

Abstract
Melanoma often metastasizes to bone where it is exposed to high concentrations of TGF-β. Constitutive Smad signaling occurs in human melanoma. Because TGF-β promotes metastases to bone by several types of solid tumors including breast cancer, we hypothesized that pharmacologic blockade of the TGF-β signaling pathway may interfere with the capacity of melanoma cells to metastasize to bone. In this study, we tested the effect of a small molecule inhibitor of TGF-β receptor I kinase (TβRI), SD-208, on various parameters affecting the development and progression of melanoma, both in vitro and in a mouse model of human melanoma bone metastasis. In melanoma cell lines, SD-208 blocked TGF-β induction of Smad3 phosphorylation, Smad3/4-specific transcription, Matrigel invasion and expression of the TGF-β target genes PTHrP, IL-11, CTGF and RUNX2. To assess effects of SD-208 on melanoma development and metastasis, nude mice were inoculated with 1205Lu melanoma cells into the left cardiac ventricle and drug was administered by oral gavage on prevention or treatment protocols. SD-208 (60mg/kg/day), started 2 days before tumor inoculation prevented the development of osteolytic bone metastases compared with vehicle. In mice with established bone metastases, the size of osteolytic lesions was significantly reduced after 4 weeks treatment with SD-208 compared to vehicle-treated mice. Our results demonstrate that therapeutic targeting of TGF-β may prevent the development of melanoma bone metastases and decrease the progression of established osteolytic lesions.
INTRODUCTION
Malignant melanoma represents the most aggressive and lethal of skin cancers, with an incidence in constant augmentation and a very poor prognosis due to limited therapy. Melanoma has become the first cause of death by cancer for patients aged 20–35. About 60,000 new cases and approximately 8000 deaths were expected in the USA for year 2007 (1). Up to 50% of patients with advanced melanoma develop bone metastases (2).
TGF-β has been implicated in the pathogenesis of melanoma (3) as well as bone metastases due to breast cancer (4). Increased production of TGF-β coupled with resistance to the growth inhibitory effects of TGF-β is a characteristic of various neoplasms, including melanoma (3, 5). In select epithelial malignancies, disruption of TGF-β-dependent signaling and transcription has been associated with resistance to TGF-β-induced growth inhibition (6).
TGF-β binds to the TGF-β type II receptor, TβRII, a receptor with constitutive kinase activity which then transphosphorylates TGF-β receptor type I (TβRI) in its Serine/Glycine-rich domain onto serine and threonine residues. This, in turn, induces TβRI kinase activity, leading to the phosphorylation of Smad2 and Smad3, proteins that constantly shuttle between the nucleus and cytoplasm (7). Upon phosphorylation in their C-terminal domain, Smad2/3 associate with Smad4 to form heterocomplexes that accumulate in the nucleus and function as transcription factors to activate transcription of target genes, binding to their 5′ regulatory regions either directly or indirectly via association with other transcription factors (3, 8–11).
The contribution of TGF-β to breast cancer bone metastasis has been well described as a feed-forward cycle whereby tumors homing favorably to bone secrete osteolytic factors such as PTHrP and IL-11. The resulting activation of osteoblasts and osteoclasts causes the degradation of the bone matrix and subsequent release of soluble factors, including TGF-β, which, in turn, exacerbate tumor cells to produce more osteoclast-activating factors (4, 12, 13). A bone metastasis signature of TGF-β-inducible genes was identified from highly bone metastatic MDA-MB-231 breast cancer cells, comprising CTGF, CXCR4, MMP-1 and IL-11, which, when overexpressed in non-metastatic cells, induced them to metastasize to bone (14, 15). In human melanoma cells, we have shown previously that Smad-dependent transcription is intact and does not correlate with proliferative responses to TGF-β (16). We characterized autocrine effects of TGF-β on melanoma by overexpressing the inhibitory Smad7, in the highly metastatic melanoma line 1205Lu, in which there is strong autoactivation of the Smad pathway (16). Stable expression of Smad7 reduced constitutive Smad2/3 phosphorylation and Smad3/Smad4-driven gene transactivation in response to TGF-β, associated with reduced tumorigenicity, MMP-2 and MMP-9 secretion by melanoma cells (17).
Our recent work also implicates a similar role for TGF-β in bone metastases due to melanoma. Specifically, we demonstrated that stable Smad7 overexpression in melanoma was associated with reduced expression of a similar set of bone metastasis-specific genes, including IL-11, PTHrP, CXCR4 and CTGF, as well as significant reduction in the capacity of melanoma cells to establish bone metastases in a nude mouse model (18). Taken together, these results suggested that inhibition of TGF-β/Smad signaling in melanoma is a worthy target for therapeutic intervention, consistent with a recent reports of efficacy for TGF-β blockade in models of breast carcinoma, pancreatic adenocarcinoma, myeloma, and glioma (19–22). We could not, however, rule out that Smad7 may also exert some of its anti-metastatic action independently from its role as a TGF-β signaling inhibitor.
The rationale to block TGF-β signaling as therapy to treat and prevent melanoma bone metastasis is substantial. First, TGF-β is abundant in bone. It is released as a consequence of osteoclastic bone resorption and stimulates tumor production of pro-metastatic factors. Overexpression of Smad7 blocks this action and reduces the capacity for bone metastases. Furthermore, melanoma cells secrete abundant amounts of, and activate, TGF-β (16, 23) that likely affect stromal and cancer cells and contribute to the metastatic process (3). Finally, host stromal cells may also secrete TGF-β when in contact with tumor cells (24). To directly interfere with the vicious cycle of bone metastasis driven by TGF-β, we examined the efficacy of systemic targeting of the Smad pathway with the small-molecule TGF-β receptor type I (TβRI) kinase inhibitor, SD-208 (25).
MATERIALS AND METHODS
Cell cultures and reagents
Human melanoma cell lines WM852, and 1205Lu, kind gifts from Dr. M. Herlyn, Wistar Institute, Philadelphia, PA, have been previously described (16, 26, 27). They were grown in a composite medium (W489) consisting of three parts MCDB153 and one part L15 supplemented with 4% fetal calf serum (FCS) and antibiotics. The melanoma cell lines 888-mel and 501-mel (28) were grown in RPMI medium supplemented with 10% FCS and antibiotics. All cells were grown at 37°C,5% CO2 in a humidified atmosphere. The Smad3/4-specific reporter plasmid (CAGA)9-MLP-luc, an artificial SMAD3/4-specific reporter construct consisting of nine repeats of the SMAD3/4-specific recognition sequence, CAGA, cloned upstream of the SV40 minimal promoter and driving the expression of the luciferase gene (29), was a gift from S. Dennler (Institut Curie). The pRL-TK vector was from Promega (Madison, WI). TGF-β1 was purchased from R&D Systems Inc. (Minneapolis, MN). The TβRI inhibitor SD-208 was obtained from Scios, Inc., South San Francisco, CA.
Biochemical methods
Protein extraction and Western blotting were performed as previously described (30). Anti-Smad3 and anti-β-actin were from Zymed (San Francisco, CA) and Sigma-Aldrich, respectively. The rabbit anti-phospho-Smad2/3 antibody (31) was a generous gift from Dr. Edward Leof (Mayo Clinic College of Medicine, Rochester, MN). Secondary anti-mouse and anti-rabbit HRP-conjugated antibodies were from Santa-Cruz Biotechnology Inc. (Santa-Cruz, CA).
Cells transfections and luciferase assays
Melanoma cells were seeded in 24-well plates and transfected at approximately 70–80% confluency with the polycationic compound Fugene™ (Roche Diagnostics, Indianapolis, IN, USA) in fresh medium containing 1% FCS. 4h after transfection, cells were pre-incubated with SD-208 for 1h, followed by a 16-h incubation in the absence or presence of TGF-β. Cells were then rinsed twice with phosphate-buffered saline and lysed in passive lysis buffer (Promega). Luciferase activities were determined with a Dual-Glo luciferase assay kit according to the manufacturer’s protocol (Promega).
Invasion assays
Tissue culture Transwell™ inserts (8-μm pore size, Falcon, Franklin Lakes, NJ) were coated for 3h with 10 μg of growth factor-reduced Matrigel™ (Biocoat, BD Biosciences, San Jose, CA) in 100μl of PBS at 37°C. The chambers were air-dried for 16 h, the Matrigel™ barrier was then reconstituted with 100 μl DMEM for 24 h at 37°C, and chambers were placed into 24-well dishes containing 750 μl of W489 medium supplemented with 0,1% FCS. Cells (5×104) were added to the upper well of each chamber in 500 μl of serum-free W489. Invasion was measured after a 24h-incubation period, cells on the upper surface of the filter were wiped off with a cotton swab, and the cells on the underside of the membrane were fixed, stained with Diff-Quik™ (Dade Behring, Düdingen, Switzerland) and counted by bright-field microscopy at x200 in six random fields. Results are expressed as the means ± S.E.M. of three independent experiments.
Animal experiments
The animal protocols for bone metastasis experiments were approved by the Institutional Animal Care and Use Committee at the University of Virginia in Charlottesville. The protocol for subcutaneous xenograft of tumor cells in mice was approved by the Institutional Animal Care and Use Committee of Indiana University. All were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Subcutaneous tumor inoculation into the right flank ventricle was performed on anesthetized mice positioned laterally. Briefly, the skin was punctured using a 26-gauge needle attached to a 1-ml syringe containing suspended tumor cells. Tumor cells (2×106 in 0.1 ml of PBS) were inoculated slowly over 20 seconds. After palpable tumor is formed the mice were randomly divided into 2 groups (n=10) to receive either vehicle or SD-208 (60mg/kg/d). Treatment was given by daily gavage for 14 days continuously. Tumor growth was measured using a digital caliper every 2 days and tumor volume was calculated using the formula for ovoid tumor volume 4/3π × L/2 (W/2)2, where L and W equal mid-axis length and width, respectively.
Athymic female nude mice 4 weeks of age were housed in laminar flow isolated hoods. Water supplemented with vitamin K and autoclaved mouse chow were provided ad libitum. For the therapeutic protocol, 3 groups of mice (n=12/group) were inoculated with 1205Lu cells and received either vehicle or SD-208, 20 or 60mg/kg/day, after lesion detection on X-ray 12 days after inoculation. The drug was administered by daily gavage. In parallel we established a preventive protocol whereby a fourth group of mice (n=12) received SD-208 60mg/kg 2 days before 1205Lu tumor inoculation and the drug was further administered by daily gavage until termination of the experiment. The same vehicle-treated group served as a control for both protocols. Radiographs were taken under mouse anesthesia mixture (30% ketamine and 20% xylazine in 0.9% NaCl). Tumor inoculation into the left cardiac ventricle was performed on anesthetized mice positioned ventral side up, as described previously (32). Briefly, the left cardiac ventricle was punctured percutaneously using a 26-gauge needle attached to a 1-ml syringe containing suspended tumor cells. Visualization of bright red blood entering the hub of the needle in a pulsatile fashion indicated a correct position in the left cardiac ventricle. Tumor cells (105 in 0.1 ml of PBS) were inoculated slowly over 1 min. Micewere followed by radiography for the development of bone lesions throughout the experiments. Mice were x-rayed in a prone and lateral position using a Digital Faxitron MX-20 with digital camera (Faxitron x-ray, Wheeling, IL), as described previously (32). Radiographs where taken at 1X magnification and when a lesion was suspected, additional images with higher magnification (4X) were taken. Images were saved and lesion areas were measured and analyzed using MetaMorph software (Molecular Devices, Downingtown. PA).
Bone histology and histomorphometry: forelimbs, hindlimbs and vertebral spines were removed from mice at the time of experimental termination. Tissues were fixed in 10% neutral buffered formalin for 48h, decalcified in 10% EDTA for 2 weeks, processed using an automated tissue processor (Excelsior, Thermoelectric), and embedded in paraffin. Mid-sagittal 3.5 μm sections were stained with hematoxylin and eosin with orange G and phloxine to visualize new bone, and with TRAP stain to visualize osteoclast. All sections were viewed on a Leica DM LB compound microscope outfitted with a Q-Imaging Micropublisher Cooled CCD color digital camera (Vashaw Scientific Inc., Washington, DC). Images were captured and analyzed using MetaMorph software (Universal Imaging Corporation). Tumor burden, defined as area of bone occupied by the tumor, was calculated at the tibia, femur and humerus at 50X magnification on H&E stained sections as previously described (4). Total bone area defined as area of both cortical and trabecular bone was calculated at the same sites using standard bone histomorphometry nomenclature (33). Total tumor area, total bone area and osteoclast number/mm of tumor/bone interface were measured in midsections of tibiae and femora without knowledge of experimental groups. Osteoblast (OB/HPF) and osteoclast count (OCL/HFP) in non tumor-bearing mice was performed on spines at 200x. All histomorphometric analysis was performed using MetaMorph software (Universal imaging corporation, Downington, PA).
RNA extraction and gene expression analysis
In vitro experiments: Total RNA was isolated using an Rneasy™ kit (Qiagen GmbH, Hilden Germany). In vivo experiments: The femur and tibiae from mice were dissected and cleaned from adhering tissues. The cartilage ends were cut off and the tumor cells in the marrow cavity were flushed out using cold PBS in a syringe with a sterile needle. After centrifugation, cells were resuspended in TRIzol (Invitrogen, San Diego, CA). Following the addition of chloroform and centrifugation, total RNA was further isolated using an Rneasy™ kit, according to the manufacturer’s instructions. Genomic DNA contaminations were eliminated by DNAse I treatment. Five hundreds nanograms (in vitro experiments) or 3μg (in vivo experiments) of RNA from each sample was reverse transcribed using the Superscript II reverse transcriptase (Invitrogen) following the manufacturer’s instructions. The resulting cDNAs were then processed for real-time PCR using SYBR Green technology. Reactions were carried out in a MyiQ Single-Color Real-Time PCR Detection System (BioRad) for 40 cycles (95°C for 15 sec/60°C for 30 sec/72°C for 30 sec) after an initial 15-min incubation at 95°C. Primers were as follows for in vitro experiments, PTHrP (sense, 5′-actcgctctgcctggttaga-3′′; anti-sense, 5′-ggaggtgtcagacaggtggt-3′); IL-11 (sense, 5′-tgaagactcggctgtgacc-3′; anti-sense, 5′-cctcacggaaggactgtctc-3′); CTGF (sense, 5′-gctaccacatttcctacctagaaatca-3′; 5′-gacagtccgtcaaaacagattgtt-3′); RUNX2 (sense, 5′-gtttccagcaggtagctgag-3′; anti-sense, 5′-cttggccctccattgtaaga-3′). Target gene expression was normalized against the endogenous control genes RPL32 (sense, 5′-tcaggtgatcttcccacctc-3′; anti-sense, 5′-accacatcccatatccctca-3′). Samples were analyzed in triplicate and data were analyzed using the ΔΔCt method as described previously (34). Bone metastases were detected in mouse bone marrow by real-time RT-PCR, with primers specific for human RPL32 (sense, 5′-tcaggtgatcttcccacctc-3′; anti-sense, 5′-accacatcccatatccctca-3′). The absence of amplification product after 40 cycles of PCR monitored in real-time was considered as absence of human melanoma cells in analyzed bones and non-establishment of bone metastases.
Statistical analyses
Differences in osteolytic lesion areas and tumor volume between groups were determined by two-way analysis of variance (ANOVA). Statistical analysis for bone and tumor Histomorphometry were done by one-way ANOVA followed by Tukey’s multiple comparison post-test. Differences in tumor take between the groups were done by one-way ANOVA followed by Newman-Keuls Multiple Comparison Test. All results were expressed as mean ± SEM, and p<0.05 was considered significant (GraphPad Prism).
RESULTS
TβRI blockade with SD-208 inhibits TGF-β-induced Smad3 phosphorylation and Smad3/4-dependent gene transcription in human melanoma cells
To determine whether SD-208 had a direct effect on Smad3 phosphorylation induced by TGF-β in melanoma cells, 1205Lu human melanoma cell cultures were incubated with various concentrations of SD-208 prior to TGF-β stimulation. Smad3 and P-Smad3 levels were estimated by Western blot analysis. As expected, TGF-β induced a rapid and dramatic elevation of P-Smad3 levels (Figure 1A, lane 2 vs. lane 1). SD-208 dose-dependently inhibited such activation (Figure 1A, lanes 3–10). A 0.5μM concentration of SD-208 was sufficient to abrogate Smad3 phosphorylation in response to TGF-β (Figure 1A, lane 6).
Figure 1. TβRI blockade with SD-208 inhibits Smad3 phosphorylation and Smad3/4-specific gene transcription in 1205Lu human melanoma cells.
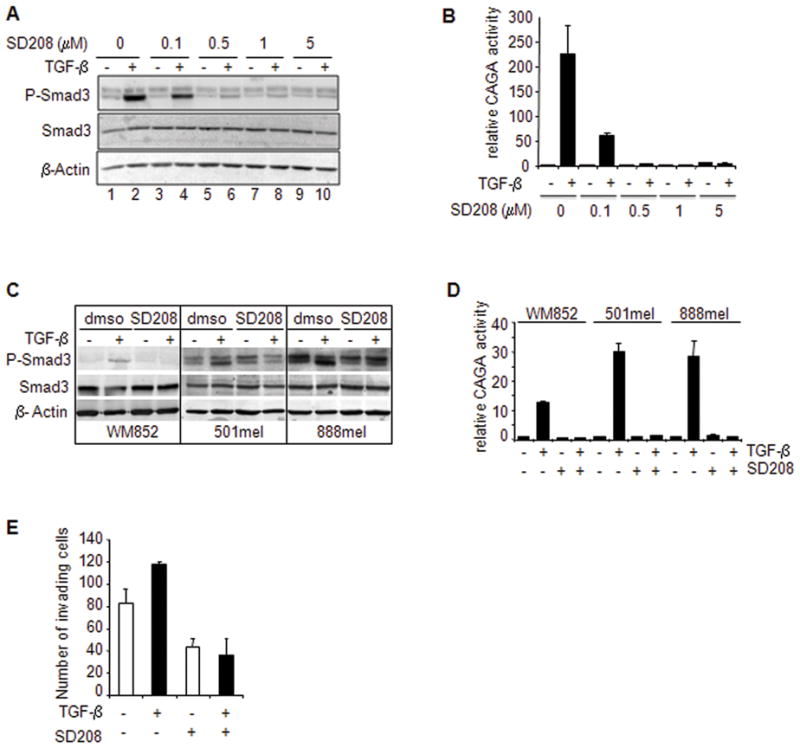
A, Sub-confluent 1205Lu human melanoma cell cultures were incubated in fresh medium containing 1% serum for 2 hours. SD-208 was then added fro a 1-h pre-incubation before addition of TGF-β (5 ng/ml) for 30 min. P-Smad3 and Smad3 contents were determined in 60 μg of whole cell lysates by Western blotting using luminescence. An antibody raised against β-actin was used for normalization. Representative results from one of three experiments with highly similar results are shown. B, subconfluent 1205Lu melanoma cell cultures were transfected with 0.4 μg (CAGA)9-MLP-luc vector together with 0.2 μg pRL-TK Renilla luciferase expression vector. Four hours post-transfection, cultures were incubated with either vehicle or SD-208 at increasing concentrations prior to stimulation with TGF-β (5 ng/mL). Luciferase activities were measured in cell extracts 20 h post-transfection. Results, expressed as fold-induction over control without TGF-β, are the mean +/− SE of three independent experiments. C, P-Smad3 and Smad3 protein levels in WM852, 501mel and 888mel melanoma cell lines under the same experimental conditions as described for 1205Lu cells in panel A. D, Smad3/4-specific transcription in WM852, 501mel and 888mel melanoma cell lines under the same experimental conditions as described for 1205Lu cells in panel B. E, Matrigel™ invasion of 1205Lu cells left untreated vs. treated with TGF-β (5ng/ml) in the absence or presence of SD208 (1μM). Results are the mean ± s.e.m. of two independent experiments, each performed with triplicate dishes.
Next, SD-208 was tested for its ability to affect TGF-β-driven transactivation of the SMAD3/4-specific reporter construct (CAGA)9-MLP-luc. As shown in (Figure 1B), and as expected from previous observations, TGF-β efficiently transactivated (CAGA)9-MLP-luc, up to 100-fold above control values. A 60 min preincubation of 1205Lu human melanoma cells with increasing concentrations of SD-208 resulted in a dose-dependent inhibition of TGF-β-induced SMAD3/4-specific reporter transactivation. Again, similar to what was observed in the case of TGF-β-induced Smad3 phosphorylation, the 0.5μM concentration of SD-208 achieved complete inhibition of TGF-β effect. Similar results were obtained in three additional human melanoma cell lines, WM852, 501mel and 888mel (Figure 1C and 1D).
SD-208 inhibits Matrigel™ invasion by human melanoma cells
We previously demonstrated that Smad7 overexpression in 1205Lu human melanoma cells inhibits the capacity to penetrate Matrigel™, suggesting that the TGF-β pathway is directly implicated in the invasive phenotype of melanoma cells (17). However, with this approach, we could not rule out a possible effect of Smad7 on invasion that would be independent from its inhibitory effect on TGF-β signaling. As shown in Figure 1E, exogenous TGF-β increased the capacity of 1205Lu cells to invade Matrigel™. On the other hand, pharmacologic inhibition of TGF-β signaling with SD-208 reduced the capacity of 1205Lu human melanoma cells to invade Matrigel™ by approx. 60%, and abrogated the stimulatory effect of exogenous TGF-β on the invasive capacity of melanoma cells.
SD-208 inhibits TGF-β-induced expression of osteolytic genes by 1205Lu human melanoma cells
The capacity of cancer cells to metastasize to bone is largely dependent on the expression of a limited set of genes considered a specific signature for bone metastasis (15). This set of genes, initially identified in breast carcinoma cells, is also activated in melanoma (18). Incubation of cultured 1205Lu human melanoma cells with TGF-β resulted in a rapid and sustained upregulation of the expression of PTHrP, IL-11, CTGF and Runx2 (Figure 2). SD-208, added 30 min. prior to TGF-β addition, reduced IL-11 basal mRNA steady-state levels, and inhibited TGF-β enhanced expression of all genes surveyed (Figure 2). Together, these data indicate that SD-208 efficiently prevents high expression of osteolytic genes by melanoma cells exposed to TGF-β.
Figure 2. SD-208 inhibits TGF-β-induced expression of osteolytic genes by 1205Lu human melanoma cells.
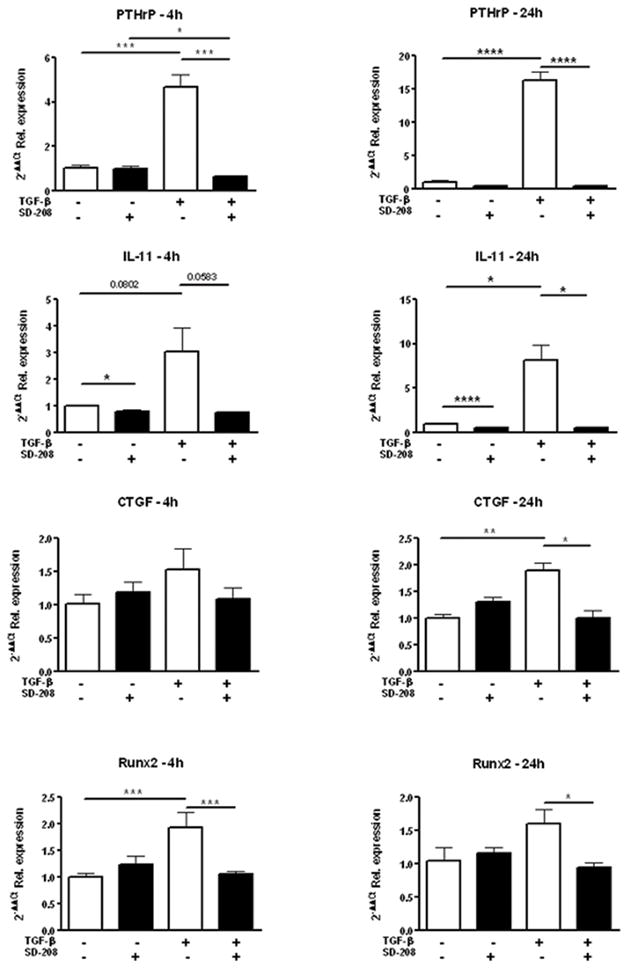
Sub-confluent 1205Lu human melanoma cell cultures were incubated in fresh medium containing 1% serum for 2 hours. SD-208 was then added for a 1-h pre-incubation before addition of TGF-β (5 ng/ml) for either 4 or 24h, at which time-points RNA was extracted and gene expression for PTHrP, IL-11, CTGF and RUNX2 was examined by quantitative RT-PCR.
SD-208 exerts potent anti-metastatic activity in vivo
1205Lu cells exhibit high autonomous, ligand-induced, constitutive TGF-β/Smad signaling and a strong transcriptional response to exogenous TGF-β (16). Moreover, they are highly invasive in vitro, tumorigenic in vivo (17), and form metastases, mainly osteolytic, when inoculated into the left cardiac ventricle of nude mice, an experimental model that addresses the process of metastasis from entry of tumor cells into the arterial circulation to the establishment of bone metastasis, and tumor-bone interactions (18). These represent an ideal human melanoma cell line for testing the efficacy of systemic administration of a TβRI/ALK5 inhibitor.
Therapeutic administration of SD-208 attenuates melanoma bone metastasis growth and increases survival in mice
In a first set of experiments, mice were inoculated 1205Lu melanoma cells. All mice (n=36) inoculated with parental 1205Lu cells developed osteolytic bone metastases detectable by X-ray 12 days post tumor inoculation. At that stage, mice were randomly divided into three groups of 12: one received vehicle alone by daily gavage (control group), while mice from the two other groups received SD-208, either 20mg/kg/day (group 20) or 60 mg/kg/day (group 60). This experimental approach was identified as therapeutic protocol.
All mice treated with vehicle alone had to be euthanized by week 5 as the metastatic burden rapidly increased. No statistically significant difference was observed between group 20 and the control group. However, group 60 exhibited a highly significant reduction of lesion area at week 5, as determined by quantitative computerized image analysis of osteolytic lesion area on radiographs (Figure 3A and 3B).
Figure 3. SD-208 inhibits the development of 1205lu bone metastases in a therapeutic protocol.
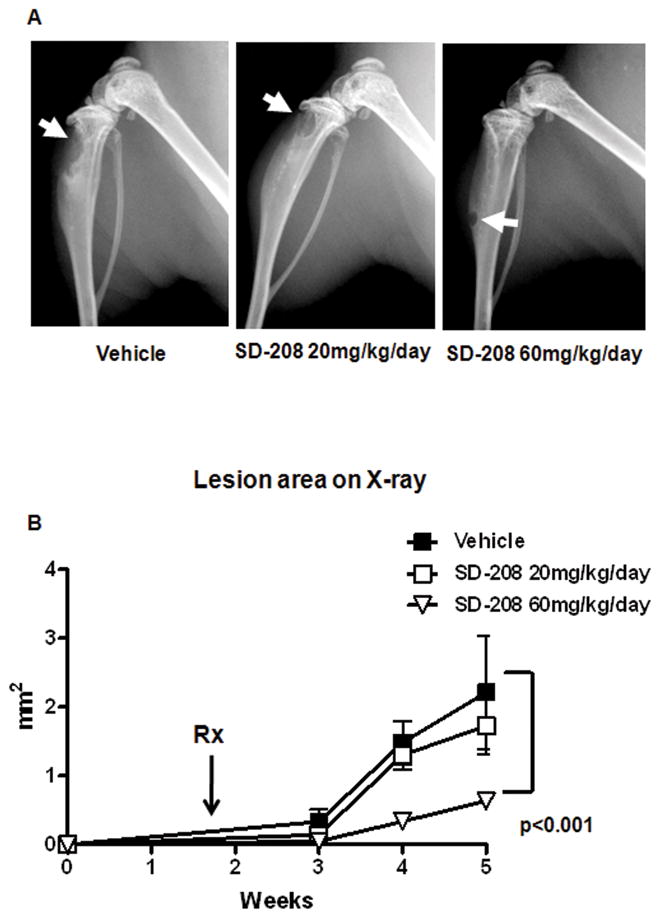
Mice were inoculated with 1205Lu cells. 12 days later, osteolytic lesions were detected by X-ray and mice divided into 3 identical groups (n=12/group) that received either vehicle or SD-208, 20 or 60mg/kg/day, administered by daily gavage. A, Representative x-ray shows a dramatic reduction in lesion area with SD-208 60 mg/kg/day, 5 weeks after inoculation. B, Quantitative analysis of lesion area showed a significant reduction in lesion area in mice treated with the highest dose of SD-208. There was a slight, non-significant, reduction in lesion area with SD-208 20 mg/kg/day.
Preventive administration of SD-208 inhibits 1205Lu human melanoma metastasis to bone by reducing tumor take
In a second set of experiments, SD-208, 60mg/kg/day was administered by gavage two days prior to 1205Lu melanoma cell inoculation to mice (n=12). The control group (n=12) received vehicle 2 days prior to inoculation of tumor cells. Daily gavage with either vehicle or SD-208 was continued for the duration of the experiment (5 weeks). As shown in (Figure 4A and 4B), SD-208 preventive treatment significantly reduced the development of osteolytic bone metastases, as determined by quantitative computerized image analysis of osteolytic lesion area on radiographs. This effect was greater than that observed in the therapeutic protocol (Figure 4C). To determine whether reduced osteolytic lesion area may result from either reduced establishment of metastases, or from reduced metastasis growth with no changes in the number of metastases, metastases were collected form mice by bone marrow flushing and species-specific quantitative PCR using human RPL32 was used to detect whether human tumor was established in bone (tumor take). As shown in Figure 4D, no significant difference in tumor take was observed between vehicle and mice receiving SD-208 in the therapeutic protocol, consistent with the fact that inoculation of all mice occurred before SD-208 administration, the later taking place once metastases are established. Thus, in the therapeutic protocol, reduced osteolytic area results from slower growth of established metastases.
Figure 4. SD-208 inhibits the development and progression of 1205lu bone metastases in a preventive protocol.
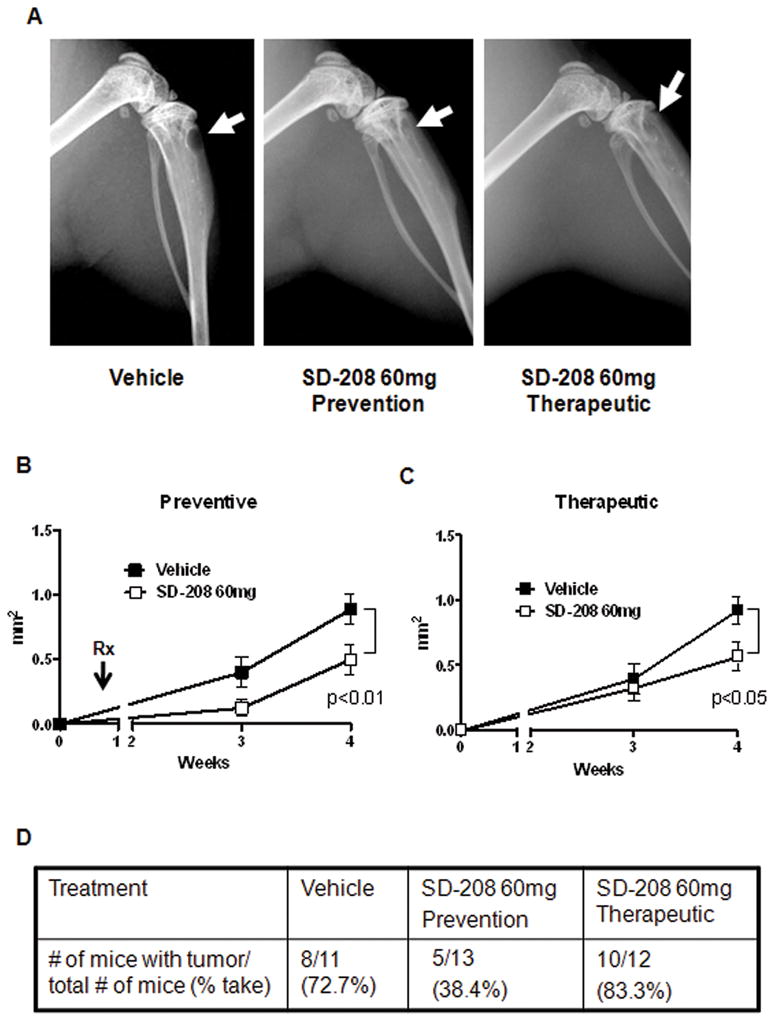
A, Representative X-ray shows a reduction in lesion area with SD-208 (60mg/kg/day) both in a preventive as well as therapeutic protocol (see corresponding Materials and Methods section for details). B and C, quantitative analysis of lesion area over a 4-week period in the preventive (B) and therapeutic (C) protocols. Note that both experimental approaches show a significant reduction in lesion area in mice treated with SD-208. D, Assessment of tumor take in the preventive vs. therapeutic protocol: note the significant reduction in tumor take in mice preventively treated with SD-208.
In contrast, in the preventive protocol tumor take was dramatically reduced tumor in mice receiving SD-208 (p<0.02). Thus, preventive TβRI kinase inhibition antagonizes the establishment of bone metastases by 1205Lu human melanoma cells. Such effect is likely additive to the inhibitory activity of SD-208 on metastasis growth, as identified in the therapeutic protocol.
Histomorphometric analysis showed a similar reduction in tumor burden (Figure 5A and 5B) as well as a corresponding increase in total bone area with both the 20 and 60 mg/kg dose (Figure 5C). This was accompanied by a reduction in osteoclast number at the tumor bone interface in mice treated with SD-208 (Figure 5D). Bone histomorphometry in non tumor-bearing bones showed a reduction in osteoclast number with increase in osteoblast number in mice treated with SD-208 (Figure 5E and 5F) compared with vehicle.
Figure 5. SD-208 inhibits osteolytic tumor area and affects osteoclast and osteoblast numbers.
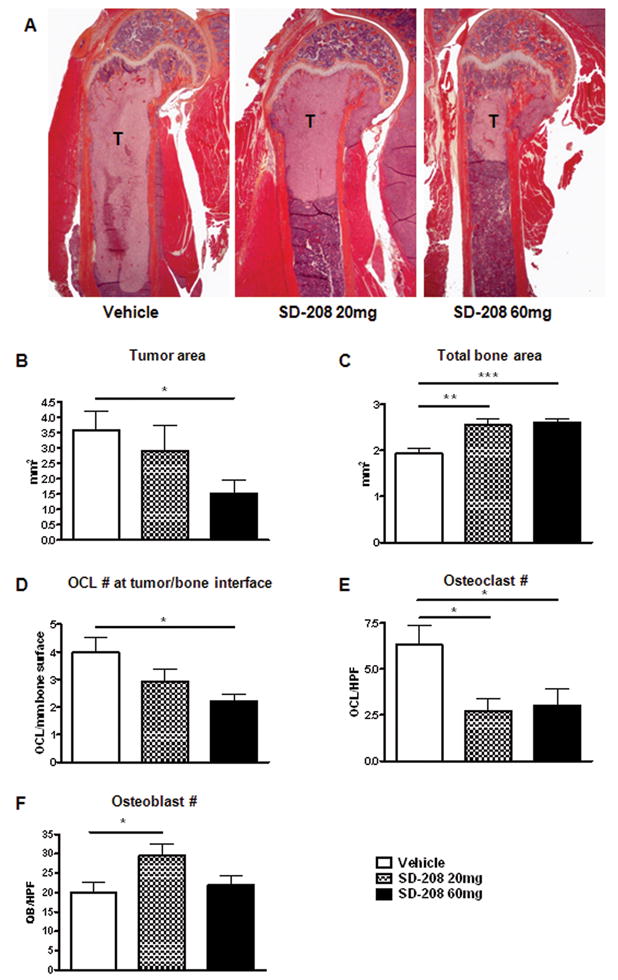
A, Representative histological section from the humerus shows a reduction in tumor area in mice treated with SD-208. B, Quantitative histomorphometry shows a significant reduction in tumor area in mice treated with SD-208 60mg, accompanied by an increase in total bone area (C) and reduction in osteoclast number at the tumor/bone interface (D). E, SD-208 reduced total osteoclast number while increasing osteoblast number (F) in non tumor-bearing bones.
Therapeutic administration of SD-208 has no effect on the growth of subcutaneous xenografts of 1205Lu melanoma cells
In another set of experiments, mice were inoculated subcutaneously with 1205Lu melanoma cells. All mice (n=20) inoculated with 1205Lu cells developed palpable tumor 5 days after inoculation. Mice were randomly divided into two groups of 10 to receive either vehicle or SD-208 (60 mg/kg/day) for 2 weeks. There was no statistically significant difference in tumor volume between treated and untreated mice (p=0.234).
DISCUSSION
TGF-β is important in both the pathophysiology of melanoma and that of bone metastases due to breast cancer. Our data indicate that TGF-β, abundant in bone matrix, also promotes melanoma bone metastasis, likely through induction of prometastatic and osteolytic genes: PTHrP, IL-11, CTGF and RUNX2, as well as to directly promote invasion. These data are consistent with our previous data in which overexpression of Smad7 in the 1205Lu melanoma line reduced invasion and bone metastases (18). In that setting, the effects of TGF-β blockade were on the tumor cells only; the host effects were indirect, a result of Smad7 to block TGF-β induction of invasion and prometastatic and osteolytic gene expression. Here, we show that systemic inhibition of TGF-β signaling has similar effects to inhibit the establishment and progression of bone metastases. However, systemic TGF-β blockade also had direct effects on the host to increase bone mass by reducing osteoclastic bone resorption and increasing osteoblast activity, even at sites distant from bone metastases. The latter data are consistent with recent publications showing that blockade of TGF-β signaling has direct effects to reduce osteoclast formation and bone resorption as well as to increase osteoblast differentiation and bone formation (35–37). Similar to these melanoma bone metastases studies, SD-208 was previously shown to increase survival following orthotopic implantation of glioma cells (25), while inhibition of TGF-β by another small-molecule TβRI kinase inhibitor decreased breast cancer metastases to lungs and skeleton in mice (19). SD-208 was also effective to prevent breast cancer bone metastases and improve survival in MDA-MB-231 model (36). Here we showed that SD-208 significantly reduced osteolytic lesion area and decreased tumor burden in mice associated with increased survival in a dose-dependent manner. We show for the first time that in the preventive setting TGF-β signaling blockade reduced both the establishment and progression of tumor in bone, as indicated by measurement of human tumor marker RPL32. Further, in the therapeutic setting, TGF-β signaling blockade reduced the progression of established bone metastases. In contrast, systemic inhibition of TGF-β signaling has no effect on the growth of established subcutaneous tumors. The latter results differ from those obtained upon stable Smad7 overexpression in melanoma cells prior to xenograft transplantation, which resulted in a significant delay in tumor growth (17). We recently found that stable Smad7 overexpression has profound effect on N-cadherin stability which generates strong heterotypic cell-cell interactions that prevent tumor development (38). It is likely that SD-208 administered to mice after tumors are palpable will not recapitulate the mechanisms by which TGF-β signaling blockade by Smad7 delays tumor growth in the xenograft model.
An possible mediator of TGF-β signaling required for melanoma bone metastasis may be the transcription factor GLI2, recently identified as a direct target of TGF-β signaling (39, 40), whose knockdown in melanoma cells dramatically reduces their capacity to form bone metastases (41). GLI2 expression was associated with the most aggressive tumors in patients with melanoma. We also found that GLI2 promotes melanoma cell invasiveness through downregulation of E-cadherin (41), a phenomenon that has been associated with progression of melanoma from radial to vertical growth phase, a critical event leading to metastatic spreading (42). In the experiments presented in this report, SD-208 consistently reduced basal and TGF-β induced GLI2 expression in melanoma cells (not shown), which thus could account, at least in part, for the inhibitory effect of SD-208 on melanoma bone metastasis.
In summary, we have demonstrated that the TβRI kinase inhibitor SD-208 antagonizes TGF-β signaling and gene responses in vitro. Activation of osteolytic genes by TGF-β in human melanoma cells is thus largely prevented. Furthermore, we have established that systemic administration of the TβRI inhibitor prevents the establishment and growth of osteolytic bone metastases in a prevention protocol whereby SD-208 is administered two days prior to tumor cell inoculation. Most importantly, we also demonstrate that SD-208 is capable of inhibiting the growth of established osteolytic bone metastases, providing novel evidence that systemic blockade of TGF-β signaling may be an effective treatment for skeletal metastases in malignant melanoma.
Acknowledgments
The authors are thankful to Drs. Meenhard Herlyn (Wistar Institute, Philadelphia, PA; Sylviane Dennler (INSERM U697) and Jean-Michel Gauthier (Glaxo-Wellcome, Les Ulis, France), Edward Leof (Mayo Clinic, Rochester, MN), and Peter ten Dijke (Leyden, The Netherlands) for providing us with reagents essential for these studies. This work was supported by the National Institutes of Health, National Cancer Institute (R01CA69158) and the U.S. Army Medical Research and Materiel Command, Department of Defense (award number 0410920) to T.A.G., the Emile and Henriette Goutière donation as well as grants from INSERM, Ligue Nationale Contre le Cancer (LNCC, Comité des Yvelines), Cancéropole Ile-de-France, and INCa, France (to A.M.). D.J. was the recipient of a post-doctoral fellowship awarded by Fondation pour la Recherche Médicale (France).
References
- 1.Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007. CA Cancer J Clin. 2007;57:43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 2.Akslen LA, Hove LM, Hartveit F. Metastatic distribution in malignant melanoma. A 30-year autopsy study. Invasion Metastasis. 1987;7:253–63. [PubMed] [Google Scholar]
- 3.Javelaud D, Alexaki VI, Mauviel A. Transforming growth factor-beta in cutaneous melanoma. Pigment Cell Melanoma Res. 2008;21:123–32. doi: 10.1111/j.1755-148X.2008.00450.x. [DOI] [PubMed] [Google Scholar]
- 4.Yin JJ, Selander K, Chirgwin JM, et al. TGF-beta signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J Clin Invest. 1999;103:197–206. doi: 10.1172/JCI3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Massague J. TGFbeta in Cancer. Cell. 2008;134:215–30. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Massague J. G1 cell-cycle control and cancer. Nature. 2004;432:298–306. doi: 10.1038/nature03094. [DOI] [PubMed] [Google Scholar]
- 7.Reguly T, Wrana JL. In or out? The dynamics of Smad nucleocytoplasmic shuttling. Trends Cell Biol. 2003;13:216–20. doi: 10.1016/s0962-8924(03)00075-8. [DOI] [PubMed] [Google Scholar]
- 8.Javelaud D, Mauviel A. Mammalian transforming growth factor-betas: Smad signaling and physio-pathological roles. Int J Biochem Cell Biol. 2004;36:1161–5. doi: 10.1016/S1357-2725(03)00255-3. [DOI] [PubMed] [Google Scholar]
- 9.Feng XH, Derynck R. Specificity and versatility in tgf-beta signaling through Smads. Annu Rev Cell Dev Biol. 2005;21:659–93. doi: 10.1146/annurev.cellbio.21.022404.142018. [DOI] [PubMed] [Google Scholar]
- 10.ten Dijke P, Hill CS. New insights into TGF-beta-Smad signalling. Trends Biochem Sci. 2004;29:265–73. doi: 10.1016/j.tibs.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 11.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 12.Kozlow W, Guise TA. Breast cancer metastasis to bone: mechanisms of osteolysis and implications for therapy. J Mammary Gland Biol Neoplasia. 2005;10:169–80. doi: 10.1007/s10911-005-5399-8. [DOI] [PubMed] [Google Scholar]
- 13.Kakonen SM, Selander KS, Chirgwin JM, et al. Transforming growth factor-beta stimulates parathyroid hormone-related protein and osteolytic metastases via Smad and mitogen-activated protein kinase signaling pathways. J Biol Chem. 2002;277:24571–8. doi: 10.1074/jbc.M202561200. [DOI] [PubMed] [Google Scholar]
- 14.Minn AJ, Kang Y, Serganova I, et al. Distinct organ-specific metastatic potential of individual breast cancer cells and primary tumors. J Clin Invest. 2005;115:44–55. doi: 10.1172/JCI22320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kang Y, Siegel PM, Shu W, et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003;3:537–49. doi: 10.1016/s1535-6108(03)00132-6. [DOI] [PubMed] [Google Scholar]
- 16.Rodeck U, Nishiyama T, Mauviel A. Independent regulation of growth and SMAD-mediated transcription by transforming growth factor beta in human melanoma cells. Cancer Res. 1999;59:547–50. [PubMed] [Google Scholar]
- 17.Javelaud D, Delmas V, Moller M, et al. Stable overexpression of Smad7 in human melanoma cells inhibits their tumorigenicity in vitro and in vivo. Oncogene. 2005;24:7624–9. doi: 10.1038/sj.onc.1208900. [DOI] [PubMed] [Google Scholar]
- 18.Javelaud D, Mohammad KS, McKenna CR, et al. Stable overexpression of Smad7 in human melanoma cells impairs bone metastasis. Cancer Res. 2007;67:2317–24. doi: 10.1158/0008-5472.CAN-06-3950. [DOI] [PubMed] [Google Scholar]
- 19.Bandyopadhyay A, Agyin JK, Wang L, et al. Inhibition of pulmonary and skeletal metastasis by a transforming growth factor-beta type I receptor kinase inhibitor. Cancer Res. 2006;66:6714–21. doi: 10.1158/0008-5472.CAN-05-3565. [DOI] [PubMed] [Google Scholar]
- 20.Azuma H, Ehata S, Miyazaki H, et al. Effect of Smad7 expression on metastasis of mouse mammary carcinoma JygMC(A) cells. J Natl Cancer Inst. 2005;97:1734–46. doi: 10.1093/jnci/dji399. [DOI] [PubMed] [Google Scholar]
- 21.Ijichi H, Chytil A, Gorska AE, et al. Aggressive pancreatic ductal adenocarcinoma in mice caused by pancreas-specific blockade of transforming growth factor-beta signaling in cooperation with active Kras expression. Genes Dev. 2006;20:3147–60. doi: 10.1101/gad.1475506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muraoka RS, Dumont N, Ritter CA, et al. Blockade of TGF-beta inhibits mammary tumor cell viability, migration, and metastases. J Clin Invest. 2002;109:1551–9. doi: 10.1172/JCI15234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rodeck U, Bossler A, Graeven U, et al. Transforming growth factor beta production and responsiveness in normal human melanocytes and melanoma cells. Cancer Res. 1994;54:575–81. [PubMed] [Google Scholar]
- 24.Dennler S, Mauviel A, Verrecchia F. TGF-beta and Stromal Influences over Local Tumor Invasion. In: Jakowlew Sonia B., editor. Transforming Growth Factor-beta in Cancer Therapy. II. Humana Press; 2008. pp. 531–51. [Google Scholar]
- 25.Uhl M, Aulwurm S, Wischhusen J, et al. SD-208, a novel transforming growth factor beta receptor I kinase inhibitor, inhibits growth and invasiveness and enhances immunogenicity of murine and human glioma cells in vitro and in vivo. Cancer Res. 2004;64:7954–61. doi: 10.1158/0008-5472.CAN-04-1013. [DOI] [PubMed] [Google Scholar]
- 26.MacDougall JR, Kobayashi H, Kerbel RS. Responsiveness of normal, dysplastic melanocytes and melanoma cells from different lesional stages of disease progression to the growth inhibitory effects of TGF-beta. Mol Cell Diff. 1993;1:21–40. [Google Scholar]
- 27.Rodeck U, Melber K, Kath R, et al. Constitutive expression of multiple growth factor genes by melanoma cells but not normal melanocytes. J Invest Dermatol. 1991;97:20–6. doi: 10.1111/1523-1747.ep12477822. [DOI] [PubMed] [Google Scholar]
- 28.Moore R, Champeval D, Denat L, et al. Involvement of cadherins 7 and 20 in mouse embryogenesis and melanocyte transformation. Oncogene. 2004;23:6726–35. doi: 10.1038/sj.onc.1207675. [DOI] [PubMed] [Google Scholar]
- 29.Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier JM. Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. Embo J. 1998;17:3091–100. doi: 10.1093/emboj/17.11.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Javelaud D, Laboureau J, Gabison E, Verrecchia F, Mauviel A. Disruption of basal JNK activity differentially affects key fibroblast functions important for wound healing. J Biol Chem. 2003;278:24624–8. doi: 10.1074/jbc.M301942200. [DOI] [PubMed] [Google Scholar]
- 31.Daniels CE, Wilkes MC, Edens M, et al. Imatinib mesylate inhibits the profibrogenic activity of TGF-beta and prevents bleomycin-mediated lung fibrosis. J Clin Invest. 2004;114:1308–16. doi: 10.1172/JCI19603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guise TA, Yin JJ, Taylor SD, et al. Evidence for a causal role of parathyroid hormone-related protein in the pathogenesis of human breast cancer-mediated osteolysis. J Clin Invest. 1996;98:1544–9. doi: 10.1172/JCI118947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parfitt AM, Drezner MK, Glorieux FH, et al. Bone histomorphometry: standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res. 1987;2:595–610. doi: 10.1002/jbmr.5650020617. [DOI] [PubMed] [Google Scholar]
- 34.Bartholin L, Wessner LL, Chirgwin JM, Guise TA. The human Cyr61 gene is a transcriptional target of transforming growth factor beta in cancer cells. Cancer Lett. 2007;246:230–6. doi: 10.1016/j.canlet.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 35.Mohammad KS, Chen CG, Balooch G, et al. Pharmacologic inhibition of the TGF-beta type I receptor kinase has anabolic and anti-catabolic effects on bone. PLoS One. 2009;4:e5275. doi: 10.1371/journal.pone.0005275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dunn LK, Mohammad KS, Fournier PG, et al. Hypoxia and TGF-beta drive breast cancer bone metastases through parallel signaling pathways in tumor cells and the bone microenvironment. PLoS One. 2009;4:e6896. doi: 10.1371/journal.pone.0006896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tang Y, Wu X, Lei W, et al. TGF-beta1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat Med. 2009;15:757–65. doi: 10.1038/nm.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Divito KA, Trabosh VA, Chen YS, et al. Smad7 restricts melanoma invasion by restoring N-cadherin expression and establishing heterotypic cell-cell interactions in vivo. Pigment Cell Melanoma Res. 2010;23:795–808. doi: 10.1111/j.1755-148X.2010.00758.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dennler S, Andre J, Alexaki I, et al. Induction of sonic hedgehog mediators by transforming growth factor-beta: Smad3-dependent activation of Gli2 and Gli1 expression in vitro and in vivo. Cancer Res. 2007;67:6981–6. doi: 10.1158/0008-5472.CAN-07-0491. [DOI] [PubMed] [Google Scholar]
- 40.Dennler S, Andre J, Verrecchia F, Mauviel A. Cloning of the human GLI2 Promoter: transcriptional activation by transforming growth factor-beta via SMAD3/beta-catenin cooperation. J Biol Chem. 2009;284:31523–31. doi: 10.1074/jbc.M109.059964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alexaki VI, Javelaud D, Van Kempen LC, et al. GLI2-Mediated Melanoma Invasion and Metastasis. J Natl Cancer Inst. 2010;102:1148–59. doi: 10.1093/jnci/djq257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gruss C, Herlyn M. Role of cadherins and matrixins in melanoma. Curr Opin Oncol. 2001;13:117–23. doi: 10.1097/00001622-200103000-00006. [DOI] [PubMed] [Google Scholar]