

Abstract
Neuroblastoma is the most common extracranial solid tumor encountered in children, and continues to carry a dismal prognosis. Focal adhesion kinase (FAK) has been shown to be upregulated in a number of human tumors and is related to tumor virulence and patient prognosis. We have demonstrated FAK expression in human neuroblastoma cell lines and tumors, and have shown that FAK is important for neuroblastoma tumor cell viability. We have also demonstrated that FAK inhibition through a number of different methods results in decreased neuroblastoma survival both in vitro and in vivo. The current review addresses the merit of further exploring FAK inhibition as a novel treatment for neuroblastoma.
Keywords: C4, FAK, neuroblastoma, NVP-TAE226, Y15
Neuroblastoma
Neuroblastoma remains the most common extra-cranial solid tumor in children and infants. This neuroendocrine tumor arises from neural crest cells, and can therefore be found in many locations throughout the body. The adrenal glands are the most common site for the tumor to arise, but neuroblastoma may be present in sympathetic neural tissue anywhere in the abdomen, pelvis, chest or neck. Over 50% of neuroblastoma occurs in children under the age of 2 years [1]. Unfortunately, there is no distinct set of symptoms characterizing neuroblastoma, frequently resulting in delays in diagnosis until the disease is in an advanced stage or widely metastatic.
Neuroblastoma is responsible for over 15% of all pediatric cancer deaths [2]. Long term overall survival rates for children diagnosed with neuroblastoma are less than 55% despite advances in surgery, chemotherapy, and radiation therapy. Because the majority of children have disseminated disease at diagnosis, even with appropriate treatment, the 3-year disease-free survival rate remains less than 20% [3]. Those children with the worst prognosis are those who have relapsed disease and tumors that are resistant to known chemotherapeutic protocols. In order to improve outcomes for these children, novel therapies for the treatment of neuroblastoma are desperately needed.
Neuroblastoma and cellular adhesion
Tumor cell motility and invasiveness clearly affects the virulence of neuroblastoma. Amplification of the MYCN oncogene is the most significant adverse prognostic indicator for human neuroblastoma and is present in approximately 20% of these tumors [4, 5]. MYCN expression correlates with higher relapse rates and tenacity of tumor cells [6, 7]. Zaizen et al illustrated the correlation between MYCN and invasive potential [8]. Using six neuroblastoma cell lines having varying levels of MYCN oncogene amplification, they performed matrigel invasion assays. They also utilized digital image analysis aimed at comparing changes in cell morphology to evaluate cellular motility. Their results confirmed that the neuroblastoma cell lines with amplification of the MYCN oncogene (IMR-32, GOTO, DZ) had a higher degree of invasion and motility than the non-amplified cell lines (NB-69, SK-N-SH) [8].
The mechanisms by which MYCN promotes an aggressive phenotype are not completely understood. In a study by Vasudevan, a cDNA microarray platform was utilized to discover gene products that were substantially up-regulated or down-regulated as a result of MYCN amplification [9]. MYCN led to up-regulation of multiple cell cycle-related genes, including MCM4, MCM7 and CDC2. Interestingly, genes that modulate cell-cell interactions and motility were down-regulated, including class I and II major histocompatibility complex (MHC) molecules, integrin subunit beta I, laminin, and many interferon-responsive genes including interferon-inducible protein 9-27. These results indicated that multiple gene targets contribute to the relationship between increased motility, and therefore tumor aggressiveness, and MYCN in neuroblastoma.
If tumor cells are to successfully metastasize, they must overcome their usual cell-cell or cell-extracellular matrix interactions. It would follow that a better understanding of the association between cellular adhesion and MYCN amplification is important in neuroblastoma metastasis. In a recent study, Ma and others reported that MYCN amplification correlates with levels of microRNA-9 (miR-9) in breast cancer cells. MicroRNA-9 is associated with increased cell motility and invasiveness through the targeting of the metastasis suppressor protein, E-cadherin [10]. More specifically to neuroblastoma, in 1995, Terpe and others reported that MYCN oncogene amplification was inversely correlated with the expression of the cell adhesion molecule, CD44s [11]. Akeson and Bernards showed that rat neuroblastoma cells transfected with a MYCN expression vector have significant reductions in mRNA and protein expression of neural cell adhesion molecule (NCAM) [12], a specific cell-cell adhesion molecule that is associated with an unfavorable prognostic phenotype in advanced stage neuroblastoma [13]. A number of studies have focused upon the relationship between MYCN and integrin expression. Research with the human neuroblastoma cell line SK-N-SH, showed that transfection of these cells with MYCN resulted in a decreased expression of the β1 integrin subunit [14]. These cells were noted to display more aggressive tumor growth when injected into nude mice [15]. These same investigators found that in addition to the downregulation of the β1 subunit, the α2 and α3 integrin subunits are also downregulated both at the RNA and protein levels in the face of MYCN [16]. Decreased expression of the integrin subunits allowed the cells to alter the cell-extracellular matrix association and survive as rounded, loose cellular aggregates. Also, these integrin subunits have been shown to be responsible for cellular interactions with laminin and collagen [17], so the loss of attachment to these extracellular matrix molecules confers an increased capability for migration. Finally, Tanaka and Fukuzawa demonstrated that MYCN overexpression in neuroblastoma cells in vitro resulted in decreased expression of α1 integrin, leading to decreased attachment and increased migratory activity of these neuroblastoma cells [18]. These studies demonstrate that cellular adhesion is inversely related to MYCN amplification, and is clearly implicated in the aggressive nature of neuroblastoma.
Focal adhesion kinase and neuroblastoma
Focal adhesion kinase (FAK) is a nonreceptor protein kinase that impacts a number of cell signaling pathways, including proliferation, cellular adhesion & migration [19]. Due to the importance of motility in neuroblastoma virulence, FAK has become a significant target of investigation. The first investigations involving FAK and neuroblastoma cell lines focused upon neurite outgrowth and neural differentiation more than the cancer aspect of neuroblastoma. For instance, Bozzo and colleagues were among the first to study FAK in neuroblastoma cell lines [17]. They showed that adhesion of SH-SY5Y neuroblastoma cells to laminin and collagen type IV caused tyrosine phosphorylation of various proteins in the 100-130 kDa range. Using protein specific antibodies, it was determined that focal adhesion kinase was among those kinases that were phosphorylated. When this phosphorylation was blocked with genistein, a specific tyrosine kinase inhibitor, neurite outgrowth in these cells was strongly inhibited. Other early studies of FAK expression in neuroblastoma involved the use of SH-SY5Y neuroblastoma cells to investigate the relationship between insulin-like growth factor −1 (IGF-1) and FAK activation in the role of neuronal morphology [20]. In these studies, the investigators determined that treatment of SH-SY5Y cells with IGF-1 caused lamellipodial advance, which signaled cell migration. They also noted that IGF-1 treatment of these cells resulted in FAK phosphorylation. Using these findings, in a later study, Kim and Feldman noted that mannitol-induced cellular detachment of SH-EP neuroblastoma cells was associated with FAK dephosphorylation that could be abrogated by treatment with IGF-1 [21]. Next, the investigators from this group utilized okadaic acid, a serine phosphatase inhibitor, to block the tyrosine phosphorylation of FAK in SH-EP neuroblastoma cells to study changes in cell morphology. Unlike their findings with mannitol, the okadaic acid-induced changes in cell morphology and FAK phosphorylation were not reversed by IGF-1 treatment [22]. Although these studies focused upon the relationship between growth factors and FAK, they revealed an association between the loss of FAK phosphorylation and apoptosis in neuroblastoma cells.
Wu and colleagues showed that FAK catalytic activity is important in NB8 neuroblastoma cell motility [23]. They found that these cells express α5β1integrin as do advanced stage human neuroblastoma tumors. This integrin subunit confers advanced migratory potential for these cells, and the authors demonstrated with the use of small hairpin RNA directed to FAK, that FAK phosphorylation was critical to the function of α5β1integrin [23]. This study provides a further connection between FAK and aggressive neuroblastomas.
FAK has been shown to be upregulated in a number of human tumors including breast [24, 25], pancreatic [26], and ovarian cancers [27]. Recently, FAK expression was investigated in human neuroblastoma tumors. Immunohistochemical staining revealed FAK to be present in 73% of neuroblastoma tumor specimens examined [28]. The researchers also noted that FAK staining correlated with FAK mRNA abundance as detected by real time quantitative PCR. In addition, FAK staining was significantly increased in INSS Stage IV tumors with amplification of the MYCN oncogene [28]. As previously discussed, MYCN is known to mediate signals involving cell adhesion, and since FAK is involved with adhesion, it could be hypothesized that FAK may be influenced by this oncogene. In fact, Beierle and others demonstrated through dual luciferase assays, chromatin immunoprecipitation and electrophoretic mobility shift assays that MYCN functions as a transcription factor for FAK. They showed that MYCN binds to the FAK promoter and augments FAK expression in vitro and in vivo [29]. The FAK-MYCN interaction augments the utility to seek out therapies to target FAK expression in neuroblastoma.
Lymphangiogenesis is important in the metastatic spread of human tumors including neuroblastoma [30]. Vascular endothelial growth factor receptor-3 (VEGFR-3) is a receptor tyrosine kinase that is critical in tumor lymphangiogenesis and the formation of tumor metastasis in a number of human cancers including breast [31], non-small cell lung [32], and prostate cancer [33]. Using PCR and immunohistochemical staining, VEGFR-3 has been detected in both human neuroblastoma cell lines and tumor specimens [30, 34, 35]. We have also found VEGFR-3 expression in human neuroblastoma cell lines (Fig. 1). Recently, Garces demonstrated through immunoprecipitation and confocal microscopy that VEGFR-3 interacts with FAK in human breast cancer cell lines [36]. Preliminary studies have been performed that demonstrate that these two kinases, FAK and VEGFR-3, interact in neuroblastoma cell lines also. SK-N-AS neuroblastoma cells were stained with fluorescent-tagged antibodies to FAK and VEGFR-3, and confocal microscopy was employed to determine colocalization of the stains. There was obvious colocalization of FAK and VEGFR-3 in this neuroblastoma cell line as detected by confocal microscopy (Fig. 2). Since metastatic disease portends a worse outcome in neuroblastoma, and lymphangiogenesis, VEGFR-3, and FAK all seem to be related, it would follow that FAK is relevant to neuroblastoma, and would be a potential target for the development of novel therapies.
Figure 1. VEGFR-3 protein is present in human neuroblastoma cell lines.
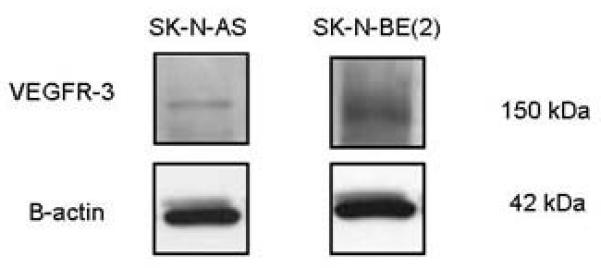
Western blotting was used to detect the presence of VEGFR-3 protein in cell lysates from SK-N-AS and SK-N-BE2 human neuroblastoma cell lines. The protein was seen in both cell lines, but is differentially expressed. Blots were stripped and probed for β-actin as an internal control for equal protein loading.
Figure 2. Focal adhesion kinase (FAK) and vascular endothelial growth factor receptor-3 (VEGFR-3) interact in human neuroblastoma cells.
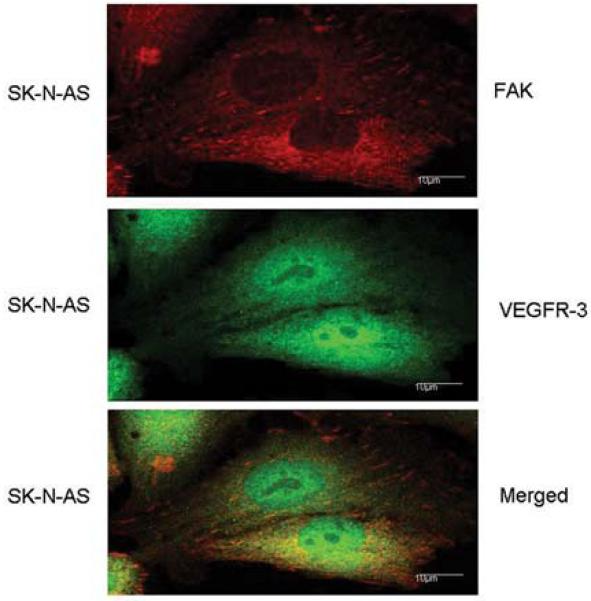
Immunofluorescence staining and confocal microscopy were employed to detect the presence of FAK and VEGFR-3 in SK-N-AS neuroblastoma cells. The cells were stained with fluorescent-tagged antibodies to FAK (top panel, red) and VEGFR-3 (middle panel, green). When the two figures were merged with confocal microscopy, colocalization of FAK and VEGFR-3 (bottom panel, yellow) was detected.
Targeting FAK in neuroblastoma
Focal adhesion kinase is expressed in neuroblastoma tumor cells and is more prominent in tumors with the more aggressive phenotypes [28, 29]. Inhibition of FAK is associated with decreased proliferation and propensity for invasion in many adult carcinomas, such as breast and pancreas [37-39]. One method that has been utilized to block FAK is AdFAK-CD. AdFAK-CD is an adenoviral construct containing the carboxy-terminal domain of FAK [40]. AdFAK-CD is analogous to FAK related non-kinase (FRNK), which has been shown to lead to decreased phosphorylation, and decreased activity, of p125FAK. Xu illustrated a loss of cellular adhesion and apoptosis in BT474 breast cancer cells that were transformed with AdFAK-CD [40]. AdFAK-CD has been successfully utilized to inhibit FAK in neuroblastoma [41]. After treatment with AdFAK-CD, MYCN+ and MYCN− isogenic neuroblastoma cells exhibited cellular detachment and a decline in proliferation and viability that was much more pronounced in the MYCN+ cell line, suggesting that the phenotypically more aggressive tumor cells (MYCN+) were more sensitive to FAK inhibition. In addition, this study also demonstrated that dual inhibition of FAK and Src results in cellular detachment, decreased viability and increased apoptosis in both the MYCN− and MYCN+ neuroblastoma tumor cells [41].
Another method of FAK inhibition is the use of small inhibiting RNA’s (siRNA). In a study using PANC1, MIAPaCa2 and BxPC3 pancreatic adenocarcinoma cell lines, siRNA interruption of FAK expression resulted in augmentation of the cytotoxic effect of gemcitabine, with elevated levels of apoptosis at lower gemcitabine concentrations [42]. SiRNA methods have also been utilized to interfere with FAK in neuroblastoma cell lines. In an isogenic MYCN+ / MYCN− neuroblastoma cell line, studies were undertaken to interrogate the effects of blocking FAK using siRNA [29]. SiRNA treatment of the isogenic MYCN+ / MYCN− neuroblastoma cell lines effectively abrogated the expression of FAK, and resulted in decreased cellular viability. As noted with the AdFAK-CD treatment, the effects of FAK knockdown were more pronounced in the MYCN+ cell line, those cells with higher levels of FAK activity. These FAK-targeted investigations have provided data to lead to further investigation with small molecule inhibitors of FAK.
Various inhibitors of FAK expression have been described, including NVP-TAE226 (TAE226), 1,2,4,5-benzenetetramine tetrahydrochloride (Y15), and PF-573,228 (Table 1). NV-TAE226 (TAE226) is a potent inhibitor of FAK that has bioavailability when administered orally. This small molecule inhibits the phosphorylation of FAK, thereby effectively decreasing FAK activity [43]. In multiple types of cancer, including esophageal [44], glioma [43] and ovarian cancer [45], treatment with TAE226 causes dose-dependent apoptosis. When SHEP and SK-N-AS neuroblastoma cell lines were treated with TAE226 at 10μM concentration, there was a significant decrease in cell survival, and cell cycle studies with flow cytometry revealed that TAE226 treatment lead to cell cycle arrest [46]. Recent data show that TAE226 treatment of SK-N-BE(2) neuroblastoma cell lines results in a decline in cellular invasion and migration (Figs. 3, 4). Judging from these data, the inhibition of FAK with TAE226 has desirable anti-tumor effects in multiple neuroblastoma cell lines.
Table 1.
| Inhibitor Name | Chemical Name | Structure | Company |
|---|---|---|---|
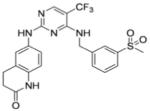
| PF 573,228 | 6-(4-(3-(methylsulfonylbenzylamino) -5-(trifluoromethyl) pyrimidin- 2-ylamino)-3, 4-dihydroquinolin- 2(1H)-one |
|
Pfizer |
| NVP-TAE226 | (2-[5-Chloro-2-[2-methoxy-4- (4-morpholinyl)phenylamino] pyrimidin-4-ylamino]-N- methylbenzamide |

|
Novartis |
| Y15 | 1,2,4,5-Benzenetetraamine tetrahydrochloride |
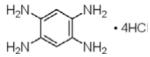
|
Sigma |
| C4 | Chloropyramine hydrochloride |
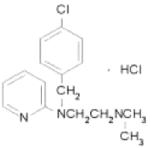
|
Sigma |
| PF-0562271 | N-Methyl-N-(3-{[2-(2-oxo-2,3- dihydro-1H-indol-5-ylamino)-5- trifluoromethyl-pyrimidin-4-ylamino]- methyl}-pyridin-2-yl)methanesulfonamide |
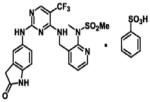
|
Pfizer |
| PF-04554878 | Unknown | Pfizer | |
| GSK2256098 | Unknown | GlaxoSmithKline |
Figure 3. Inhibition of FAK with TAE226 leads to decreased cell invasion in SK-N-BE(2) neuroblastoma cells.

SK-N-BE(2) neuroblastoma cells were treated with TAE226 in various concentrations and matrigel invasion assays were performed. After 24 hours, the cells were fixed, stained, and counted. There was a significant decrease in the number of invading cells (4582 ± 183 vs. 2219 ± 113 cells, p<0.001, control vs. TAE226) seen with the lowest TAE226 treatment of 0.5 μM, which continued with increasing concentrations.
Figure 4. TAE226 treatment decreases cell migration in SK-N-BE(2) neuroblastoma cell line.

SK-N-BE(2) neuroblastoma cells were plated onto culture dishes and allowed to attach for 24 hours. They were then treated with TAE226 in various concentrations and a standard scratch was created with a 2mm pipette tip through the monolayer of cells. Digital photos were obtained to determine the area of the scratch remaining after 48 hours. The area of closure of the scratch relative to the initial scratch area was significantly diminished with TAE226 treatment. The cells showed significantly less migration beginning with the lowest concentration of TAE226, 0.5 μM (0.764 ± 0.05 vs. 0.593 ± .04, p<0.05, control vs. TAE226), and continued this finding with the increasing concentrations of TAE226.
Golubovskaya and others have recently described a small molecule inhibitor of FAK, 1,2,4,5-benzenetetraamine tetrahydrochloride (Y15) (Table 1) [47]. Y15 was found through in silico screening to bind to, and block, the tyrosine 397 phosphorylation site of FAK, inhibiting FAK activity [47]. These investigators showed that inhibition of FAK with 1,2,4,5-benzenetetraamine tetrahydrochloride resulted in decreased breast cancer tumor growth [47] and pancreatic cancer tumor growth both in vitro and in nude mouse models [48]. In addition, a recent study by Perry showed that inhibition of FAK with 1,2,4,5-benzenetetraamine tetrahydrochloride resulted in decreased adhesion in the sarcoma cell lines HT-1080, KHOS-240S and A-673 [49]. In the neuroblastoma cell lines SK-N-AS and SK-N-BE(2) (MYCN nonamplified and amplified, respectively), treatment with 1,2,4,5-benzenetetraamine tetrahydrochloride resulted in decreased cellular attachment and viability, and increased apoptosis in vitro as evaluated by trypan blue exclusion, Hoechst 33258 staining, and immunoblotting [50]. Two neuroblastoma tumor lines with MYCN amplification (SK-N-BE(2) and WAC2) exhibited decreased tumor growth in a nude mouse xenograft model after 30 mg/kg/day of 1,2,4,5-benzenetetraamine tetrahydrochloride treatment [50]. Notably, as seen with other methods of FAK inhibition, the affects of 1,2,4,5-benzenetetraamine tetrahydrochloride treatment were more pronounced in the MYCN amplified neuroblastoma cell lines [50]. An important finding has been that normal human ganglion cells (SKP cells) are not affected by 1,2,4,5-benzenetetraamine tetrahydrochloride treatment, even at high concentrations (Fig. 5). Due to the successful in vivo suppression of tumor growth and the lack of impact on normal tissues, 1,2,4,5-benzenetetraamine tetrahydrochloride is a logical candidate for further testing in neuroblastoma.
Figure 5. 1,2,4,5-benzentetraamine tetrahydrochloride (Y15) treatment of SKP cells does not significantly affect cellular viability.

The SKP, normal human ganglion cells, and SK-N-BE(2), human neuroblastoma cell lines, were treated with varying concentrations of Y15. Alamar Blue assays were utilized to detect cell viability. Even at concentrations as low as 2 μM the Y15 significantly decreased the viability of the neuroblastoma cell line, SK-N-BE(2) (shaded triangles). In contrast, Y15 treatment had no effect upon the viability of the SKP cell line (shaded boxes), even at concentrations as high as 25 μM. This study demonstrates the specificity of Y15 to the neuroblastoma cancer cells, and the lack of effect upon normal cell lines.
Finally, the interruption of protein-protein interactions involving FAK has become another area of focus for FAK targeting in cancer. After Garces and others demonstrated the unique interaction between FAK and VEGFR-3, they were able to show that interruption of this interaction resulted in decreased breast cancer cell survival [36]. They utilized a small peptide, AV3, indentified through phage display experiments, to interrupt the FAK–VEGFR-3 interaction. AV3 has been utilized in studies with neuroblastoma cell lines. SK-N-AS, MYCN non-amplified with low VEGFR-3 and FAK, and IMR-32, MYCN amplified with high VEGFR-3 and FAK, were treated with control scrambled peptide and the AV3 peptide. Apoptosis was measured using Hoechst staining and reported as percent apoptotic cells. There was a marked increase in apoptosis in the MYCN amplified IMR-32 cells with AV3 treatment compare to the SK-N-AS cell line, suggesting that the MYCN amplified cells were more susceptible to interruption of the FAK-VEGFR-3 interaction (Fig. 6). Successful inhibition of the FAK-VEGFR-3 interaction has also been achieved using a small molecule inhibitor, chloropyramine hydrochloride (C4) (Table 1) [51]. Kurenova and others demonstrated in both in vitro and in vivo models that C4 inhibition of the FAK-VEGFR-3 interaction leads to decreased tumor cell survival and tumor growth in breast cancer cells [51]. Preliminary studies with C4 in neuroblastoma cell lines have also found decreased tumor cell survival in vitro and decreased tumor growth in vivo. Nude mice with SK-N-BE(2) flank tumors were treated with vehicle or 60 mg/kg/day of chloropyramine hydrochloride (C4). Tumors from mice treated with C4 weighed 2.5 times less than those from mice treated with vehicle only (Fig. 7). These preliminary data show that blocking the FAK-VEGFR-3 interaction may have utility in decreasing tumor growth in neuroblastoma.
Figure 6. Treatment with AV3 peptide leads to increased apoptosis in the IMR-32 human neuroblastoma cell line.

SK-N-AS, MYCN non-amplified with low VEGFR-3 and FAK, and IMR-32, MYCN amplified with high VEGFR-3 and FAK, human neuroblastoma cell lines were treated with AV3 peptide, scrambled peptide, or vehicle (control). Cells were stained with Hoechst 33342 to detect apoptosis. Cells were counted and apoptosis was reported as percent apoptotic cells. There was a marked increase in apoptosis in the MYCN amplified IMR-32 cells with AV3 treatment compare to the non-amplified SK-N-AS cell line with AV3 treatment. Apoptosis was not affected in either cell line by the scrambled peptide or vehicle alone.
Figure 7. Treatment of SK-N-BE(2) tumor xenografts in a nude mouse model with chloropyramine hydrochloride (C4) results in a significant decrease in tumor weight.

SK-N-BE(2) neuroblastoma cells (2.5 × 106 cells) were injected into the right flank of nude mice. When tumors became palpable (100 mm3) mice were treated with intraperitoneal injections of C4 (60 mg/kg/day) or vehicle. After 21 days, the tumors were harvested and weighed. The tumors from the C4 treated animals weighed 2.5 times less than the tumors from control treated animals.
Conclusions
Neuroblastoma continues to be an aggressive and devastating tumor. FAK inhibition has been shown to decrease viability and invasion of neuroblastoma both in vitro and in vivo. Clinical trials of small molecule FAK inhibitors are underway with many adult tumors. One of the first clinically available FAK inhibitors, PF-00562271 (Table 1), was assessed with a Phase I trial between December 2005 and April 2009. One hundred six patients with head and neck, prostate and pancreas neoplasms were enrolled. The drug was well tolerated, and, in addition, 17% of the patients had stable disease for more than 6 cycles [52]. Patients are currently being enrolled in Phase I trials involving two other FAK inhibitors, PF-04554878 (Table 1) and GSK2256098 (Table 1) [53]. Additional investigations of the mechanisms involved with FAK inhibition in neuroblastoma both in vitro and in vivo will be necessary before clinical studies can become a reality for these patients. In addition, further studies are needed to elucidate the effectiveness of FAK inhibition when combined with other treatment modalities. FAK inhibition is an exciting novel therapy on the horizon for children with this disease.
Acknowledgements
Projects described in this review were funded in part by the Children’s Neuroblastoma Cancer Foundation and St. Baldrick’s Foundation (EAB). LG and EAB are supported in part by grants from the National Cancer Institute, Grant Numbers T32CA091078 and K08CA118178, respectively. The content of this review is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health. The authors wish to thank the UAB High Resolution Imaging Facility for assistance with confocal microscopy and Jerry E. Stewart and Shawn Williams for their outstanding technical assistance.
Footnotes
Conflict of Interest The authors have no conflicts of interest.
References
- [1].Spix C, Pastore G, Sankila R, Stiller CA, Steliarova-Foucher E. Neuroblastoma incidence and survival in European children (1978-1997): report from the Automated Childhood Cancer Information System project. Eur. J. Cancer. 2006;42(13):2081–2091. doi: 10.1016/j.ejca.2006.05.008. [DOI] [PubMed] [Google Scholar]
- [2].Maris JM, Matthay KK. Molecular biology of neuroblastoma. J. Clin. Oncol. 1999;17(7):2264–2279. doi: 10.1200/JCO.1999.17.7.2264. [DOI] [PubMed] [Google Scholar]
- [3].Pearson AD, Pinkerton CR, Lewis IJ, Imeson J, Ellershaw C, Machin D, Group, E. N. S. Group), C. s. C. a. L. G. C. f. U. K. C. s. C. S. High-dose rapid and standard induction chemotherapy for patients aged over 1 year with stage 4 neuroblastoma: a randomised trial. Lancet Oncol. 2008;9(3):247–256. doi: 10.1016/S1470-2045(08)70069-X. [DOI] [PubMed] [Google Scholar]
- [4].Stanton L, Schwab M, Bishop J. Nucleotide sequence of the human N-myc gene. Proc. Natl. Acad. Sci. U. S. A. 1986;83(6):1772–1786. doi: 10.1073/pnas.83.6.1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Brodeur G, Seeger R, Schwab M, Varmus H, Bishop J. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science. 1984;224(4653):1121–1124. doi: 10.1126/science.6719137. [DOI] [PubMed] [Google Scholar]
- [6].Schweigerer L, Breit S, Wenzel A, Tsunamoto K, Ludwig R, Schwab M. Augmented MYCN expression advances the malignant phenotype of human neuroblastoma cells: evidence for induction of autocrine growth factor activity. Cancer Res. 1990;50(14):4411–4416. [PubMed] [Google Scholar]
- [7].Fredlund E, Ringnér M, Maris JM, Påhlman S. High Myc pathway activity and low stage of neuronal differentiation associate with poor outcome in neuroblastoma. Proc. Natl. Acad. Sci. U. S. A. 2008;105(37):14094–14099. doi: 10.1073/pnas.0804455105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zaizen Y, Taniguchi S, Suita S. The role of cellular motility in the invasion of human neuroblastoma cells with or without N-myc amplification and expression. J. Pediatr. Surg. 1998;33(12):1765–1770. doi: 10.1016/s0022-3468(98)90281-0. [DOI] [PubMed] [Google Scholar]
- [9].Vasudevan SA, Nuchtern JG, Shohet JM. Gene profiling of high risk neuroblastoma. World J. Surg. 2005;29(3):317–324. doi: 10.1007/s00268-004-7820-7. [DOI] [PubMed] [Google Scholar]
- [10].Ma L, Young J, Prabhala H, Pan E, Mestdagh P, Muth D, Teruya-Feldstein J, Reinhardt F, Onder TT, Valastyan S, Westermann F, Speleman F, Vandesompele J, Weinberg RA. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat. Cell Biol. 2010;12(3):247–256. doi: 10.1038/ncb2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Terpe HJ, Christiansen H, Gonzalez M, Berthold F, Lampert F. Differentiation and prognosis of neuroblastoma in correlation to the expression of CD44s. Eur. J. Cancer. 1995;31A(4):549–552. doi: 10.1016/0959-8049(95)00061-m. [DOI] [PubMed] [Google Scholar]
- [12].Akeson R, Bernards R. N-myc down regulates neural cell adhesion molecule expression in rat neuroblastoma. Mol. Cell. Biol. 1990;10(5):2012–2016. doi: 10.1128/mcb.10.5.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Korja M, Jokilammi A, Salmi TT, Kalimo H, Pelliniemi TT, Isola J, Rantala I, Haapasalo H, Finne J. Absence of polysialylated NCAM is an unfavorable prognostic phenotype for advanced stage neuroblastoma. BMC Cancer. 2009;9:57. doi: 10.1186/1471-2407-9-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Judware R, Culp LA. Over-expression of transfected N-myc oncogene in human SKNSH neuroblastoma cells down-regulates expression of beta 1 integrin subunit. Oncogene. 1995;11(12):2599–2607. [PubMed] [Google Scholar]
- [15].Judware R, Lechner R, Culp LA. Inverse expressions of the N-myc oncogene and beta 1 integrin in human neuroblastoma: relationships to disease progression in a nude mouse model system. Clin. Exp. Metastasis. 1995;13(2):123–133. doi: 10.1007/BF00133617. [DOI] [PubMed] [Google Scholar]
- [16].Judware R, Culp LA. Concomitant down-regulation of expression of integrin subunits by N-myc in human neuroblastoma cells: differential regulation of alpha2, alpha3 and beta1. Oncogene. 1997;14(11):1341–1350. doi: 10.1038/sj.onc.1200955. [DOI] [PubMed] [Google Scholar]
- [17].Bozzo C, Defilippi P, Silengo L, Tarone G. Role of tyrosine phosphorylation in matrix-induced neurite outgrowth in human neuroblastoma cells. Exp. Cell. Res. 1994;214(1):313–322. doi: 10.1006/excr.1994.1263. [DOI] [PubMed] [Google Scholar]
- [18].Tanaka N, Fukuzawa M. MYCN downregulates integrin alpha1 to promote invasion of human neuroblastoma cells. Int. J. Oncol. 2008;33(4):815–821. [PubMed] [Google Scholar]
- [19].Gabarra-Niecko V, Schaller M, Dunty J. FAK regulates biological processes important for the pathogenesis of cancer. Cancer Metastasis Rev. 2003;22(4):359–374. doi: 10.1023/a:1023725029589. [DOI] [PubMed] [Google Scholar]
- [20].Leventhal P, Shelden E, Kim B, Feldman E. Tyrosine phosphorylation of paxillin and focal adhesion kinase during insulin-like growth factor-I-stimulated lamellipodial advance. J Biol. Chem. 1997;272(8):5214–5218. doi: 10.1074/jbc.272.8.5214. [DOI] [PubMed] [Google Scholar]
- [21].Kim B, Feldman E. Insulin-like growth factor I prevents mannitol-induced degradation of focal adhesion kinase and Akt. J. Biol. Chem. 2002;277(30):27393–27400. doi: 10.1074/jbc.M201963200. [DOI] [PubMed] [Google Scholar]
- [22].Kim B, van Golen C, Feldman E. Degradation and dephosphorylation of focal adhesion kinase during okadaic acid-induced apoptosis in human neuroblastoma cells. Neoplasia. 2003;5(5):405–416. doi: 10.1016/s1476-5586(03)80043-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wu L, Bernard-Trifilo J, Lim Y, Lim S, Mitra S, Uryu S, Chen M, Pallen C, Cheung N, Mikolon D, Mielgo A, Stupack D, Schlaepfer D. Distinct FAK-Src activation events promote alpha5beta1 and alpha4beta1 integrin-stimulated neuroblastoma cell motility. Oncogene. 2008;27(10):1439–1448. doi: 10.1038/sj.onc.1210770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Cance W, Harris J, Iacocca M, Roche E, Yang X, Chang J, Simkins S, Xu L. Immunohistochemical analyses of focal adhesion kinase expression in benign and malignant human breast and colon tissues: correlation with preinvasive and invasive phenotypes. Clin. Cancer Res. 2000;6(6):2417–2423. [PubMed] [Google Scholar]
- [25].Lark A, Livasy C, Dressler L, Moore D, Millikan R, Geradts J, Iacocca M, Cowan D, Little D, Craven R, Cance W. High focal adhesion kinase expression in invasive breast carcinomas is associated with an aggressive phenotype. Mod. Pathl. 2005;18(10):1289–1294. doi: 10.1038/modpathol.3800424. [DOI] [PubMed] [Google Scholar]
- [26].Furuyama K, Doi R, Mori T, Toyoda E, Ito D, Kami K, Koizumi M, Kida A, Kawaguchi Y, Fujimoto K. Clinical significance of focal adhesion kinase in resectable pancreatic cancer. World J. Surg. 2006;30(2):219–226. doi: 10.1007/s00268-005-0165-z. [DOI] [PubMed] [Google Scholar]
- [27].Judson P, He X, Cance W, Van Le L. Overexpression of focal adhesion kinase, a protein tyrosine kinase, in ovarian carcinoma. Cancer. 1999;86(8):1551–1556. doi: 10.1002/(sici)1097-0142(19991015)86:6<1551::aid-cncr23>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- [28].Beierle E, Massoll N, Hartwich J, Kurenova E, Golubovskaya V, Cance W, McGrady P, London W. Focal adhesion kinase expression in human neuroblastoma: immunohistochemical and real-time PCR analyses. Clin. Cancer Res. 2008;14(11):3299–3305. doi: 10.1158/1078-0432.CCR-07-1511. [DOI] [PubMed] [Google Scholar]
- [29].Beierle E, Trujillo A, Nagaram A, Kurenova E, Finch R, Ma X, Vella J, Cance W, Golubovskaya V. N-MYC regulates focal adhesion kinase expression in human neuroblastoma. J. Biol. Chem. 2007;282(17):12503–12516. doi: 10.1074/jbc.M701450200. [DOI] [PubMed] [Google Scholar]
- [30].Lagodny J, Jüttner E, Kayser G, Niemeyer C, Rössler J. Lymphangiogenesis and its regulation in human neuroblastoma. Biochem. Biophys. Res. Commun. 2007;352(2):571–577. doi: 10.1016/j.bbrc.2006.11.062. [DOI] [PubMed] [Google Scholar]
- [31].van Iterson V, Leidenius M, von Smitten K, Bono P, Heikkilä P. VEGF-D in association with VEGFR-3 promotes nodal metastasis in human invasive lobular breast cancer. Am. J. Clin. Pathol. 2007;128(5):759–766. doi: 10.1309/7FXVRMXF58PVRJUH. [DOI] [PubMed] [Google Scholar]
- [32].Saintigny P, Kambouchner M, Ly M, Gomes N, Sainte-Catherine O, Vassy R, Czernichow S, Letoumelin P, Breau J, Bernaudin J, Kraemer M. Vascular endothelial growth factor-C and its receptor VEGFR-3 in non-small-cell lung cancer: concurrent expression in cancer cells from primary tumour and metastatic lymph node. Lung Cancer. 2007;58(2):205–213. doi: 10.1016/j.lungcan.2007.06.021. [DOI] [PubMed] [Google Scholar]
- [33].Jennbacken K, Vallbo C, Wang W, Damber J. Expression of vascular endothelial growth factor C (VEGF-C) and VEGF receptor-3 in human prostate cancer is associated with regional lymph node metastasis. Prostate. 2005;65(2):110–116. doi: 10.1002/pros.20276. [DOI] [PubMed] [Google Scholar]
- [34].Beierle E, Dai W, Langham MJ, Copeland E. r., Chen M. Expression of VEGF receptors in cocultured neuroblastoma cells. J. Surg. Res. 2004;119(1):56–65. doi: 10.1016/j.jss.2004.01.002. [DOI] [PubMed] [Google Scholar]
- [35].Beierle E, Dai W, Langham MJ, Copeland E. r., Chen M. VEGF receptors are differentially expressed by neuroblastoma cells in culture. J. Pediatr. Surg. 2003;38(3):514–521. doi: 10.1053/jpsu.2003.50091. [DOI] [PubMed] [Google Scholar]
- [36].Garces C, Kurenova E, Golubovskaya V, Cance W. Vascular endothelial growth factor receptor-3 and focal adhesion kinase bind and suppress apoptosis in breast cancer cells. Cancer Res. 2006;66(3):1446–1454. doi: 10.1158/0008-5472.CAN-05-1661. [DOI] [PubMed] [Google Scholar]
- [37].Golubovskaya V, Beviglia L, Xu L, Earp H. r., Craven R, Cance W. Dual inhibition of focal adhesion kinase and epidermal growth factor receptor pathways cooperatively induces death receptor-mediated apoptosis in human breast cancer cells. J Biol Chem. 2002;277(41):38978–38987. doi: 10.1074/jbc.M205002200. [DOI] [PubMed] [Google Scholar]
- [38].Golubovskaya V, Zheng M, Zhang L, Li J, Cance W. The direct effect of focal adhesion kinase (FAK), dominant-negative FAK, FAK-CD and FAK siRNA on gene expression and human MCF-7 breast cancer cell tumorigenesis. BMC Cancer. 2009;9:280. doi: 10.1186/1471-2407-9-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zheng D, Golubovskaya V, Kurenova E, Wood C, Massoll N, Ostrov D, Cance W, Hochwald S. A novel strategy to inhibit FAK and IGF-1R decreases growth of pancreatic cancer xenografts. Mol Carcinog. 2010;49(2):200–209. doi: 10.1002/mc.20590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Xu L, Yang X, Bradham C, Brenner D, Baldwin AJ, Craven R, Cance W. The focal adhesion kinase suppresses transformation-associated, anchorage-independent apoptosis in human breast cancer cells. Involvement of death receptor-related signaling pathways. J Biol Chem. 2000;275(39):30597–30604. doi: 10.1074/jbc.M910027199. [DOI] [PubMed] [Google Scholar]
- [41].Beierle E, Ma X, Trujillo A, Kurenova E, Cance W, Golubovskaya V. Inhibition of focal adhesion kinase and src increases detachment and apoptosis in human neuroblastoma cell lines. Mol Carcinog. 2010;49(3):224–234. doi: 10.1002/mc.20592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Duxbury MS, Ito H, Benoit E, Zinner MJ, Ashley SW, Whang EE. RNA interference targeting focal adhesion kinase enhances pancreatic adenocarcinoma gemcitabine chemosensitivity. Biochem Biophys Res Commun. 2003;311(3):786–792. doi: 10.1016/j.bbrc.2003.10.060. [DOI] [PubMed] [Google Scholar]
- [43].Shi Q, Hjelmeland AB, Keir ST, Song L, Wickman S, Jackson D, Ohmori O, Bigner DD, Friedman HS, Rich JN. A novel low-molecular weight inhibitor of focal adhesion kinase, TAE226, inhibits glioma growth. Mol Carcinog. 2007;46(6):488–496. doi: 10.1002/mc.20297. [DOI] [PubMed] [Google Scholar]
- [44].Wang ZG, Fukazawa T, Nishikawa T, Watanabe N, Sakurama K, Motoki T, Takaoka M, Hatakeyama S, Omori O, Ohara T, Tanabe S, Fujiwara Y, Shirakawa Y, Yamatsuji T, Tanaka N, Naomoto Y. TAE226, a dual inhibitor for FAK and IGF-IR, has inhibitory effects on mTOR signaling in esophageal cancer cells. Oncol Rep. 2008;20(6):1473–1477. [PubMed] [Google Scholar]
- [45].Halder J, Lin YG, Merritt WM, Spannuth WA, Nick AM, Honda T, Kamat AA, Han LY, Kim TJ, Lu C, Tari AM, Bornmann W, Fernandez A, Lopez-Berestein G, Sood AK. Therapeutic efficacy of a novel focal adhesion kinase inhibitor TAE226 in ovarian carcinoma. Cancer Res. 2007;67(22):10976–10983. doi: 10.1158/0008-5472.CAN-07-2667. [DOI] [PubMed] [Google Scholar]
- [46].Beierle EA, Trujillo A, Nagaram A, Golubovskaya VM, Cance WG, Kurenova EV. TAE226 inhibits human neuroblastoma cell survival. Cancer Invest. 2008;26(2):145–151. doi: 10.1080/07357900701577475. [DOI] [PubMed] [Google Scholar]
- [47].Golubovskaya V, Nyberg C, Zheng M, Kweh F, Magis A, Ostrov D, Cance W. A small molecule inhibitor, 1,2,4,5-benzenetetraamine tetrahydrochloride, targeting the y397 site of focal adhesion kinase decreases tumor growth. J Med Chem. 2008;51(23):7405–7416. doi: 10.1021/jm800483v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Hochwald S, Nyberg C, Zheng M, Zheng D, Wood C, Massoll N, Magis A, Ostrov D, Cance W, Golubovskaya V. A novel small molecule inhibitor of FAK decreases growth of human pancreatic cancer. Cell Cycle. 2009;8(15):2435–2443. doi: 10.4161/cc.8.15.9145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Perry BC, Wang S, Basson MD. Extracellular pressure stimulates adhesion of sarcoma cells via activation of focal adhesion kinase and Akt. Am J Surg. 2010;200(5):610–614. doi: 10.1016/j.amjsurg.2010.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Beierle E, Ma X, Stewart J, Nyberg C, Trujillo A, Cance W, Golubovskaya V. Inhibition of focal adhesion kinase decreases tumor growth in human neuroblastoma. Cell Cycle. 2010;9(5):1005–1015. doi: 10.4161/cc.9.5.10936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Kurenova E, Hunt D, He D, Magis A, Ostrov D, Cance W. Small molecule chloropyramine hydrochloride (C4) targets the binding site of focal adhesion kinase and vascular endothelial growth factor receptor 3 and suppresses breast cancer growth in vivo. J Med Chem. 2009;52(15):4716–4724. doi: 10.1021/jm900159g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Molckovsky A, Siu LL. First-in-class, first-in-human phase I results of targeted agents: highlights of the 2008 American society of clinical oncology meeting. J Hematol Oncol. 2008;1:20. doi: 10.1186/1756-8722-1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Schultze A, Fiedler W. Therapeutic potential and limitations of new FAK inhibitors in the treatment of cancer. Expert Opin Investig Drugs. 2010;19(6):777–788. doi: 10.1517/13543784.2010.489548. [DOI] [PubMed] [Google Scholar]