulted in near to complete inactivation of FadD [13]. The fadD5 gene in M. tuberculosis H37Rv was disrupted by homologous recombination by a two-step counter-selection method [14]. Briefly, two sets of primers were designed to both amplify the fadD5 gene and introduce base pair substitutions into the highly conserved fatty acid binding motif. The first purified PCR product was digested by HindIII and KpnI and inserted into the similarly digested p2NIL vector. The second purified PCR product was digested by KpnI and NotI and inserted into the similarly digested p2NIL vector containing the first PCR product. The method of Parish and Stoker was followed to construct the final delivery vector, generate the mutants, and select the mutants [14].
The nucleotide substitutions in the fadD5 mutant strain were ascertained by 1) sequencing the mutated fadD5, and 2) Southern blotting with the restriction enzyme, KpnI, to cleave the mutated FACS motif (see Fig. 2 legend for additional details). Western blots were performed with anti-Mce1A and anti-Mce1D antibodies raised in rabbits [11] on fadD5 mutant whole cell lysates to ensure that mce genes downstream of fadD5 were expressed and hence the mutations had no polar effects.
Fig. 2. Southern blot analysis of the fadD5 mutant strain and growth kinetics in standard medium.
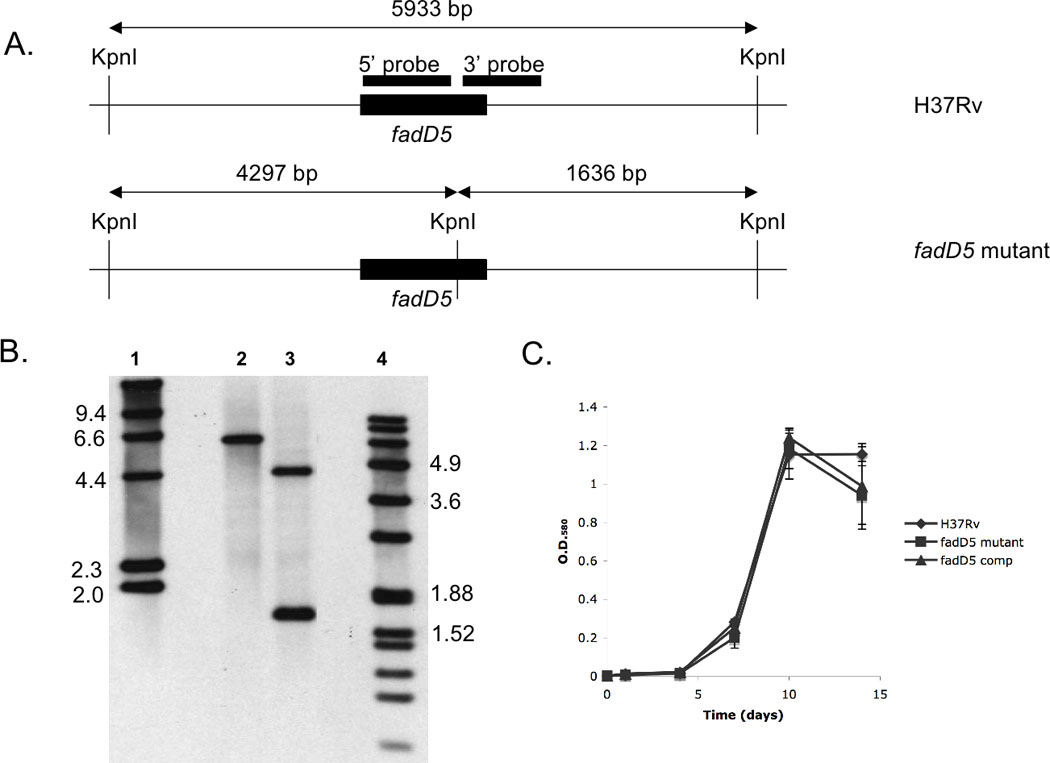
(A) Restriction digestion sites within and surrounding fadD5 genomic region. The 5’ and 3’ probes (black boxes) represent the regions recognized in the Southern blot analysis of H37Rv WT (top) and fadD5 mutant (bottom) genomic DNA. (B) Southern blot analysis of the WT (lane 2) and fadD5 mutant genomic DNA (lane 3) cleaved by the restriction enzyme, KpnI. The lanes are flanked by the digoxigenin-labeled molecular mass standards, II (lane 1) and VII (lane 4) (Roche Diagnostics). (C) Growth kinetics of the WT, fadD5 mutant, and fadD5 complemented strains in 7H9 medium. Cultures were inoculated in triplicate at O.D.580 0.004 and optical densities were measured at 1,4,7,10, and 14 days post-infection.
We created the fadD5 complemented strain by amplifying the 1665-bp fadD5 gene from H37Rv genomic DNA and introducing PacI and KpnI restriction sites at the 5’ and 3’ ends of the fragment, respectively. The fadD5 product was inserted into the integrating vector, pMVGS [15] by the knock-in strategy [9] to produce pMVGS-fadD5.
Mouse infections
Eight-week old C57BL/6 mice (Jackson Laboratories) were infected with the above three M. tuberculosis strains via the aerosol route with the Inhalation Exposure System (Glas-Col). The inoculum doses were assessed from harvest of the right lungs of three mice (per infection) 24 hours post-infection (p.i.). Mice were infected with a dose of 95–128 bacilli per lung for the infections. At 1, 18, 42, 100, and 168 days p.i., the right lungs from four mice of each group were collected, homogenized in PBS-Tween-80 (0.05%), diluted appropriately, and plated onto 7H11 agar plates (supplemented as above). At each time point, the bacterial load of each organ was determined by colony forming unit (cfu) enumeration.
Histology
The mouse left lungs were fixed in 10% formalin in neutral (PBS) buffer and were embedded in paraffin, sectioned, and stained for histology with either hematoxylin and eosin (H&E) or the Ziehl-Neelson technique. Three sections were obtained from each lung from four mice at days 42 and 168 p.i. A veterinary pathologist, blinded to the sources of the sections, performed the histopathology analysis of the lung sections from mice infected for 42 and 168 days.
Determination of mouse morbidity
Two groups of five mice each, infected with either the WT or fadD5 mutant strains, were followed until they displayed characteristic moribund features prior to death (i.e., weight loss, failure to groom, ruffled fur, and lethargy). After this assessment, mice were anaesthetized and then euthanized by cervical dislocation. The health of the mice was monitored daily by the above veterinary staff.
RAW264.7 macrophage-like cell infection
The RAW264.7 murine macrophage-like cell line (ATCC) was maintained in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen) at 37°C in a 5% CO2 humidified incubator. The cells (105 cells mL−1) were plated in 12-well plates, activated with 30 units mL−1 of recombinant IFN-γ (eBioscience), and incubated overnight.
The cells were infected at a multiplicity of infection (m.o.i.) of 1:10 (cells:bacteria). Lipopolysaccharide (1 µg mL−1; Sigma) was used as a positive control and uninfected cells served as a negative control. At 6, 24, 48, and 72 hours post-infection, cells were washed three times with chilled PBS to remove extracellular bacteria, and then lysed with PBS/0.1% Triton X-100. Appropriately diluted cell lysates were plated on 7H11 agar plates (supplemented as above) for cfu enumeration.
Cytokine production by infected macrophage-like cells
RAW264.7 macrophage-like cells were cultured in DMEM/FBS medium for up to 72 hours in the presence or absence of M. tuberculosis. Supernatants were collected at 6, 24, 48, and 72 hours p.i., and passed through a 0.2 µm filter (Fisher) to remove extracellular bacteria. The concentrations of TNFα, IL-6, and MCP-1 in the supernatants were quantified by ELISA, according to the manufacturer’s protocols (eBioscience). The mean of three cytokine values measured for each supernatant (using three microplate wells) were given as the final cytokine values.
Statistics
Mouse survival was compared by Kaplan-Meier curves. The mean cfu counts of M. tuberculosis recovered from lungs of each mouse group (four per group) were compared by the Student’s t-test and were considered significant at p < 0.05.
RESULTS
Functional disruption of the fadD5 gene
The substitutions in the predicted FACS motif of fadD5 were confirmed by Southern blot analysis (Fig. 2A–B) and PCR (not shown). Previously, we observed that the deletion of a gene in the mce1 operon resulted in the polar disruption of the downstream operon genes [9]. In contrast, Western blot analysis of the cell extracts of the fadD5 mutant indicated that the expression of the downstream genes, mce1A and mce1D, were unaffected by this mutation (not shown). The WT, fadD5 mutant, and fadD5 complemented strains exhibited similar growth profiles in aerated 7H9 liquid medium (Fig. 2C), which supports the previous finding that fadD5 is non-essential in vitro [16, 17].
Growth of the fadD5 mutant in the presence of fatty acid substrates
To evaluate if FadD5 contributed to fatty acid catabolism, we measured the growth of the M. tuberculosis strains in Sauton’s minimal medium containing glycerol, oleate, palmitate, caprylate, or mycolic acid as the sole carbon source. All strains grew at similar rates over a two-week period in complete Sauton’s medium containing glycerol or Sauton’s modified medium containing oleate, palmitate, or caprylate as the sole carbon source (Fig. 3A–D). However, the fadD5 mutant did not reach the growth density achieved by WT or fadD5 complemented strains in Sauton’s minimal medium containing mycolic acid as the sole source of carbon (Fig. 3E). At days 10 and 14, O.D.580 observed with the mutant was 0.20 +/−0.001 and 0.25 +/−0.006, respectively, compared to 0.31 +/−0.003 and 0.33+/−0.005 with the WT (p<0.00015, p<0.0005) and 0.28+/−0.029 and 0.32+/−0.014 with the complemented strain (p<0.03, p<0.007). The mce1 operon mutant maintained growth comparable to WT in minimal medium containing mycolic acid (Fig. 3F).
Fig. 3. Growth of M. tuberculosis strains in a single carbon source medium.
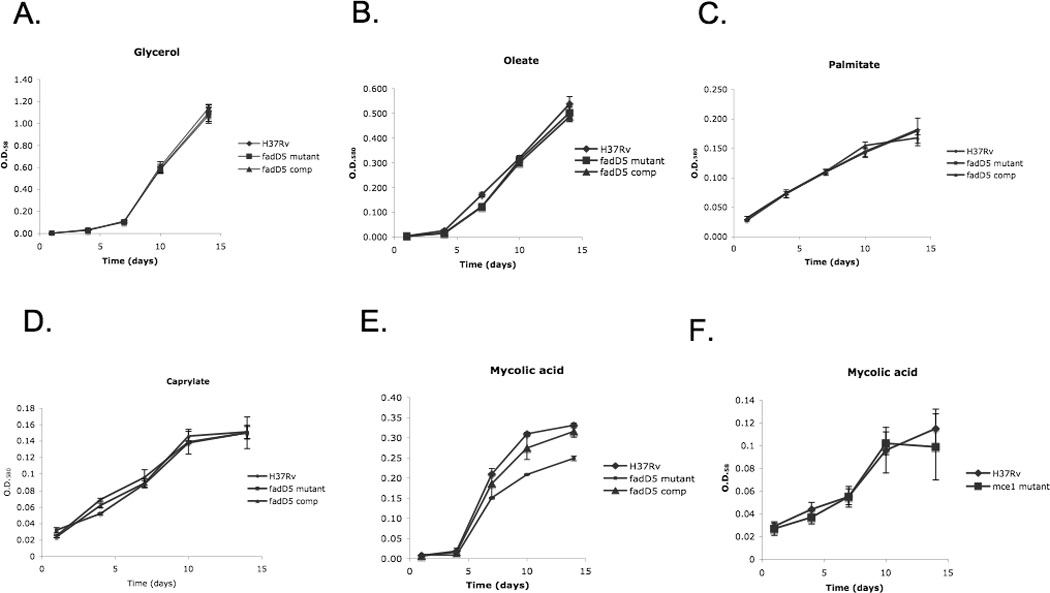
Growth kinetics of WT, fadD5 mutant, and fadD5 complemented strains in (A) complete Sauton’s medium (with glycerol), incomplete Sauton’s medium (without glycerol), and with (B) oleate, (C) palmitate, (D) caprylate, or (E) mycolic acid. Growth kinetics comparing WT and mce1 operon mutant in incomplete Sauton’s medium with (F) mycolic acid. The average of three O.D.580 measurements per strain was plotted for each time point.
Growth of the fadD5 mutant strain in macrophages in vitro
We compared the growth of the fadD5 mutant to WT in activated RAW264.7 macrophage-like cells. At each interval spanning 72 hours, the fadD5 mutant grew at roughly the same rate and achieved similar cell density as WT (Fig. 4A).
Fig. 4. Growth and cytokine induction of macrophage-like RAW264.7 cells infected with M. tuberculosis strains.
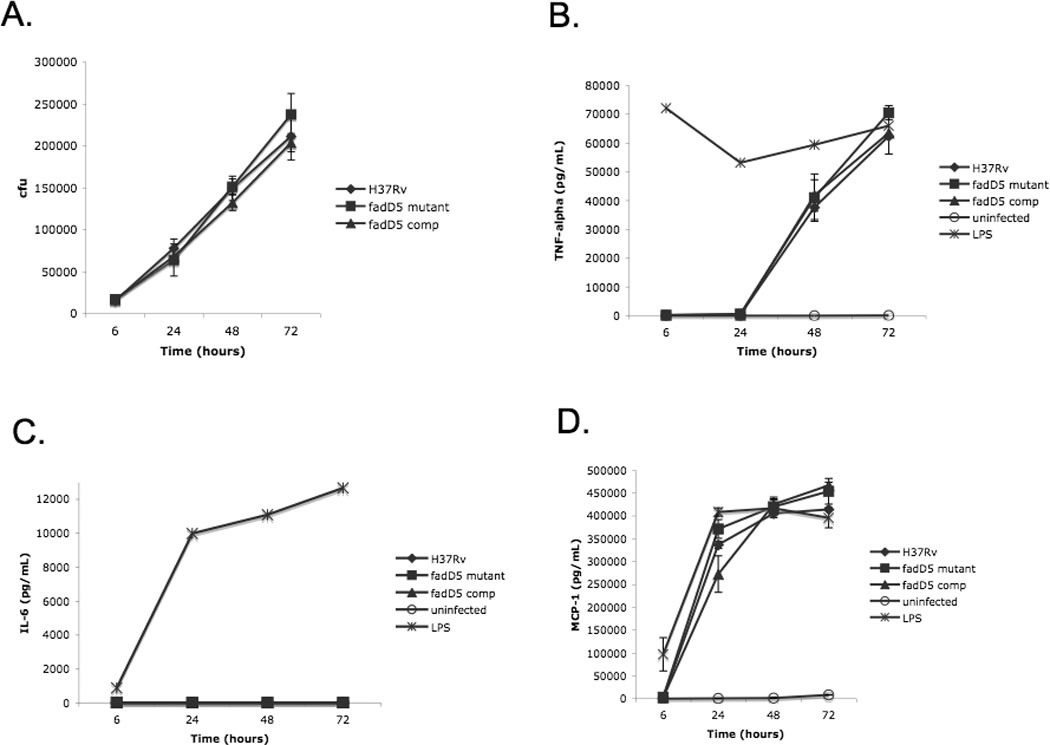
(A) Recovery of WT, fadD5 mutant, and fadD5 complemented strains from macrophage-like RAW264.7 cells (n=3 per strain; m.o.i. 10:1) over a 72 hr period. Uninfected RAW cells served as a negative control, while LPS-infected RAW cells served as a positive control. At each time point, supernatants were filtered and collected for ELISA analysis. The production and release into the supernatant of (B) TNFα, (C) IL-6, and (D) MCP-1 were measured.
In addition, cytokine production by the RAW264.7 macrophage-like cells was measured (Fig. 4B–D). TNFα, IL-6, and MCP-1 were produced in similar quantities by either fadD5-infected cells or WT-infected cells. IL-6 production was considerably low for all three strains.
Attenuated growth of the fadD5 mutant strain in mice
The bacterial loads recovered from the mouse lungs at 18, 42, 100, and 168 days were significantly higher in the WT-infected mice compared to those infected with the fadD5 mutant strain (p<0.015 at 168 day time point, Student’s t-test) (Fig. 5). The fadD5 mutant recorded a lower rate of replication throughout the course of infection compared to WT and fadD5-complemented strains. Also, the fadD5 mutant never attained the plateau phase of infection typically achieved by WT M. tuberculosis in C57BL/6 mouse lungs, which fluctuated around 106 and 107 bacilli per lung. Instead, the fadD5 mutant established a plateau phase at levels one to two logs lower for the duration of the infection. The complemented mutant set its plateau phase at the intermediate range between that of the WT and fadD5 mutant.
Fig. 5. Recovery of M. tuberculosis strains from infected C57BL/6 mouse lungs.
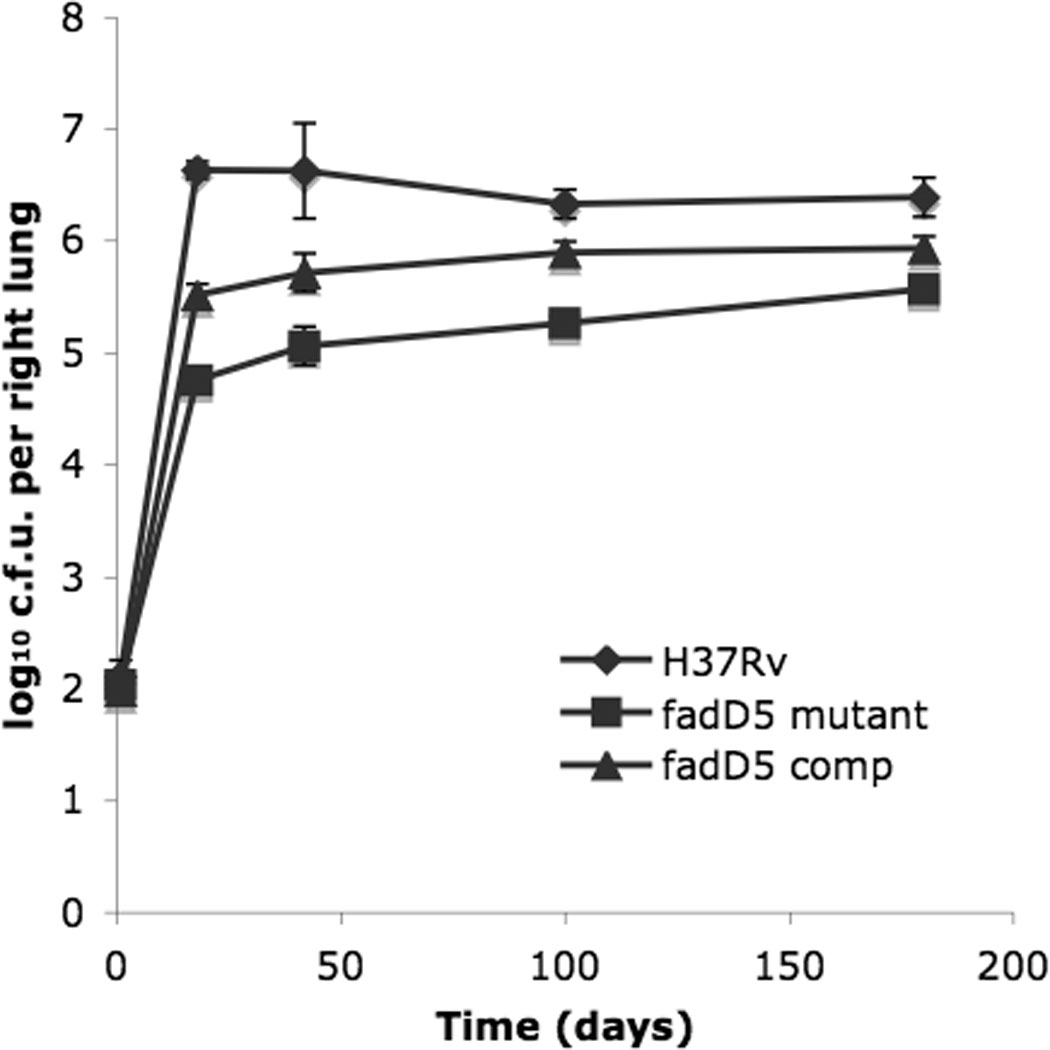
Mice were infected with a dose of 95–128 bacilli per lung with WT, fadD5 mutant, and fadD5 complemented strains. Bacilli were recovered (n=4 per strain) at the indicated time points and enumerated on 7H11 agar plates.
Mouse survival analysis
The attenuated growth of the fadD5 mutant strain was similarly reflected in the survival kinetics of the infected mice; the fadD5 mutant-infected mice survived an average of 166 days longer than WT-infected mice (Fig. 6). The median survival time was 297 days for the WT-infected mice, 357 days for complement strain-infected mice, and 485 days for the fadD5 mutant-infected mice (p<0.05 for comparison of mice infected with WT or complemented strain vs fadD5 mutant, Mantel-Cox log-rank test).
Fig. 6. Survival kinetics of C57/BL6 mice infected with M. tuberculosis strains.
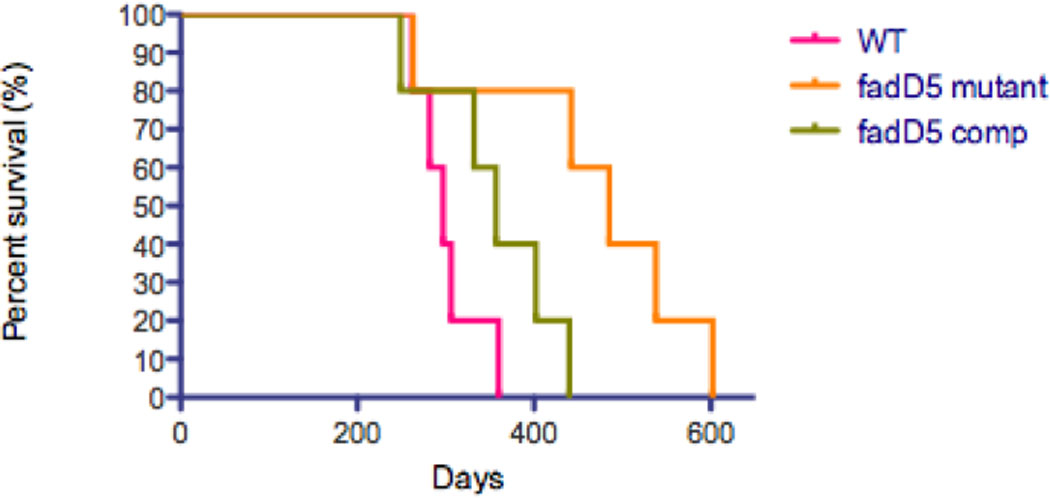
Percent survival of mice after aerosol infection with the WT and complemented strains compared against fadD5-mutant infected mice (5 mice per group). Kaplan-Meier curve was generated by GraphPad Prism software (p<0.05 for comparison of mice infected with WT or complemented strain vs fadD5-infected mice, Mantel-Cox log-rank test).
Histopathology of fadD5 mutant-infected mouse lungs
Gross lung examination revealed fewer numbers of granulomatous lesions in fadD5 mutant-infected mouse lungs compared to WT and fadD5 complemented-infected lungs by day 42 and maintained throughout the course of infection until day 168 p.i. (Fig. 7A–C). The fadD5 mutant-infected mouse lungs were also consistently smaller in size than WT-infected lungs.
Fig. 7. M. tuberculosis-infected gross mouse lungs and histology at 168 days p.i.
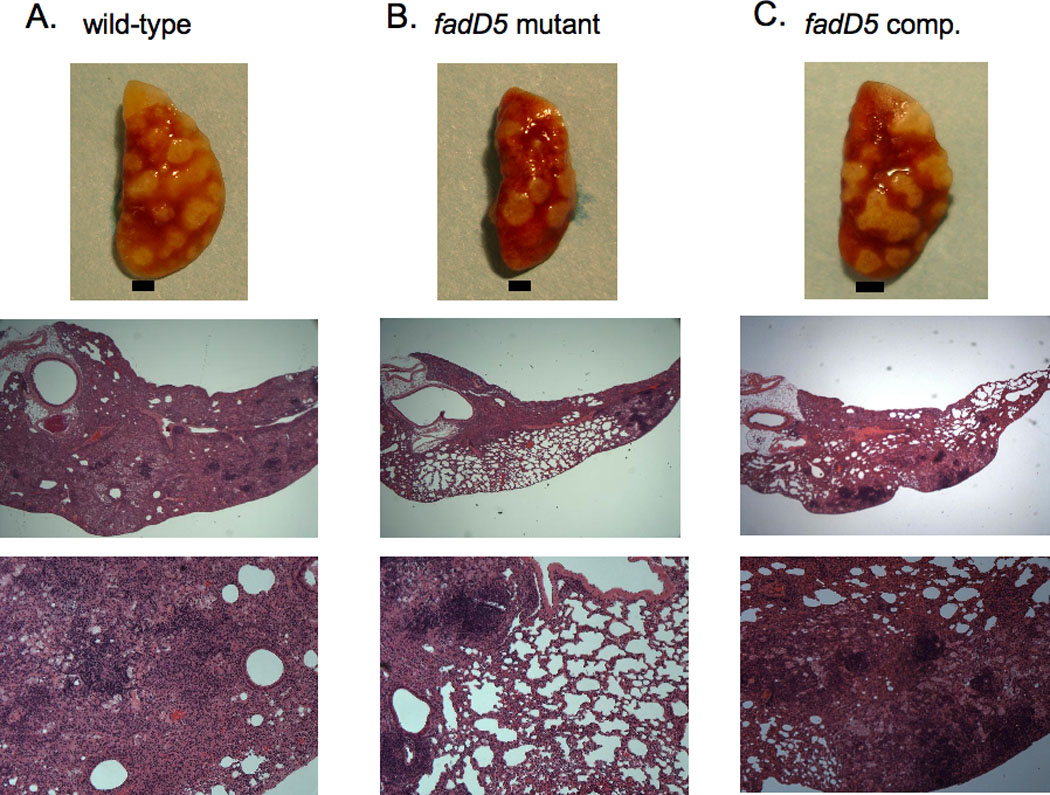
Gross lung pathology of mice infected with WT (A), fadD5 mutant (B), or fadD5 complemented (C) strains. Each black bar represents 1.5mm. Histological sections of H&E-stained lungs of mice infected with (A) WT, (B) fadD5 mutant, or (C) fadD5 complemented strains. Magnified at x25 (middle figures) and x100 (bottom figures).
The histological examination of the lungs after 168 days of infection revealed lesions in >90% of the lung parenchyma in mice infected with the WT strain vs. <50% in those infected with the fadD5 mutant (Fig. 7). The veterinary pathologist, blinded to the study, reported that fadD5 mutant-infected lung sections displayed fewer markers of disease (i.e., necrosis) compared to WT-infected lung sections at both day 42 p.i. and day 168 p.i (Fig. 7A–B). By day 168 p.i., fadD5 mutant-infected lung sections revealed the absence of necrosis, while WT-infected lung sections were noted to have highly diffuse coalescing nodules with mild necrosis. The fadD5 complemented-infected lung sections displayed lesions that were intermediate in severity between that of WT- and fadD5 mutant-infected lungs (Fig. 7C).
DISCUSSION
The five amino acid substitutions in the putative FACS motif of FadD5 diminished the ability of M. tuberculosis to grow in mycolic acid as a sole carbon source. This method of disrupting a gene avoids the potential polar effects of gene disruption, especially of a gene belonging to an operon. These findings support the importance of the highly conserved FACS motif for the function of FadD, and that M. tuberculosis FadD5 uses fatty acid as a substrate. Most significantly, these amino acid substitutions in the putative FACS motif attenuated the growth of M. tuberculosis in vivo, and not in vitro, suggesting that FadD5 is required for maintaining M. tuberculosis viability in a mammalian host.
Among the 36 fadD genes in M. tuberculosis, one subset expresses fatty acyl-AMP ligases (FAAL) associated with lipid synthesis, while the other subset expresses FACL associated with lipid degradation [18]; fadD5 falls into the latter category due to the lack of a trypsin cleavage site characteristic of FAAL. FadD5 exhibits a 45% overall identity to the well-studied FACS FadD in E. coli, which is responsible for the coupled import and activation of long-chain fatty acids for further metabolism in the β-oxidation pathway [13, 19]. E. coli FadD and M. tuberculosis FadD5 share a 52% homology at the putative FACS motif [13].
In vitro, glucose and glycerol is processed through the TCA cycle to deliver carbon and energy for M. tuberculosis [20]. However, a shift from carbohydrate to lipid metabolism is believed to occur in vivo [21, 22]. The fatty acid metabolic isoenzymes, isocitrate lyase 1/2 (ICL1/2) are the first set of enzymes acting in the glyoxylate shunt, and their activity allows M. tuberculosis to utilize fatty acids as a sole carbon source in vitro [23]. In vivo, ICL1/2 are vital for the bacterium’s survival throughout the course of infection; the double mutant Δicl1 Δicl2 is unable to replicate in mice and is rapidly cleared from mouse lungs and spleens [23]. However, unlike these icl mutants, the fadD5 mutant replicated in mice at a lower rate than WT and was never cleared of infection. The fadD5 mutant persisted during the plateau phase at a set point lower than that observed with WT (Fig. 5).
Compared to WT, the fadD5 mutant in minimal liquid medium containing mycolic acid as the sole carbon source showed diminished growth, while other fatty acids oleate and palmitate (other LCFAs) or caprylate (a short-chain fatty acid) as the sole carbon source had no effect. However, the fadD5 mutant was not completely inhibited in mycolic acid-containing medium, suggesting that mutated FadD5 retains residual enzymatic activity, or that M. tuberculosis has an alternative pathway for mycolic acid catabolism. It should be noted that the difference between this mutant and WT strains is in only 5 amino acids, and we were still able to detect this phenotypic difference related to fatty acid utilization in vitro associated with virulence difference in vivo. The observation that the mce1 operon mutant (which expresses intact FadD5) was able to grow normally in mycolic acid suggests that an alternative pathway exists for mycolic acid importation. The phylogenomic and genetic analyses of the mce operons strongly suggest that these operons comprise ABC lipid importers [24, 25]. Indeed, one member mce4 was recently shown to serve as a possible cholesterol importer system [26].
Complementation of the fadD5 mutant strain with the pMVGS-fadD5 partly restored the activity of FadD5 in these studies. The mutant strain was complemented by integrating the pMVGS-fadD5 plasmid into the host chromosome. The incomplete restoration of the WT phenotype in the complemented strain may be due to the expression of fadD5 under a non-native promoter (glutamate synthase) control.
To our knowledge, this is the first study directly examining the role of a FACS predicted to be involved in lipid degradation in M. tuberculosis. Among the FadD paralogues that have been examined are those involved in lipid synthesis, such as FadD23 in sulfolipid-1 synthesis [27], FadD26 and FadD28 in phthiocerol dimycocerosate synthesis [28, 29], and FadD32 in mycolic acid synthesis [30]. The diverse functions of FadDs lend support to the theory that the FadD paralogues are not functionally redundant, but rather have unique roles in lipid metabolism. Indeed, the FadD paralogues are thought to process a variety of fatty acids [5], particularly given that fatty acids of different lengths are generated during the β-oxidation cycle [31]. The decreased growth of the fadD5 mutant in the presence of mycolic acid as the sole carbon source raises a rather intriguing possibility that this gene product may play a role in recycling mycolic acids released from M. tuberculosis that die in vivo.
Acknowledgments
This project was supported by a grant from the National Institutes of Health, R01AI035266.
Footnotes
The authors do not have a commercial or other association that might post a conflict of interest (e.g., pharmaceutical stock ownership, consultancy, advisory board membership, relevant patents, or research funding).
Keystone Symposia, Tubercolosis: Biology, Pathology and Therapy; January 2009; Keystone, Colorado; Abstract #188
American Society for Microbiology 108th General Meeting; June 2008; Boston, Massachusetts; Abstract #U-042
REFERENCES
- 1.Parrish NM, Dick JD, Bishai WR. Mechanisms of latency in Mycobacterium tuberculosis. Trends Microbiol. 1998;6:107–112. doi: 10.1016/s0966-842x(98)01216-5. [DOI] [PubMed] [Google Scholar]
- 2.Corbett EL, Watt CJ, Walker N, et al. The growing burden of tuberculosis: global trends and interactions with the HIV epidemic. Arch Intern Med. 2003;163:1009–1021. doi: 10.1001/archinte.163.9.1009. [DOI] [PubMed] [Google Scholar]
- 3.Dye C, Scheele S, Dolin P, Pathania V, Raviglione MC. Consensus statement. Global burden of tuberculosis: estimated incidence, prevalence, and mortality by country. WHO Global Surveillance and Monitoring Project. JAMA. 1999;282:677–686. doi: 10.1001/jama.282.7.677. [DOI] [PubMed] [Google Scholar]
- 4.Betts JC, Lukey PT, Robb LC, McAdam RA, Duncan K. Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol Microbiol. 2002;43:717–731. doi: 10.1046/j.1365-2958.2002.02779.x. [DOI] [PubMed] [Google Scholar]
- 5.Cole ST, Brosch R, Parkhill J, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 6.Tekaia F, Gordon SV, Garnier T, Brosch R, Barrell BG, Cole ST. Analysis of the proteome of Mycobacterium tuberculosis in silico. Tuber Lung Dis. 1999;79:329–342. doi: 10.1054/tuld.1999.0220. [DOI] [PubMed] [Google Scholar]
- 7.Lima P, Sidders B, Morici L, et al. Enhanced mortality despite control of lung infection in mice aerogenically infected with a Mycobacterium tuberculosis mce1 operon mutant. Microbes Infect. 2007;9:1285–1290. doi: 10.1016/j.micinf.2007.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kameda K, Nunn WD. Purification and characterization of acyl coenzyme A synthetase from Escherichia coli. J Biol Chem. 1981;256:5702–5707. [PubMed] [Google Scholar]
- 9.Shimono N, Morici L, Casali N, et al. Hypervirulent mutant of Mycobacterium tuberculosis resulting from disruption of the mce1 operon. Proc Natl Acad Sci U S A. 2003;100:15918–15923. doi: 10.1073/pnas.2433882100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Uchida Y, Casali N, White A, Morici L, Kendall LV, Riley LW. Accelerated immunopathological response of mice infected with Mycobacterium tuberculosis disrupted in the mce1 operon negative transcriptional regulator. Cell Microbiol. 2007;9:1275–1283. doi: 10.1111/j.1462-5822.2006.00870.x. [DOI] [PubMed] [Google Scholar]
- 11.Chitale S, Ehrt S, Kawamura I, et al. Recombinant Mycobacterium tuberculosis protein associated with mammalian cell entry. Cell Microbiol. 2001;3:247–254. doi: 10.1046/j.1462-5822.2001.00110.x. [DOI] [PubMed] [Google Scholar]
- 12.Casali N, White AM, Riley LW. Regulation of the Mycobacterium tuberculosis mce1 operon. J Bacteriol. 2006;188:441–449. doi: 10.1128/JB.188.2.441-449.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Black PN, Zhang Q, Weimar JD, DiRusso CC. Mutational analysis of a fatty acyl-coenzyme A synthetase signature motif identifies seven amino acid residues that modulate fatty acid substrate specificity. J Biol Chem. 1997;272:4896–4903. doi: 10.1074/jbc.272.8.4896. [DOI] [PubMed] [Google Scholar]
- 14.Parish T, Stoker NG. Use of a flexible cassette method to generate a double unmarked Mycobacterium tuberculosis tlyA plcABC mutant by gene replacement. Microbiology. 2000;146 (Pt 8):1969–1975. doi: 10.1099/00221287-146-8-1969. [DOI] [PubMed] [Google Scholar]
- 15.Mougous JD, Petzold CJ, Senaratne RH, et al. Identification, function and structure of the mycobacterial sulfotransferase that initiates sulfolipid-1 biosynthesis. Nat Struct Mol Biol. 2004;11:721–729. doi: 10.1038/nsmb802. [DOI] [PubMed] [Google Scholar]
- 16.Sassetti CM, Boyd DH, Rubin EJ. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol. 2003;48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- 17.Lamichhane G, Zignol M, Blades NJ, et al. A postgenomic method for predicting essential genes at subsaturation levels of mutagenesis: application to Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2003;100:7213–7218. doi: 10.1073/pnas.1231432100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trivedi OA, Arora P, Sridharan V, Tickoo R, Mohanty D, Gokhale RS. Enzymatic activation and transfer of fatty acids as acyl-adenylates in mycobacteria. Nature. 2004;428:441–445. doi: 10.1038/nature02384. [DOI] [PubMed] [Google Scholar]
- 19.Nunn WD. A molecular view of fatty acid catabolism in Escherichia coli. Microbiol Rev. 1986;50:179–192. doi: 10.1128/mr.50.2.179-192.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boshoff HI, Barry CE. A low-carb diet for a high-octane pathogen. Nat Med. 2005;11:599–600. doi: 10.1038/nm0605-599. [DOI] [PubMed] [Google Scholar]
- 21.McKinney JD, Honer zu Bentrup K, Muñoz-Elias EJ, et al. Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase. Nature. 2000;406:735–738. doi: 10.1038/35021074. [DOI] [PubMed] [Google Scholar]
- 22.Dubnau E, Chan J, Mohan VP, Smith I. Responses of Mycobacterium tuberculosis to growth in the mouse lung. Infect Immun. 2005;73:3754–3757. doi: 10.1128/IAI.73.6.3754-3757.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muñoz-Elias EJ, McKinney JD. Mycobacterium tuberculosis isocitrate lyases 1 and 2 are jointly required for in vivo growth and virulence. Nat Med. 2005;11:638–644. doi: 10.1038/nm1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Casali N, Riley LW. A phylogenomic analysis of the Actinomycetales mce operons. BMC Genomics. 2007;8:60. doi: 10.1186/1471-2164-8-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Joshi SM, Pandey AK, Capite N, Fortune SM, Rubin EJ, Sassetti CM. Characterization of mycobacterial virulence genes through genetic interaction mapping. Proc Natl Acad Sci U S A. 2006;103:11760–11765. doi: 10.1073/pnas.0603179103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pandey AK, Sassetti CM. Mycobacterial persistence requires the utilization of host cholesterol. Proc Natl Acad Sci U S A. 2008 doi: 10.1073/pnas.0711159105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lynett J, Stokes RW. Selection of transposon mutants of Mycobacterium tuberculosis with increased macrophage infectivity identifies fadD23 to be involved in sulfolipid production and association with macrophages. Microbiology. 2007;153:3133–3140. doi: 10.1099/mic.0.2007/007864-0. [DOI] [PubMed] [Google Scholar]
- 28.Camacho LR, Constant P, Raynaud C, et al. Analysis of the phthiocerol dimycocerosate locus of Mycobacterium tuberculosis. Evidence that this lipid is involved in the cell wall permeability barrier. J Biol Chem. 2001;276:19845–19854. doi: 10.1074/jbc.M100662200. [DOI] [PubMed] [Google Scholar]
- 29.Sirakova TD, Fitzmaurice AM, Kolattukudy P. Regulation of expression of mas and fadD28, two genes involved in production of dimycocerosyl phthiocerol, a virulence factor of Mycobacterium tuberculosis. J Bacteriol. 2002;184:6796–6802. doi: 10.1128/JB.184.24.6796-6802.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Portevin D, de Sousa-D'Auria C, Montrozier H, et al. The acyl-AMP ligase FadD32 and AccD4-containing acyl-CoA carboxylase are required for the synthesis of mycolic acids and essential for mycobacterial growth: identification of the carboxylation product and determination of the acyl-CoA carboxylase components. J Biol Chem. 2005;280:8862–8874. doi: 10.1074/jbc.M408578200. [DOI] [PubMed] [Google Scholar]
- 31.Ehrt S, Schnappinger D. Mycobacterium tuberculosis virulence: lipids inside and out. Nat Med. 2007;13:284–285. doi: 10.1038/nm0307-284. [DOI] [PubMed] [Google Scholar]