

Abstract
Rationale
The development of the cardiac outflow tract (OFT) and great vessels is a complex process that involves coordinated regulation of multiple progenitor cell populations. Among these populations, neural crest cells make important contributions to OFT formation and aortic arch remodeling. While numerous signaling pathways, including Notch, have been implicated in this process, the role of epigenetics in OFT development remains largely unexplored.
Objective
As histone deacetylases (Hdacs) play important roles in the epigenetic regulation of mammalian development, we have investigated the function of Hdac3, a class I Hdac, during cardiac neural crest development.
Methods and Results
Using two neural crest drivers, Wnt1-Cre and Pax3Cre, we show that loss of Hdac3 in neural crest results in perinatal lethality and cardiovascular abnormalities, including interrupted aortic arch type B, aortic arch hypoplasia, double outlet right ventricle and ventricular septal defect. Affected embryos are deficient in aortic arch artery smooth muscle during mid-gestation, despite intact neural crest cell migration and preserved development of other cardiac and truncal neural crest derivatives. The Hdac3-dependent block in smooth muscle differentiation is cell autonomous and is associated with downregulation of the Notch ligand Jagged1, a key driver of smooth muscle differentiation in the aortic arch arteries.
Conclusions
These results indicate that Hdac3 plays a critical and specific regulatory role in the neural crest-derived smooth muscle lineage and in formation of the OFT.
Keywords: Hdac3, congenital heart disease, smooth muscle, cardiac development
Mammalian cardiac outflow tract (OFT) and aortic arch morphogenesis is characterized by the tightly coupled development of multiple cell types. The complexity of this process is underscored by the prevalence of congenital anomalies of great vessel patterning in humans.1 The heart, OFT and aortic arch are formed by several developmentally distinct cell populations, including cardiomyocytes (derived from the first and second heart fields), vascular smooth muscle cells (derived from the second heart field and neural crest), and endothelial cells.2 These populations of cells interact within the maturing pharyngeal arches - a dynamic milieu in which cross-regulation between these cell types, as well as the pharyngeal endoderm and ectoderm, results in the coordinated development of the OFT, great vessels and neighboring structures.3, 4 Many of the processes that regulate cell fate decisions and patterning during OFT development are also active in vascular remodeling in adult disease states in humans.5
The OFT initially develops as a single vessel, the truncus arteriosus, comprised of myocardium and smooth muscle cells emerging from a single, unseptated ventricle. Neural crest cells, which make up the bulk of the pharyngeal arch mesenchyme, contribute to the smooth muscle of the OFT and also condense to form much of the smooth muscle of the aortic arch arteries - a series of paired vessels that connect the developing truncus arteriosus to the dorsal aortae.6 The aortic arch arteries are then extensively remodeled, eventually giving rise to the mature aortic arch and several great vessels. Neural crest cells also contribute to septation and rotation of the OFT, with secondary effects on septation of the ventricles. These processes are highly dependent on the migration, expansion and differentiation of cardiac neural crest in response to cues from neighboring cells.1
Murine models of congenital cardiovascular abnormalities have shed new light on the interplay between cell populations in the pharyngeal arches, as well as on the pathogenesis of human congenital cardiac disease. For instance, human mutations in components of the Notch signaling pathway can result in Alagille syndrome, a heterogeneous disorder that can include cardiac OFT defects. It has been demonstrated in mice that active Notch signaling in both neural crest cells and the second heart field is critical for OFT morphogenesis.7, 8 Notch signaling from endothelial cells to neural crest in the pharyngeal mesenchyme via the Notch ligand Jagged1 is required to initiate smooth muscle differentiation in the aortic arch arteries; in the absence of such signaling, mice develop cardiac abnormalities reminiscent of those found in Alagille patients.7
In addition to Notch, other signaling modalities including the Wnt, FGF and BMP pathways, as well as activation of the MAPK pathway by integrin signaling have been shown to be important in OFT formation or remodeling.4, 9-12 However, in spite of our increasing understanding of the signaling pathways that influence OFT formation, the mechanisms by which progenitor cells become competent to respond to external stimuli and the means by which these stimuli are coupled to changes in gene expression remain largely unknown.
Class I Hdacs are important regulators of transcription, via both chromatin-mediated (epigenetic) transcriptional repression and direct deacetylation of key transcriptional regulators.13, 14 Class I Hdacs include Hdac1, Hdac2, Hdac3 and Hdac8. During embryogenesis, these proteins are widely expressed, but play specific roles as modulators of early developmental processes and organogenesis.15-19 As such, class I Hdacs are intriguing candidates for studying the regulation of gene expression during cardiac neural crest development.
Interestingly, neural crest-specific deletion of Hdac1, Hdac2 or Hdac8 alone does not affect patterning of the great vessels.19 Hdac3, however, is unique among class I Hdacs in that it associates with NCoR or SMRT in a well-characterized transcriptional repressor complex.20 Global deletion of Hdac3 results in lethality at the gastrulation stage.15 In a tissue-specific context, Hdac3 has been shown to regulate differentiation in osteoblastic and myocytic precursors.21, 22 Additionally, Hdac3 has been shown to regulate cell cycle progression in cell lines and cardiomyocytes.15, 23-26 Hdac3 also plays important roles in metabolic regulation in multiple tissues, including the heart, through its action as a transcriptional repressor.15, 25, 27, 28
In order to study the role of Hdac3 in cardiac neural crest, we genetically inactivated Hdac3 in premigratory neural crest cells. Using in vivo and ex vivo approaches, we show that Hdac3 plays a critical role in smooth muscle differentiation of neural crest cells.
Materials and Methods
Mice
Wnt1-Cre, Pax3Cre, Hdac3flox and Z/EG mice were maintained on mixed CD1/B6/129 genetic backgrounds, separated by 3-6 generations of incrossing from pure parental backgrounds.29-32 The University of Pennsylvania Institutional Animal Care and Use Committee approved all animal protocols.
Histology, immunofluorescence, and in situ hybridization
These techniques were performed as previously described.33 Mutant and littermate control embryos were generated from Wnt1-Cre; Hdac3f/+; Z/EG, Wnt1-Cre; Hdac3f/+ or Pax3Cre/+; Hdac3f/+ animals crossed to Hdac3f/+, Hdac3f/+; Z/EG or Hdac3f/f animals, respectively.
Neural Tube Explant Assays
Mutant embryos were obtained from crosses in which Wnt1-Cre; Hdac3f/+; Z/EG males were crossed to Hdac3f/f females, and age-matched control embryos were generated from Wnt1-Cre; Z/EG males crossed to WT females. Control and mutant embryos were dissected in parallel in a blinded manner. E9.5 embryos were dissected in sterile Hank’s balanced salt solution (HBSS) supplemented with 1% penicillin/streptomycin. The neural tube from the otic placode to first dorsal root ganglion was dissected and incubated in 0.75mg/mL type I collagenase (Worthington biochemical) in HBSS for 20 minutes at 37°C. Using tungsten needles, the neural tube was then microdissected from the surrounding mesenchyme, split in half longitudinally, and plated on glass chamber slides pre-coated with 200μg/mL fibronectin (Roche). Explants were incubated for 48 hours at 37°C and 5% CO2 in DMEM supplemented with 2% horse serum and 1% penicillin/streptomycin. Following fixation and immunostaining, each GFP+ cell that had delaminated from the neural tube was scored as SMA-positive or SMA-negative.
Statistics
The chi-square test and student’s 2-tailed t test were used to ascertain differences between groups. A x2 or p-value of less than 0.05 was considered significant.
Results
Hdac3 is expressed by neural crest and is efficiently deleted in premigratory neural crest by Wnt1-Cre
Immunofluorescence of staged embryos shows that Hdac3 is widely expressed at E10.5, including in the dorsal neural tube, where neural crest cells are specified before delaminating and migrating throughout the embryo (Figure 1A). Hdac3 is also expressed by neural crest derivatives, including dorsal root ganglia (DRG) (Figure 1A), pharyngeal arch mesenchyme (Figure 1B), conotruncal cushions of the developing outflow tract (Online Figure IA) and chromaffin cells of the adrenal medulla (Online Figure IB).
Figure 1. Wnt1-Cre efficiently deletes Hdac3 in premigratory neural crest cells and neural crest derivatives.
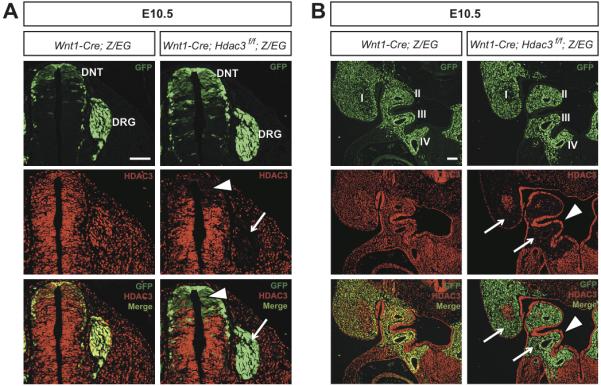
A & B, Immunohistochemistry for GFP and Hdac3 A, Frontal sections of the dorsal neural tube (DNT) and dorsal root ganglia (DRG) at E10.5. Wnt1-Cre derivatives marked by GFP expression are found in the control (left) and mutant DNT and DRG. Mutants exhibit efficient deletion of Hdac3 in premigratory neural crest cells in the DNT (arrowhead) and in postmigratory cells in the DRG (arrow), while control embryos show Hdac3 expression in both regions. B, Frontal sections of the pharyngeal arches at E10.5. Neural crest cells expressing GFP populate the pharyngeal arches of control (left) and mutant embryos. In control embryos, Hdac3 is expressed throughout the pharyngeal arch region. Mutant embryos show deletion of Hdac3 in the neural crest-derived pharyngeal mesenchyme (arrows), with retention of Hdac3 expression in ectoderm and pharyngeal endoderm (arrowheads). Roman numerals denote pharyngeal arch number. Scale bars: 100μm.
The Wnt1-Cre transgene is expressed by premigratory neural crest cells as early as E8.75.29 We used Wnt1-Cre and a floxed Hdac3 allele (Hdac3f) to delete Hdac3 in premigratory neural crest cells and their derivatives, and we used the Z/EG reporter to lineage trace neural crest cells in both control and mutant embryos. In this lineage tracing strategy, Cre mediates a recombination event that results in the constitutive expression of GFP in all derivatives of Wnt1-Cre-expressing cells. Immunostaining for GFP revealed expression in the dorsal neural tube in both Wnt1-Cre; Hdac3f/f (termed Hdac3Wnt1NCKO); Z/EG and Wnt1-Cre; Z/EG control embryos (Figure 1A). In E10.5 Hdac3Wnt1NCKO embryos, the GFP-positive cells in the dorsal neural tube show loss of Hdac3 protein (Figure 1A), indicating efficient Cre-mediated recombination in neural crest. Lineage tracing analysis further demonstrated that neural crest cells appropriately populate the DRG, pharyngeal arches, conotruncus, and adrenal glands in Hdac3Wnt1NCKO; Z/EG embryos, despite efficient deletion of Hdac3 in all of these tissues (Figure 1A,B, Online Figure IA,B). In the pharyngeal arches of mutant embryos, loss of Hdac3 protein is specific to the neural crest-derived mesenchyme, while expression is retained in ectoderm and pharyngeal endoderm (Figure 1B). Taken as a whole, these results indicate that Wnt1-Cre efficiently deletes Hdac3 specifically in neural crest cells and in neural crest derivatives, and that cardiac neural crest specification, migration and survival are grossly intact in the absence of Hdac3.
Loss of Hdac3 in neural crest results in perinatal lethality and severe cardiovascular and thymus abnormalities
Hdac3Wnt1NCKO embryos are found at expected Mendelian ratios in late gestation and are viable until birth (Table 1). However, these mice uniformly die at P0 (Table 1). As neural crest cells make important contributions to the development of the cardiac OFT, we sought to analyze OFT morphology in Hdac3Wnt1NCKO embryos. Neural crest gives rise to the smooth muscle of the aortic arch from its origin to the ductus arteriosus and large proportions of the smooth muscle in the great arteries. This smooth muscle is critical for vascular integrity during development. In several mutant embryos, we observed complete absence of the preductal aortic arch (Figure 2A versus 2B), a condition known as interrupted aortic arch (IAA) type B in humans. Other mutants demonstrated aortic arch hypoplasia (Figure 2C). Both IAA type B and aortic arch hypoplasia are rare cardiac abnormalities in humans, although both are commonly found in patients with DiGeorge syndrome and other neurocristopathies.34
Table 1.
Hdac3Wnt1NCKO mice exhibit perinatal lethality.
| Wnt1-Cre; Hdac3f/+ x Hdac3f/+ | ||||
|---|---|---|---|---|
| E16.5-E18.5 | # Observed | Observed | #Expected | Expected |
| Hdac3 +/+ | 9 | 0.12 | 9 | 0.13 |
| Hdac3 f/+ | 17 | 0.23 | 19 | 0.25 |
| Hdac3 f//f | 8 | 0.11 | 9 | 0.13 |
| Wnt1-Cre; Hdac3 +/+ | 7 | 0.09 | 9 | 0.13 |
| Wnt1-Cre; Hdac3 f/+ | 23 | 0.31 | 19 | 0.25 |
| Wnt1-Cre; Hdac3f/f | 11 | 0.15 | 9 | 0.13 |
| Total | 75 | |||
| Chi Sq | 0.82 | |||
| P1 | # Observed | Observed | # Expected | Expected |
| Hdac3 +/+ | 10 | 0.14 | 9 | 0.13 |
| Hdac3 f/+ | 26 | 0.36 | 18 | 0.25 |
| Hdac3 f//f | 10 | 0.14 | 9 | 0.13 |
| Wnt1-Cre; Hdac3 +/+ | 9 | 0.12 | 9 | 0.13 |
| Wnt1-Cre; Hdac3 f/+ | 18 | 0.25 | 18 | 0.25 |
| Wnt1-Cre; Hdac3f/f | 0 | 0 | 9 | 0.13 |
| Total | 73 | |||
| Chi Sq | 0.03 | |||
Figure 2. Late gestational Hdac3Wnt1NCKO embryos exhibit severe cardiovascular abnormalities.
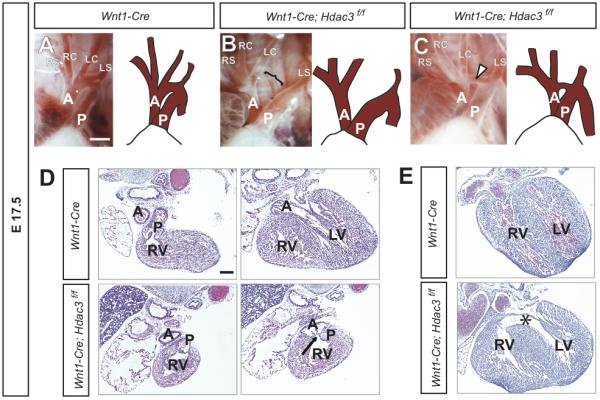
A-C. Gross images and drawings of E17.5 aortic arches. A, A control embryo, showing normal patterning of the great vessels in late gestation. B, A mutant embryo demonstrating a complete discontinuity of the aortic arch (open bracket), similar to interrupted aortic arch type B in humans. C, A mutant embryo showing hypoplasia of the aortic arch (arrowhead). A, aorta; P, pulmonary trunk; RS, right subclavian artery; RC, right common carotid artery; LC, left common carotid artery; LS, left subclavian artery. D, Sequential H&E-stained slides from the same control (top) and mutant hearts. In the control heart, the pulmonary trunk arises from the right ventricle (RV) while the aorta arises from the left ventricle (LV). In this mutant, both the pulmonary trunk and the aorta (arrow) arise from the right ventricle, indicating a double outlet right ventricle. E, A control embryo (top) with an intact ventricular septum and a littermate mutant with a large ventricular septal defect (*). The ventricular septal defect is the only cardiac abnormality in this mutant. Scale bars: A-C, 1mm. D,E, 300μm.
In addition to contributing directly to the aortic arch and great arteries, neural crest cells are involved in septation and rotation of the OFT, and directly give rise to the smooth muscle of the aorticopulmonary septum. Failure of septation can result in persistent truncus arteriosus (PTA), while a combination of incomplete septation and deficient outflow tract rotation can result in double outlet right ventricle (DORV). We observed examples of PTA and DORV in Hdac3Wnt1NCKO embryos (Figure 2D and data not shown). It is thought that neural crest cells in the developing aorticopulmonary septum signal to adjacent myocardium during the formation of the intraventricular septum. Consistent with the role of neural crest-derived smooth muscle in ventricular septation, we have observed severe ventricular septal defects (VSDs) in many Hdac3Wnt1NCKO embryos (Figure 2E). Table 2 summarizes the cardiovascular abnormalities observed in late gestation Hdac3Wnt1NCKO embryos.
Table 2.
Late gestational cardiovascular and thymic abnormalities in Hdac3Wnt1NCKO embryos.
| E16.5-18.5 | Wnt1-Cre | Wnt1-Cre; Hdac3f/f |
|---|---|---|
| IAA type B | 0% (0/6) | 32% (6/19) |
| Aortic arch hypoplasia | 0% (0/6) | 16% (3/19) |
| PTA | 0% (0/6) | 5% (1/19) |
| DORV | 0% (0/6) | 26% (5/19) |
| VSD | 0% (0/6) | 69% (11/16) |
| Thymic Dysplasia | 0% (0/6) | 100% (19/19) |
In order to provide further support for our findings, we used a distinct neural crest Cre driver, Pax3Cre, to delete Hdac3. Hdac3Pax3NCKO mice succumb by P1 (Online Table I) and exhibit a similar array of cardiovascular defects as Hdac3Wnt1NCKO embryos (Online Figure II, Online Table II). These findings support the conclusion that Hdac3 is required in cardiac neural crest.
The thymus is derived primarily from endoderm but develops in close association with the pharyngeal arches and neural crest. The thymic primordia migrate caudally through the pharyngeal mesenchyme during midgestation to final destinations in the anterior mediastinum. The thymuses of Hdac3Wnt1NCKO embryos fail to descend appropriately and are dysplastic (Online Figure III). The abnormal thymic development in Hdac3Wnt1NCKO embryos is consistent with prior studies that have documented the vital role of neural crest during thymus development, including in thymic migration, intrathymic smooth muscle formation, pericyte formation, and egress of thymocytes.35-37
Peripheral neurogenesis and development of the adrenal medulla occur normally in Hdac3Wnt1NCKO embryos
In addition to smooth muscle of the OFT, neural crest cells in the cardiac and trunk region form the neurons that populate the dorsal root, sympathetic and enteric ganglia, and the chromaffin cells of the adrenal medulla. In order to determine the requirements for Hdac3 in the differentiation of these derivatives, we first analyzed the peripheral nervous system of Hdac3Wnt1NCKO embryos. The neurofilament 2H3 is a marker of differentiated neurons. Immunostaining for 2H3 and GFP revealed that differentiated neurons populate the dorsal root ganglia and enteric ganglia of E10.5 Hdac3Wnt1NCKO; Z/EG embryos, and that these cells are neural crest-derived (Figure 3A and data not shown). Quantification of peripheral neuron staining intensity in the dorsal root ganglia revealed no significant difference between mutant and control embryos (Figure 3B). Optical projection tomography reconstructions of whole mount 2H3-stained embryos revealed normal patterning of the peripheral nervous system, including cranial nerves, dorsal root and sympathetic ganglia, in Hdac3Wnt1NCKO embryos compared to controls (Figure 3C).
Figure 3. Peripheral neurogenesis occurs normally in the absence of neural crest expression of Hdac3.

A, Immunohistochemistry for GFP and neurofilament (2H3), a marker of differentiated neurons, in frontal sections of the dorsal neural tube (DNT) and dorsal root ganglia (DRG). Both control and Mutant DRG feature differentiated, 2H3-positive neurons at E10.5 (arrowheads). B, Total neuronal area was calculated from anatomically matched sections from control and mutant embryos. Error bars represent SEM. N.S., not significantly different. C, Optical projection tomography renderings of whole mount 2H3-stained E10.5 embryos. Cranial nerves V, VII-XII (labeled) and DRG are present and patterned appropriately in mutant embryos. Scale bars: A, 100μm. B, 400μm.
The adrenal medullae of E14.5 Hdac3Wnt1NCKO; Z/EG embryos also develop normally when compared to control littermates, as determined by immunofluorescence for the neuroendocrine marker tyrosine hydroxylase (TH) and for GFP (Online Figure IV). Quantification of TH staining intensity revealed no significant abnormalities in chromaffin cell development in the absence of Hdac3 (Online Figure IV).
The aortic arch arteries of E11.5 Hdac3Wnt1NCKO embryos show decreased expression of smooth muscle markers
In midgestation, neural crest cells make up the bulk of the mesenchyme in the third, fourth, and sixth pharyngeal arches (Figure 1B, Figure 4A,B). A subset of these cells directly apposes the endothelium of the developing aortic arch arteries (Figure 4B). Between E10.5 and E11.5, these neural crest derivatives condense in response to signals from the endothelium and differentiate into the smooth muscle of the aortic arch arteries, eventually giving rise to a layer of smooth muscle actin (SMA)-positive tissue that is normally several cells thick by E11.5 (Figure 4A,C,D).33 In mutant embryos, as in littermate controls, neural crest cells appropriately populate the mesenchyme surrounding the arch arteries (Figure 4E,F,I). However, in three of four E11.5 mutant embryos, we observed profound deficiencies in smooth muscle populating the nascent arch arteries, as assessed by immunofluorescence for SMA and the additional smooth muscle marker SM22α (Figure 4E,G,H, Online Figure V). These defects are particularly pronounced in the left fourth and sixth arteries (Figure 4E,J), which give rise to the preductal aortic arch and ductus arteriosus, respectively. Of note, parts of the second arch arteries that are largely populated by non-neural crest-derived smooth muscle cells develop normally in mutants (Figure 4E,J). The 75% penetrance of gross smooth muscle deficiencies in the caudal arch arteries at E11.5 parallels the penetrance of aortic arch abnormalities observed at E17.5 (Table 2). Mid-gestational decreases in aortic arch artery smooth muscle have previously been shown to portend OFT defects,7 and we therefore hypothesized that these smooth muscle deficiencies observed at E11.5 account for the late gestational cardiovascular phenotype observed in Hdac3Wnt1NCKO embryos.
Figure 4. Hdac3Wnt1NCKO embryos display deficiencies in aortic arch artery smooth muscle at E11.5.

A-H. Immunohistochemistry for GFP and α-smooth muscle actin (SMA) in frontal sections of E11.5 pharyngeal arches. A, Representative image of a control pharyngeal arch at low magnification, showing robust layers of smooth muscle surrounding aortic arch arteries II, III, IV and VI (labeled), bilaterally. Note that the bulk of pharyngeal mesenchyme is comprised of GFP-positive neural crest derivatives. B-D, High magnification images of the left fourth arch artery in the control embryo. B, Neural crest cells populate the region directly surrounding the artery. C,D, A layer of neural crest-derived, SMA-positive smooth muscle surrounds the control artery. E, Low magnification image of a mutant pharyngeal arch, showing neural crest derivatives populating the mesenchyme. Note the decreased smooth muscle in the regions surrounding the third, fourth and sixth arteries, particularly on the left side, while a thick layer of non-neural crest-derived smooth muscle surrounds the second arteries, as in the control F-H, High magnification images of the mutant left fourth artery. F, Neural crest cells appropriately populate the region surrounding the artery. G,H, In affected mutants, few neural crest-derived cells surrounding the fourth arch artery are SMA-positive smooth muscle cells. Three of four mutant and zero of four controlembryos analyzed at this time point showed a similar pattern of staining. Roman numerals denote aortic arch artery number. I, Quantification of cells in the pharyngeal mesenchyme of aortic arches III-VI from serial sections of E11.5 control and mutant embryos. J, Quantification of SMA staining intensity of each aortic arch artery, from serial sections in control and mutant embryos. Asterisks denote p < 0.05. K, Quantification of phospho-histone H3 (pH3)- and Tunel-positive in serial sections of the pharyngeal arch mesenchyme. Scale bars: 100μm.
Hdac3-deficient neural crest cells exhibit a cell autonomous defect in smooth muscle differentiation
In order to determine the mechanism underlying the smooth muscle abnormalities in E11.5 Hdac3Wnt1NCKO embryos, we first determined rates of proliferation and apoptosis in third, fourth and sixth pharyngeal arch mesenchyme at E11.5 using phospho-histone H3 and Tunel staining, respectively. Neither proliferation nor apoptosis rates were significantly altered in Hdac3Wnt1NCKO arches versus littermate controls (Figure 4K). This suggests that neither increases in cell death in arch artery smooth muscle, nor decreased smooth muscle proliferation contribute to the defects observed. Rather, these results suggest that smooth muscle cells fail to form adequately in Hdac3Wnt1NCKO pharyngeal arches.
Notch signaling is a critical regulator of smooth muscle formation in neural crest cells, and loss of Notch signaling in the neural crest results in a similar array of OFT defects as observed in this study, with similar decreases in aortic arch artery smooth muscle at E11.5.7 The Notch ligand Jagged1 is initially expressed in the endothelial cells of the nascent aortic arch arteries at E10.5, and in a positive feedback loop, Jagged1 expression is upregulated in surrounding neural crest cells in response to Jagged1-mediated Notch activation.33, 38 In Hdac3Wnt1NCKO embryos, Jagged1 expression in the aortic arch arteries is significantly decreased at E11.5 when compared to Wnt1-Cre controls (Figure 5A). Quantification of Jagged1 staining intensity in the region surrounding each aortic arch artery indicated that deficient Jagged1 expression is most prominent in the mesenchyme surrounding the left fourth aortic arch artery (Figure 5B). This finding is consistent with our observations of decreased smooth muscle formation in this artery, and abnormal development of the left fourth arch artery derivatives in late gestation.
Figure 5. Expression of the Notch ligand Jagged1, a critical regulator of smooth muscle differentiation in the aortic arch arteries, is dysregulated in Hdac3Wnt1NCKO arches at E11.5.

A, Representative immunofluorescence for SMA and Jagged1 from frontal sections of mutant versus control left fourth and sixth arch arteries. B, Quantification of Jagged1 staining intensity in serial sections from three severely affected mutants relative to controls. Scale bar: 100μm.
Based upon our in vivo observations, we hypothesized that neural crest expression of Hdac3 is required for smooth muscle differentiation. We used an ex vivo approach to test this hypothesis. We performed neural tube explant assays using E9.5 Hdac3Wnt1NCKO; Z/EG and Wnt1-Cre; Z/EG control embryos. Genetically labeled neural crest cells were allowed to migrate from the dorsal neural tube onto a surrounding layer of fibronectin under conditions that allow for spontaneous differentiation into smooth muscle. Both control and Hdac3-deficient neural crest cells were able to migrate from the dorsal neural tube onto the fibronectin layer (Figure 6A,B). However, while control cells assume a large, flat conformation after two days in culture, mutant cells maintain a rounder phenotype (Figure 6A,B). Co-staining for GFP and SMA revealed that control cells are able to efficiently form smooth muscle in culture, while the proportion of GFP-positive cells costaining for SMA is significantly lower in mutant explant cultures (Figure 6C-G). These results suggest that Hdac3-deficient neural crest cells exhibit a primary defect in smooth muscle differentiation, consistent with the decreased smooth muscle observed in E11.5 mutant aortic arch arteries and decreased expression of the Notch ligand Jagged1.
Figure 6. Hdac3-deficient neural crest cells demonstrate a cell autonomous defect in smooth muscle differentiation.

A-F, Immunocytochemistry for GFP and α-smooth muscle actin (SMA) in neural tube explant cultures. A,B, Representative images of control and mutant neural crest cells following two days of migration and growth. Note that mutant neural crest cells (B) are small and blastic compared to control cells (A). C-F, Significant numbers of control cells stain positive for SMA (C,E) while mutant cells are rarely SMA-positive (D,F). G, Each GFP-positive cell derived from four control and four mutant embryos was scored as SMA-positive or -negative. Scale bar: 100μm.
Discussion
Class I Hdacs, including Hdac3, are widely expressed during embryogenesis and can regulate hundreds of genes affecting dozens of pathways, including those involved in cell survival, proliferation and metabolism.39 In light of their role as global modulators of gene expression, it is somewhat surprising that deletion studies have often revealed highly specific, non-redundant roles for individual class I Hdacs in various cell lineages. For instance, despite widespread expression, global deletion of Hdac2 results predominantly in cardiac abnormalities, while global deletion of Hdac8 affects patterning of the anterior calvarium and viscerocranium.19, 40
In this study, we have elucidated a specific role for Hdac3 in smooth muscle differentiation of cardiac neural crest cells during OFT and aortic arch artery morphogenesis. The specificity of this role is underscored by the lack of peripheral nervous system or adrenal abnormalities in Hdac3Wnt1NCKO mutants. The smooth muscle deficiencies observed in Hdac3Wnt1NCKO embryos lead to OFT and aortic arch artery abnormalities, such as DORV, PTA and type B IAA – abnormalities that are attributable to a failure of cardiac neural crest-derived structures to form properly.6 It is interesting to note that while these abnormalities are commonly found in human patients with neurocristopathy, additional OFT malformations often found in such patients, including retroesophageal right subclavian artery and vascular rings, were not observed in this study; this is likely because these latter two lesions are generally associated with abnormal persistence of structures which normally regress during development, rather than with the failure of such structures to form appropriately.6
In the pharyngeal arches, populations of neural crest cells, pharyngeal endoderm, epithelial cells and cardiomyocytes exist in close association with one another, and exert important regulatory roles mediating cell-cell communication. The signals between these cells exist in a delicate balance, and are instrumental for coordinating their migration, proliferation and differentiation. Cardiac OFT malformations have been described as a common endpoint for disruptions to these processes in the pharyngeal arches; indeed, numerous mouse models in which crosstalk between progenitor cell populations is disrupted result in a wide array of OFT defects.1, 4, 9-12, 33 Notch signaling via the Jagged1 ligand has previously been shown to initiate a cascade of smooth muscle differentiation in the aortic arch arteries, as well as to upregulate the expression of Jagged1 itself, in a positive feedback loop.33, 38 The decreased expression of Jagged1 in the aortic arch arteries of Hdac3Wnt1NCKO embryos suggests that Hdac3 may be required for Notch-mediated upregulation of Jagged1 and subsequent propagation of smooth muscle differentiation in the aortic arch arteries. Future work will elucidate the direct targets of Hdac3 that are responsible for its regulation of smooth muscle differentiation and Jagged1 expression, as well as the mechanism by which this regulation occurs.
Hdacs are capable of modulating gene expression in at least two ways. In the classical, epigenetic mechanism, Hdacs deacetylate histone tails at target loci; this results in down-regulation of transcription by specifically preventing the binding of bromodomain-containing transcription factors, and by inducing a change in chromatin conformation that sterically hinders the binding of additional transcription factors.13 In a second, non-epigenetic mechanism, Hdacs modulate transcription by deacetylating target transcription factors, resulting in altered stability and activity of these factors.14, 41 Additional work is needed to determine the nature of Hdac3-mediated regulation of smooth muscle differentiation, including whether this regulation occurs via direct deacetylation of transcription factor targets, or whether this effect relies on epigenetic silencing of target loci, including potential inhibitors of the Notch-Jagged1 positive feedback loop.
In addition to its relevance to congenital heart disease, smooth muscle formation is clinically important in the setting of coronary arterial stent restenosis, where proliferation of smooth muscle cells results in neointimal hyperplasia. Agents such as sirolimus, and rapamycin have been shown to be clinically useful in preventing restenosis at least in part by shifting the balance of smooth muscle cells from a proliferating, less differentiated phenotype towards a more quiescent and contractile cell type.42, 43 Many of the pathways involved in smooth muscle formation in embryogenesis are thought to be similarly involved in smooth muscle differentiation and vascular remodeling in the adult.44 While we have demonstrated a requirement of Hdac3 in smooth muscle differentiation during embryonic cardiac development, additional work is needed to determine whether Hdac3 plays an analogous role in smooth muscle formation and phenotypic modulation in the adult vasculature. Hdac inhibitors are widely used therapeutic agents and have previously been proposed as useful agents in the treatment of heart failure.40 Given a potential role of Hdac3 in modulation of vascular smooth muscle behavior, vascular effects of Hdac inhibitors in various patient populations, including those with coronary disease and vascular stents, will be interesting topics for future study.
Supplementary Material
Novelty and Significance.
What is Known?
-
-
During embryogenesis, neural crest cells make important contributions to the smooth muscle of the cardiac outflow tract (OFT) and great vessels.
-
-
Many of the signaling pathways that regulate the migration, proliferation, and differentiation of cardiac neural crest have been identified.
-
-
Abnormalities in these processes have been implicated in congenital heart disease in humans.
What New Information Does This Article Contribute?
-
-
The class I histone deacetylase Hdac3 plays a critical role in murine OFT formation
-
-
Hdac3 is required for smooth muscle differentiation in neural crest cells, but not for differentiation of other cardiac neural crest derivatives.
The neural crest is an embryonic progenitor cell population that provides a useful model for studying how cell fate decisions are made during cardiac development and in adult cardiovascular disease. While many of the signaling pathways involved in cardiac neural crest development have been elucidated, additional levels of regulation, including epigenetic changes, have not yet been examined. We Using genetic deletion of Hdac3 in mouse, we show, for the first time, that this important chromatin remodeling enzyme plays an essential role in OFT development. In the absence of neural crest expression of Hdac3, the affected embryos show a variety of OFT and intracardiac defects. We show that Hdac3 is required for smooth muscle differentiation of neural crest cells, and that loss of Hdac3 results in down-regulation of the important smooth muscle regulatory gene Jagged1 in midgestation. These findings reveal a novel level of regulation in the process of smooth muscle differentiation - a process that is of potential relevance not only to the study of congenital heart disease, but to understanding vascular remodeling in adult cardiovascular disease as well.
Acknowledgements
We thank Ashley Cohen for animal husbandry, Theresa Alenghat for generation of the Hdac3 floxed allele, Kurt Engleka for OPT imaging, and members of the Epstein lab, including Rajan Jain and Arun Padmanabhan for helpful discussion.
Sources of Funding
This work was supported by R01 HL071546, the WW Smith endowed chair, and the Spain Fund for Cardiovascular Research (J.A.E.), DK43806 and the Cox Institute for Medical Research (M.A.L), the Medical Scientist Training Program grant (N.S.), and K99-R00 HL098366 (C.M.T).
Non-standard Abbreviations and Acronyms
- OFT
outflow tract
- IAA
inturrupted aortic arch
- DORV
double outlet right ventricle
- PTA
persistent truncus arteriosus
- VSD
ventricular septal defect
- DRG
dorsal root ganglia
- SMA
smooth muscle actin
Footnotes
Disclosures
None.
Subject codes:
[6] Cardiac development
[130] Animal models of human disease
[162] Smooth muscle proliferation and differentiation
In August 2011, the average time from submission to first decision for all original research papers submitted to Circulation Research was 16 days.
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jain R, Rentschler S, Epstein JA. Notch and cardiac outflow tract development. Ann N Y Acad Sci. 2010;1188:184–190. doi: 10.1111/j.1749-6632.2009.05099.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vincent SD, Buckingham ME. How to make a heart: The origin and regulation of cardiac progenitor cells. Curr Top Dev Biol. 2010;90:1–41. doi: 10.1016/S0070-2153(10)90001-X. [DOI] [PubMed] [Google Scholar]
- 3.Park EJ, Watanabe Y, Smyth G, Miyagawa-Tomita S, Meyers E, Klingensmith J, Camenisch T, Buckingham M, Moon AM. An fgf autocrine loop initiated in second heart field mesoderm regulates morphogenesis at the arterial pole of the heart. Development. 2008;135:3599–3610. doi: 10.1242/dev.025437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guo C, Sun Y, Zhou B, Adam RM, Li X, Pu WT, Morrow BE, Moon A. A tbx1-six1/eya1-fgf8 genetic pathway controls mammalian cardiovascular and craniofacial morphogenesis. J Clin Invest. 2011;121:1585–1595. doi: 10.1172/JCI44630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoshida T, Owens GK. Molecular determinants of vascular smooth muscle cell diversity. Circ Res. 2005;96:280–291. doi: 10.1161/01.RES.0000155951.62152.2e. [DOI] [PubMed] [Google Scholar]
- 6.Stoller JZ, Epstein JA. Cardiac neural crest. Semin Cell Dev Biol. 2005;16:704–715. doi: 10.1016/j.semcdb.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 7.High FA, Zhang M, Proweller A, Tu L, Parmacek MS, Pear WS, Epstein JA. An essential role for notch in neural crest during cardiovascular development and smooth muscle differentiation. J Clin Invest. 2007;117:353–363. doi: 10.1172/JCI30070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.High FA, Jain R, Stoller JZ, Antonucci NB, Lu MM, Loomes KM, Kaestner KH, Pear WS, Epstein JA. Murine jagged1/notch signaling in the second heart field orchestrates fgf8 expression and tissue-tissue interactions during outflow tract development. J Clin Invest. 2009;119:1986–1996. doi: 10.1172/JCI38922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huh SH, Ornitz DM. Beta-catenin deficiency causes digeorge syndrome-like phenotypes through regulation of tbx1. Development. 2010;137:1137–1147. doi: 10.1242/dev.045534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang J, Chang JY, Huang Y, Lin X, Luo Y, Schwartz RJ, Martin JF, Wang F. The fgf-bmp signaling axis regulates outflow tract valve primordium formation by promoting cushion neural crest cell differentiation. Circ Res. 2010 doi: 10.1161/CIRCRESAHA.110.225318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Newbern J, Zhong J, Wickramasinghe RS, Li X, Wu Y, Samuels I, Cherosky N, Karlo JC, O’Loughlin B, Wikenheiser J, Gargesha M, Doughman YQ, Charron J, Ginty DD, Watanabe M, Saitta SC, Snider WD, Landreth GE. Mouse and human phenotypes indicate a critical conserved role for erk2 signaling in neural crest development. Proc Natl Acad Sci U S A. 2008;105:17115–17120. doi: 10.1073/pnas.0805239105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vallejo-Illarramendi A, Zang K, Reichardt LF. Focal adhesion kinase is required for neural crest cell morphogenesis during mouse cardiovascular development. J Clin Invest. 2009;119:2218–2230. doi: 10.1172/JCI38194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat Rev Genet. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.LeBoeuf M, Terrell A, Trivedi S, Sinha S, Epstein JA, Olson EN, Morrisey EE, Millar SE. Hdac1 and hdac2 act redundantly to control p63 and p53 functions in epidermal progenitor cells. Dev Cell. 2010;19:807–818. doi: 10.1016/j.devcel.2010.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Knutson SK, Chyla BJ, Amann JM, Bhaskara S, Huppert SS, Hiebert SW. Liver-specific deletion of histone deacetylase 3 disrupts metabolic transcriptional networks. EMBO J. 2008;27:1017–1028. doi: 10.1038/emboj.2008.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dovey OM, Foster CT, Cowley SM. Histone deacetylase 1 (hdac1), but not hdac2, controls embryonic stem cell differentiation. Proc Natl Acad Sci U S A. 2010;107:8242–8247. doi: 10.1073/pnas.1000478107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Montgomery RL, Davis CA, Potthoff MJ, Haberland M, Fielitz J, Qi X, Hill JA, Richardson JA, Olson EN. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 2007;21:1790–1802. doi: 10.1101/gad.1563807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ye F, Chen Y, Hoang T, Montgomery RL, Zhao XH, Bu H, Hu T, Taketo MM, van Es JH, Clevers H, Hsieh J, Bassel-Duby R, Olson EN, Lu QR. Hdac1 and hdac2 regulate oligodendrocyte differentiation by disrupting the beta-catenin-tcf interaction. Nat Neurosci. 2009;12:829–838. doi: 10.1038/nn.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haberland M, Mokalled MH, Montgomery RL, Olson EN. Epigenetic control of skull morphogenesis by histone deacetylase 8. Genes Dev. 2009;23:1625–1630. doi: 10.1101/gad.1809209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guenther MG, Barak O, Lazar MA. The smrt and n-cor corepressors are activating cofactors for histone deacetylase 3. Mol Cell Biol. 2001;21:6091–6101. doi: 10.1128/MCB.21.18.6091-6101.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schroeder TM, Kahler RA, Li X, Westendorf JJ. Histone deacetylase 3 interacts with runx2 to repress the osteocalcin promoter and regulate osteoblast differentiation. J Biol Chem. 2004;279:41998–42007. doi: 10.1074/jbc.M403702200. [DOI] [PubMed] [Google Scholar]
- 22.Zeng L, Xiao Q, Margariti A, Zhang Z, Zampetaki A, Patel S, Capogrossi MC, Hu Y, Xu Q. Hdac3 is crucial in shear- and vegf-induced stem cell differentiation toward endothelial cells. J Cell Biol. 2006;174:1059–1069. doi: 10.1083/jcb.200605113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Y, Kao GD, Garcia BA, Shabanowitz J, Hunt DF, Qin J, Phelan C, Lazar MA. A novel histone deacetylase pathway regulates mitosis by modulating aurora b kinase activity. Genes Dev. 2006;20:2566–2579. doi: 10.1101/gad.1455006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bhaskara S, Chyla BJ, Amann JM, Knutson SK, Cortez D, Sun ZW, Hiebert SW. Deletion of histone deacetylase 3 reveals critical roles in s phase progression and DNA damage control. Mol Cell. 2008;30:61–72. doi: 10.1016/j.molcel.2008.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Montgomery RL, Potthoff MJ, Haberland M, Qi X, Matsuzaki S, Humphries KM, Richardson JA, Bassel-Duby R, Olson EN. Maintenance of cardiac energy metabolism by histone deacetylase 3 in mice. J Clin Invest. 2008;118:3588–3597. doi: 10.1172/JCI35847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trivedi CM, Lu MM, Wang Q, Epstein JA. Transgenic overexpression of hdac3 in the heart produces increased postnatal cardiac myocyte proliferation but does not induce hypertrophy. J Biol Chem. 2008;283:26484–26489. doi: 10.1074/jbc.M803686200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alenghat T, Meyers K, Mullican SE, Leitner K, Adeniji-Adele A, Avila J, Bucan M, Ahima RS, Kaestner KH, Lazar MA. Nuclear receptor corepressor and histone deacetylase 3 govern circadian metabolic physiology. Nature. 2008;456:997–1000. doi: 10.1038/nature07541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun Z, Singh N, Mullican SE, Everett LJ, Li L, Yuan L, Liu X, Epstein JA, Lazar MA. Diet-induced lethality due to loss of hdac3 in heart and skeletal muscle. J Biol Chem. 2011 doi: 10.1074/jbc.M111.277707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang X, Rowitch DH, Soriano P, McMahon AP, Sucov HM. Fate of the mammalian cardiac neural crest. Development. 2000;127:1607–1616. doi: 10.1242/dev.127.8.1607. [DOI] [PubMed] [Google Scholar]
- 30.Engleka KA, Gitler AD, Zhang M, Zhou DD, High FA, Epstein JA. Insertion of cre into the pax3 locus creates a new allele of splotch and identifies unexpected pax3 derivatives. Dev Biol. 2005;280:396–406. doi: 10.1016/j.ydbio.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 31.Feng D, Liu T, Sun Z, Bugge A, Mullican SE, Alenghat T, Liu XS, Lazar MA. A circadian rhythm orchestrated by histone deacetylase 3 controls hepatic lipid metabolism. Science. 2011;331:1315–1319. doi: 10.1126/science.1198125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Novak A, Guo C, Yang W, Nagy A, Lobe CG. Z/eg, a double reporter mouse line that expresses enhanced green fluorescent protein upon cre-mediated excision. Genesis. 2000;28:147–155. [PubMed] [Google Scholar]
- 33.High FA, Lu MM, Pear WS, Loomes KM, Kaestner KH, Epstein JA. Endothelial expression of the notch ligand jagged1 is required for vascular smooth muscle development. Proc Natl Acad Sci U S A. 2008;105:1955–1959. doi: 10.1073/pnas.0709663105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morini F, Cozzi DA, Ilari M, Casati A, Cozzi F. Pattern of cardiovascular anomalies associated with esophageal atresia: Support for a caudal pharyngeal arch neurocristopathy. Pediatr Res. 2001;50:565–568. doi: 10.1203/00006450-200111000-00005. [DOI] [PubMed] [Google Scholar]
- 35.Foster K, Sheridan J, Veiga-Fernandes H, Roderick K, Pachnis V, Adams R, Blackburn C, Kioussis D, Coles M. Contribution of neural crest-derived cells in the embryonic and adult thymus. J Immunol. 2008;180:3183–3189. doi: 10.4049/jimmunol.180.5.3183. [DOI] [PubMed] [Google Scholar]
- 36.Foster KE, Gordon J, Cardenas K, Veiga-Fernandes H, Makinen T, Grigorieva E, Wilkinson DG, Blackburn CC, Richie E, Manley NR, Adams RH, Kioussis D, Coles MC. Ephb-ephrin-b2 interactions are required for thymus migration during organogenesis. Proc Natl Acad Sci U S A. 2010;107:13414–13419. doi: 10.1073/pnas.1003747107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zachariah MA, Cyster JG. Neural crest-derived pericytes promote egress of mature thymocytes at the corticomedullary junction. Science. 2010;328:1129–1135. doi: 10.1126/science.1188222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feng X, Krebs LT, Gridley T. Patent ductus arteriosus in mice with smooth muscle-specific jag1 deletion. Development. 2010;137:4191–4199. doi: 10.1242/dev.052043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Z, Zang C, Cui K, Schones DE, Barski A, Peng W, Zhao K. Genome-wide mapping of hats and hdacs reveals distinct functions in active and inactive genes. Cell. 2009;138:1019–1031. doi: 10.1016/j.cell.2009.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Trivedi CM, Luo Y, Yin Z, Zhang M, Zhu W, Wang T, Floss T, Goettlicher M, Noppinger PR, Wurst W, Ferrari VA, Abrams CS, Gruber PJ, Epstein JA. Hdac2 regulates the cardiac hypertrophic response by modulating gsk3 beta activity. Nat Med. 2007;13:324–331. doi: 10.1038/nm1552. [DOI] [PubMed] [Google Scholar]
- 41.Trivedi CM, Zhu W, Wang Q, Jia C, Kee HJ, Li L, Hannenhalli S, Epstein JA. Hopx and hdac2 interact to modulate gata4 acetylation and embryonic cardiac myocyte proliferation. Dev Cell. 2010;19:450–459. doi: 10.1016/j.devcel.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martin KA, Rzucidlo EM, Merenick BL, Fingar DC, Brown DJ, Wagner RJ, Powell RJ. The mtor/p70 s6k1 pathway regulates vascular smooth muscle cell differentiation. Am J Physiol Cell Physiol. 2004;286:C507–517. doi: 10.1152/ajpcell.00201.2003. [DOI] [PubMed] [Google Scholar]
- 43.Hegner B, Lange M, Kusch A, Essin K, Sezer O, Schulze-Lohoff E, Luft FC, Gollasch M, Dragun D. Mtor regulates vascular smooth muscle cell differentiation from human bone marrow-derived mesenchymal progenitors. Arterioscler Thromb Vasc Biol. 2009;29:232–238. doi: 10.1161/ATVBAHA.108.179457. [DOI] [PubMed] [Google Scholar]
- 44.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.