

Abstract
The present work investigates the feasibility of the design of a novel floating elementary osmotic pump tablet (FEOPT) to prolong the gastric residence of a highly water-soluble drug. Diethylcarbamazine citrate (DEC) was chosen as a model drug. The FEOPT consisted of an osmotic core (DEC, mannitol, and hydrophilic polymers) coated with a semipermeable layer (cellulose acetate) and a gas-generating gelling layer (sodium bicarbonate, hydrophilic polymers) followed by a polymeric film (Eudragit RL 30D). The effect of formulation variables such as concentration of polymers, types of diluent, and coat thickness of semipermeable membrane was evaluated in terms of physical parameters, floating lag time, duration of floatation, and in vitro drug release. The Fourier transform infrared and X-ray diffraction analysis were carried out to study the physicochemical changes in the drug excipients powder blend. The integrity of the orifice and polymeric film layer was confirmed from scanning electron microscopy image. All the developed FEOPT showed floating lag time of less than 8 min and floating duration of 24 h. A zero-order drug release could be attained for DEC. The formulations were found to be stable up to 3 months of stability testing at 40°C/75% relative humidity.
Electronic supplementary material
The online version of this article (doi:10.1208/s12249-011-9699-6) contains supplementary material, which is available to authorized users.
Key words: controlled release, diethylcarbamazine citrate, floating elementary osmotic pump tablet, polymeric film
INTRODUCTION
The application of osmotic pressure-based oral drug delivery systems is one of the most popular approaches to control drug release. The first osmotic pump tablet (OPT) was elementary osmotic pump (EOPT) developed in 1970s (1). The EOPT generally consists of a compartment including the drug and osmotic agents (osmogen) with a semipermeable membrane permeable (SPM) to an external fluid and impermeable to agents in the compartment with an orifice mechanically drilled or formed in situ. The advantages of OPT that holds a prominent place among controlled release systems are pH-independent drug release, reduced adverse reactions, improved patient compliance, and exhibiting good in vitro–in vivo correlation (2). EOPT works on the principle of osmosis, in which solvent moves from lower concentration to higher concentration of solute through SPM until equilibrium at both sides (3). Previous reports on osmotic pump describe simple osmotically controlled system comprising of drug, osmotic agent, and diluents coated with a membrane. It has also been reported by various researchers that the intrinsic water solubility of drug plays an important role in the design of an OPT. Solubility of drug should be within 50–300 mg/ml to attain zero-order release rate (4). For the drugs with poor and intermediate solubility, various osmotic pumps like multi-chamber, push pull, multi-particulate, and modified osmotic pumps have been reported (2). However, no such type of osmotic pump has been developed separately for water-soluble drugs like diethylcarbamazine citrate (DEC). Thus, in our current studies, we have developed a floating elementary osmotic pump tablet (FEOPT) of DEC using the principle of elementary osmotic pump by incorporating hydrophilic polymers in the core compartment. Floating osmotic drug delivery systems draw water from the surrounding fluid on coming in contact with the gastric environment. Gas is formed by chemical reaction of gas-generating agent with the surrounding fluid. The hydration of gel layer simultaneously helps to entrap the gas and makes the system buoyant. The buoyant tablets then release drug via the orifice due to the development of osmotic pressure gradient after entry of gastric fluid in core compartment. In the present study, FEOPT systems were investigated for the delivery of a water-soluble drug. DEC, a Biopharmaceutics Classification System class III drug (aqueous solubility 1,000 mg/ml), was chosen as a model drug (5).
The higher aqueous solubility of DEC may itself generate very high osmotic pressure within the core compartment of tablet. Thus, this property could be utilized to formulate EOPT of DEC without significant need of osmotic agent. DEC is first-line drug to control and treat lymphatic filariasis caused by Wuchereria bancrofti and Brugia malayi. DEC possesses high therapeutic window and considered to be safe up to 2,000 mg. The recommended oral dose of DEC is 100 mg thrice a day up to 12–21 days to obtain maximal medicinal effect against W. bancrofti (6,7). DEC is readily absorbed and stable throughout gastrointestinal tract (GIT) (8). Plasma t1/2 of DEC varies from 4 to 12 h depending upon urinary pH which requires frequent dosing of DEC (9). Mean resident time of DEC in blood can be increased either by preparing its sustained release formulation or by increasing urinary pH; the later task, however, is neither convenient nor clinically safe.
The normal gastrointestinal transit time varies from 6 to 8 h depending upon the fed state of GIT (10). Gastric retention of large dosage form has longer duration in fed state compared to fasting state. The formulation of once-a-day dose regimen has no relevance until it is enabled to remain in the GIT for 24 h, which provides longer absorption time (11). Formulation of an osmotic pump with gastroretentive property in a single device is always being a challenge to formulation scientists. Considering all above advantages of gastric retention and osmotic pump, we have combined both technologies and tried to develop once-a-day gastroretentive osmotic pump, i.e., FEOPT that could efficaciously deliver the DEC in a controlled manner within GIT using floatation technology that enables to retain this system within gastric region. Our extensive search has confirmed that no literatures are available for such kind of floatable osmotic drug delivery system on DEC.
Our EOPT consists of a core tablet containing the drug, diluents, swellable polymers, and osmotic agent and further coated with cellulose acetate to form a SPM. Swellable system consists of swellable polymers (hydroxypropyl methylcellulose (HPMC) and polyethylene oxide (PEO)), which swell and expand rapidly in volume by continuous imbibition of water and control the release of drug. To design and fabricate FEOPT, an EOPT may further be coated with two successive layers; first layer consisted of different proportions of gelling agents (e.g., HPMC K4M and polyvinylpyrrolidone (PVP)) and gas-generating agent (e.g., sodium bicarbonate), while second layer consisted of a polymeric film (e.g., Eudragit RL 30D) coated over gelling layer. In general, the excipients used in FEOPT formulations are generally regarded as safe within their inactive ingredient guide limits (acceptable safety limits from FDA) and are most common in various other formulations. A micropore was created at the convex vertex of final FEOPT tablet using a mechanical microdriller. The solvent (acid/buffer) imbibes through polymeric film within FEOPT, reacts with sodium bicarbonate (basic salt), and liberates CO2 gas due to chemical reaction which provides buoyancy to the whole system. The gas entrapped in gel matrix lowers the density of the system and allows the system to float in gastric fluid. The solvent crosses SPM, consistently dissolves the drug and other osmotic agents (osmogen) of the core, and creates a constant osmotic pressure to release the drug in controlled manner through the orifice. The developed formulations were evaluated in terms of various preparative parameters for the development of EOPT and FEOPT (12).
MATERIALS AND METHOD
Materials
DEC was kindly procured from Inga laboratories (Mumbai, India) as a gift sample. Hydroxypropyl methylcellulose (HPMC K4M, HPMC K100) and PEO were obtained from Torrent research centre (Ahmedabad, India). The following chemicals and excipients were purchased from commercial sources and used as received: cellulose acetate (39.8% acetylation), PVP, mannitol, castor oil, microcrystalline cellulose (MCC; Avicel PH101), dicalcium phosphate (DCP), magnesium stearate, and talc from Central Drug House (New Delhi, India). Acetone (AR grade) was purchased from Qualigens Chemicals (Mumbai, India). Disodium hydrogen orthophosphate, dibutyl phthalate, and PEG 400 and PEG 4000 were purchased from S.D. Fine Chemicals (Mumbai, India).
Method
Drug–Excipient Compatibility Studies
Fourier Transform Infrared Spectroscopy
Infrared spectra DEC, physical mixture (HPMC/PEO/DCP/MCC/PVP 3:1:1:1:0.5) and physical mixture of drug and excipients (1:1) were recorded on Fourier transform infrared (FT-IR) instrument (Shimadzu, Japan) equipped with temperature-controlled high-sensitivity deuterated l-alanine doped triglycine sulfate detector. Samples were prepared and compressed with KBr on Minipress (Jasco, Japan) to form a disk. The compressed disks were scanned over 400 to 4,000 cm−1, and characteristic peaks were recorded and evaluated (3).
X-Ray Diffraction Study
The powder X-ray diffraction (PXRD) patterns of DEC, physical mixture, and physical mixture of drug and excipients were recorded on X-ray diffractometer (Rigaku, Japan) equipped with Cu-rotating anode (radiation; λ = 1.54 nm) generated at 18 kW. Powder diffractometer operating on Bragg–Brentano geometry was fitted with a curved crystal graphite monochromator in the diffraction beam from the range of 20–40° (2θ). The powder was packed into the rotating sample holder (13).
Preparation of Core Tablets
Core tablets of DEC were prepared by wet granulation method. Different compositions (Table I) of drug and excipients (except magnesium stearate and talc) were passed through #60 sieve and mixed together for 10 min in geometric mean. Geometrical mixing was performed by placing the powder with the smallest quantity in the bottle by adding an equal amount of each of the other powders. Continuous addition of each of the remaining powder in an amount that is equal to the blend in the bottle was incorporated and mixed well after each addition to form a homogenous mixture. The blend was granulated using 5% (w/v) solution of PVP in acetone, and wet mass was passed through #36 mesh sieve. Granules were dried in an oven at 50°C for 2 h. Dried granules were lubricated with a mixture of talc and magnesium stearate (each passed through #60 mesh sieve). Lubricated blend was compressed with average weight of 500 mg on a single station tablet punching machine (Cadmach, Ahmedabad, India) fitted with 12-mm round standard concave punch (14).
Table I.
Composition for Different Batches of Elementary Osmotic Pump Tablet
| Ingredients/mg | Batches | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| I | II | III | IV | V | VI | VII | VIII | IX | X | ||
| DEC | 300 | 300 | 300 | 300 | 300 | 300 | 300 | 300 | 300 | 300 | |
| PEO WSR Coagulant | 45 | 40 | 25 | 30 | 35 | 0 | 35 | 35 | 35 | 35 | |
| PEO WSR N60K | 0 | 0 | 0 | 0 | 0 | 35 | 0 | 0 | 0 | 0 | |
| HPMC K4M | 140 | 100 | 100 | 100 | 100 | 100 | 0 | 100 | 100 | 100 | |
| HPMC K100 Premium | 0 | 0 | 0 | 0 | 0 | 0 | 100 | 0 | 0 | 0 | |
| DCP | 5 | 50 | 65 | 60 | 40 | 40 | 40 | 0 | 40 | 40 | |
| MCC | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 40 | 0 | 0 | |
| Mannitol | 0 | 0 | 0 | 0 | 15 | 15 | 15 | 15 | 15 | 15 | |
| Binder (PVP 5%, w/v) | 3.5 | 3.5 | 3.5 | 3.5 | 3.5 | 3.5 | 3.5 | 3.5 | 3.5 | 3.5 | |
| Magnesium stearate | 3.25 | 3.25 | 3.25 | 3.25 | 3.25 | 3.25 | 3.25 | 3.25 | 3.25 | 3.25 | |
| Talc | 3.25 | 3.25 | 3.25 | 3.25 | 3.25 | 3.25 | 3.25 | 3.25 | 3.25 | 3.25 | |
| Plasticizer (%, w/w of CA) | Castor oil | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 0 | 2 |
| PEG-400 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | |
| Coating thickness (μm) | 100 | 90 | 95 | 100 | 105 | 100 | 95 | 100 | 100 | 200 | |
DEC diethylcarbamazine citrate, PEO polyethylene oxide, HPMC hydroxypropyl methylcellulose, DCP dicalcium phosphate, MCC microcrystalline cellulose, PVP polyvinylpyrrolidone, CA cellulose acetate, PEG polyethylene glycol
Fabrication of Elementary Osmotic Pump Tablet
EOPT tablets were prepared by coating of core tablets with a mixture of cellulose acetate, castor oil, and acetone typically in a weight ratio of 0.2:0.004:99.79 to form SPM. Conventional laboratory coating pan (Scientific instruments, New Delhi, India) fitted with three baffles placed at angle of 120° having outer diameter of 10 cm was used. Coating process was optimized (100 tablets/batch) with following condition: preheating temperature, 50°C; preheating time, 30 min; inlet temperature, 48–50°C; outlet temperature, 38–40°C; atomizing air pressure, 1.1 bar; and spray rate, 5–10 ml/min. The coated tablets were further dried in the coating chamber for additional 30 min at 50°C to evaporate the residual moisture (15). The coated tablets were withdrawn from coating pan, when desired coating weight was achieved which was correlated with desired coating thickness.
Fabrication of Floating Elementary Osmotic Pump Tablet
FEOPT consists of two successive layers: gas-generating gelling layer followed by protective polymeric film on EOPT as shown in Fig 1. EOPT was first coated with a solution (dichloromethane/water 9:1) of sodium bicarbonate, HPMC K4M, and PVP plasticized with PEG 6000 (10%, w/w) to form gelling layer. Compositions of gelling layer were varied as represented in the Table II, to obtain minimum buoyancy lag time with maximum floatation for FEOPT. The coating was performed in conventional laboratory coating pan as described above and dried additional 30 min at 50°C to evaporate the residual moisture. The final successive protective layer was coated using solution of Eudragit RL 30D (2%, w/v) in acetone, plasticized with dibutyl phthalate (15%, w/w). Coating was continued until desired weight of Eudragit coated on the tablet as final protective film. The tablets were dried in the coating pan for additional 30 min after the desired coating was achieved. The protective polymeric film maintained the integrity of FEOPT tablets within release media, whereas gas-generating layer helps in floatation. An orifice (300 μm) was mechanically drilled on one face of (convex shape) coated tablet using microdriller (Kamlesh Engineers, Udaipur, India), up to the half thickness of tablets (16).
Fig. 1.
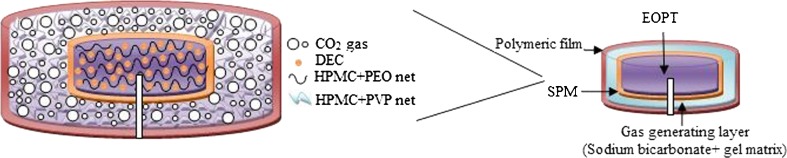
Schematic diagram of FEOPT composed of EOPT, surrounded by gas-generating layer and polymeric film drilled with an orifice. Enlarged diagram describing the formation of HPMC–PEO gel networks within the core, which entraps drug molecules and provides controlled release. Diagram also describing the mechanism of floatation, CO2 gas dispersed within the HPMC-PVP gel network of gas-generating layer
Table II.
Compositions of Gas-Generating Layer and Polymeric Film for Different Batches of Floating Elementary Osmotic Pump Tablet
| Batches | HPMC K4M (%, w/w) | PVP (%, w/w) | Sodium bicarbonate (%, w/w) | Eudragit RL 30D (%, w/w) | Floating lag time (min) | Floating duration (h) |
|---|---|---|---|---|---|---|
| V/FT 1 | 6 | 1 | 4 | 5 | 5 ± 1 | 10 ± 0.5 |
| V/FT 2 | 6 | 2 | 4 | 5 | 5 ± 1 | 12 ± 0.5 |
| V/FT 3 | 7 | 2 | 4 | 5 | 5 ± 1 | 15 ± 0.5 |
| V/FT 4 | 9 | 2 | 4 | 5 | 5 ± 1 | 15.5 ± 0.5 |
| V/FT 5 | 7 | 2 | 5 | 5 | 4 ± 1 | 18 ± 0.5 |
| V/FT 6 | 7 | 2 | 6 | 5 | 4 ± 1 | 24 ± 0.5 |
| V/FT 7 | 7 | 2 | 7 | 5 | 3 ± 1 | 16 ± 0.5 |
| V/FT 8 | 7 | 2 | 6 | 6 | 6 ± 1 | 24 ± 0.5 |
| V/FT 9 | 7 | 2 | 6 | 7 | 8 ± 1 | 24 ± 0.5 |
%, w/w denotes percent of weight of the core tablet. Floating lag time denotes time taken by the system to float, after contact with SGF. Floating time denotes the duration of the floatation
Evaluation
Granules Characterization
Angle of Repose
Angle of repose (θ) of granules was determined by funnel method. Funnel slope of 32° to the vertical axis and 3.2 cm orifice opening was used. The granules were allowed to flow through funnel freely onto the clean surface. Funnel was placed in such a height that bottom tip of funnel should not touched apex of heap of granules (17). Height and radius of cone were measured through a scale, and values were placed in Eq. 1.
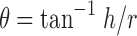 |
1 |
where θ = angle of repose, h = height of cone, and r = radius of cone base
Bulk and Tapped Density
Bulk and tapped density of granules were determined using tap density tester USP method II (ETD-1020, Electrolab, India) (18). Carr’s compressibility index and Hausner ratio were represented by Eqs. 2 and 3.
 |
2 |
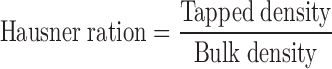 |
3 |
Evaluation of Tablet
Thickness
The thickness of six tablets was measured using digital vernier calipers (Absolute Digimatic, Mitutoyo Corp, Japan). The thickness of each tablet was deviated from ±5% of mean value.
Hardness
Hardness of tablets was determined using digital hardness tester (Campbell Electronics, Mumbai, India). The instrument was calibrated using standard weight of 5 kg.
Weight Variation Test
Twenty tablets were selected at random, and average weight was determined. Individual tablet was weighed and compared with the average weight. Percentage deviation and weight variation were calculated for all the batches (18).
Drug Content Test
Ten individual tablets were weighed and powdered. The powder (equivalent to 300 mg of DEC) was dissolved in a 100-ml volumetric flask filled with distilled water using magnetic stirrer (Eltek MS 203, India) for 24 h. Solution was filtered through Whatman filter paper no. 1, diluted suitably, and analyzed spectrophotometrically at 220 nm (Shimadzu-1700, Japan) (19).
Measurement of Coat Weight and Coat Thickness
Ten tablets were weighed and average weight was determined. Average weight of coat was calculated by deducting average weight of pre-coated tablets from the average weight of the coated tablets. Also the average diameter of 10 pre-coated and post-coated tablets (EOPT) were determined using digital vernier calipers. The difference in diameter was considered to be the thickness of the coating. It was observed that 30 and 60 mg weight gain in tablet were equivalent to 100 and 200 μm thickness of coating membrane, respectively.
In Vitro Drug Release
In vitro release study of FEOPT was determined using USP-XXIV dissolution apparatus type II (Campbell Electronics, Mumbai, India) at 50 rpm in 900 ml of simulated gastric fluid (SGF; pH = 1.2) without pepsin, maintained at temperature of 37 ± 0.5°C. At predetermined time intervals, 1 ml of aliquot was withdrawn, filtered, and analyzed spectrophotometrically at 220 nm (Shimadzu-1700, Japan) (19). Three replicates were performed (n = 3).
Buoyancy Study
The time taken by FEOPT tablet to float (lag time) and duration of floatation were determined using USP-XXIV type II apparatus, and the paddle was rotated at 50 rpm in 900 ml of SGF without pepsin at temperature of 37 ± 0.5°C (20). Three replicates were performed (n = 3).
Scanning Electron Microscopy Study
Surface morphology and effect of dissolution media on SPM was studied by scanning electron microscopy (SEM) (JEOL, JSM-6100, Japan). After 24 h of dissolution, the tablets were air-dried and placed on a spherical brass stub (12 mm diameter) with a double-backed adhesive tape. The mounted samples were sputter-coated for 5 min with gold using fine coat ion sputter (JEOL, JFC-1100, Japan) and examined under SEM (12).
Statistical Data Analysis
Results of in vitro drug release profile were expressed as mean ± standard deviation. Release profiles of various batches were compared using model independent pair-wise approach, which includes calculation of difference factor (f1) and similarity factor (f2).
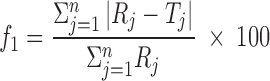 |
4 |
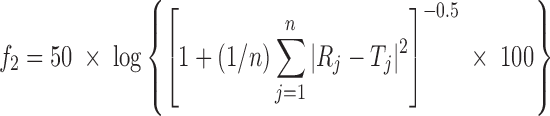 |
5 |
where n is sampling number and Rj and Tj are percent dissolved of the reference and test products at each time point j. The two release profiles are considered to be similar, if f1 value is lower than 15 (between 0 and 15) and f2 value is more than 50 (between 50 and 100) (21).
Accelerated Stability Studies
Accelerated stability studies of optimized tablets of FEOPT were carried out at 40°C/75% relative humidity for 3 months under the International Conference of Harmonization (ICH) guidelines. These tablets were packed in high-density polyethylene container and placed in stability chamber (Narang Scientific Works, New Delhi, India). The samples were withdrawn after 3 months and evaluated in terms of drug content, hardness, floating lag time, duration of floatation, and drug release (22).
Mathematical Modeling of In Vitro Release Kinetics
Various mathematical models were assessed by fitting in vitro release data of the developed batches into different mathematical models to analyze the release, and the best-fitting release mechanism was ascertained. These models were zero-order kinetics, first-order kinetics, Higuchi, and Korsemeyer–Peppas employing the following set of equations:
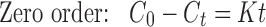 |
6 |
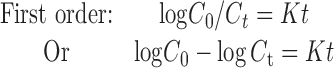 |
7 |
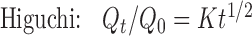 |
8 |
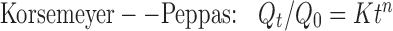 |
9 |
where C0 is the initial concentration of drug in tablet (concentration; that is to be released outside), Ct is concentration at time t present in the solution (concentration; that is released outside tablet), K is the rate constant, t is time, Qt/Q0 is the fraction of drug released, and n is release exponent in Korsmeyer–Peppas model. The value of n is used to indicate different release mechanisms. Value of n = 0.5 indicates Fickian (case I) release, >0.5 but <0.89 for non-Fickian (anomalous) release, n ≈ 1 indicates case II transport (zero-order release), and >1 indicates super-case II type of release (23).
RESULTS AND DISCUSSION
FT-IR Study
Figure 2 shows the characteristic peaks of excipients, pure drug, physical mixture of both, and after 3 months storage. The presence of vibration in the region of 3,350, 3,050, and 1,650–1,750 cm−1 for –OH, –C–H, and –C=O groups showed its presence in DEC. There was neither origin of any new characteristic peaks nor absence of any original characteristic peaks which revealed no incompatibility between drug and excipients even after 3 month storage.
Fig. 2.

FT-IR graph: a physical mixture of excipients, b pure drug, c physical mixture of both, and d after 3 month storage of physical mixture of both. Graph showing no incompatibility between drug and excipients
X-Ray Diffraction
Diffractograms of excipients, pure drug, physical mixture of both, and after 3 month storage are shown in Fig. 3. The major peaks for DEC was seen at 21°, 24°, 27°, and 32° at angle of diffraction (2θ) and remained same in physical mixture. There was no sign of formation of any new peak or absence/shift in original characteristic peak (13). The XRD data revealed crystalline nature of drug, physical compatibility between drug and excipients, and stability of FEOPT.
Fig. 3.

XRD graph: a physical mixture of excipients, b pure drug, c physical mixture of both, and d after 3 month storage of physical mixture of both
Scanning Electron Microscopy
The shape and size of the orifice were examined at magnification of ×100 and presented in Fig. 4. SEM study suggested that the helix of microdriller has created smooth orifice. The orifice and texture of SPM were found to be intact even after 24 h of dissolution.
Fig. 4.

Scanning electron microscopic photograph of orifice in SPM after 24 h dissolution
Evaluation of Optimal Formulation
Official and non-official tests of granules and tablet evaluation (bulk density, tapped density, angle of repose, compressibility index of granules, and hardness, friability, and drug content of tablet) were performed to optimize the formulation of FEOPT tablets. The batch V/FT6 was subjected for various pharmacopoeal and non-pharmacopoeal tests, results of which are listed in Table III. The lubricated granules had shown excellent free flowing characteristic as demonstrated by angle of repose (<30). Compressibility parameters like Carr’s index and Hausner ratio were 27.7%, and 1.3 also showed passable limit on the scale of flowability. Hardness (7 ± 0.5 kg/cm2) and friability (0.3%) of the tablet were found to be good and within the limit. An ideal zero-order release profile of drug should give linear equation of Ctvs. t and considered as reference. The regression coefficient (R2) value for reference should be 1. An ideal zero-order release profile is one which shows perfectly zero-order release kinetics (i.e., 100% drug release in 24 h = 4.167% drug release/h). Thus, an ideal zero-order release formulation should release 4.167% of drug per hour. Ideal zero-order theoretical profile was considered as standard to compare the release profile of all the developed batches. The difference factor (f1) and similarity factor (f2) were calculated and presented in Table IV.
Table III.
Different Evaluation Parameters for Batch V/FT6
| Parameters | Value |
|---|---|
| Bulk density of granules | 0.52 g/ml |
| Tapped density of granules | 0.72 g/ml |
| Carr’s compressibility index of granule | 27.7% |
| Hausner ratio of granules | 1.3 |
| Angle of repose (θ), pre- and lubricated granule | 39° and 26° |
| Hardness of core tablet | 7 ± 0.5 kg/cm2 |
| Friability of core tablet | 0.3% |
| Thickness of core tablet | 4.12 ± 0.05 mm |
| Thickness of EOPT tablet (SPM-coated) | 4.21 ± 0.01 mm |
| Thickness of FEOPT tablet | 5.45 ± 0.08 mm |
| Drug content | 97.98–102.36% |
Table IV.
Difference Factor (f 1) and Similarity Factor (f 2) for Different Batches of EOPT and FEOPT
| Batches | Difference factor (f 1) | Similarity factor (f 2) | Inference | |
|---|---|---|---|---|
| EOPT | FEOPT | |||
| III | 40.04 | 38.44 | Failed | |
| IV | 52.6 | 20.88 | Failed | |
| V | 8.15 | 72.08 | Good | |
| V/FT6 (50 rpm) | 3.21 | 81.20 | Very good | |
| V/FT6 (100 rpm) | 5.1 | 79.12 | Good | |
| V/FT6 (150 rpm) | 10.19 | 66.52 | Good | |
| VI | 78.21 | 24.37 | Failed | |
| VII | 63.05 | 30.33 | Failed | |
| VIII | 59.63 | 30.48 | Failed | |
| IX | 49.72 | 34.8 | Failed | |
| X | 10.15 | 62.20 | Passable | |
| V/FT6 after 3 month | 8.86 | 65.58 | Good | |
Difference factor (f 1) is proportional to average difference between two release profiles. Similarity factor (f 2) measures the closeness between the two release profiles and inversely proportional to the average squared difference between two profiles. Difference factor of 0–15 ensures minor difference between two batches. Similarity factor of 50–100 ensures sameness and performance of different batches. When two profile is identical, f 2 = 100
It was found that optimized batch V of EOPT showed desired and more controlled release compared to other batches (Fig. 5). The release from batch V/FT6 was found to be 13.6%, 29.21%, 56%, 84%, and 98% in 3, 6, 12, 18, and 24 h. Batch V/FT6 also exhibited minimum floating lag time along with the highest floating time (Table II). Exhausted shells, after dissolution, were visually observed for any imperfection or cracks in the coating. There were no visible cracks in the coating, and it remained intact in all the batches after 24 h of dissolution.
Fig. 5.

Effect of different HPMC-to-PEO WSR Coagulant ratios on drug delivered from an elementary osmotic pump tablet. Each dissolution tests were performed in triplicate
Formulation Aspects of EOPT
Drug release in our preliminary batches showed (data has not shown) faster drug release (t90% in 3 h) from SPM-coated DEC tablets (without any polymer) than formulations containing polymers. This can be ascribed to high aqueous solubility of DEC. Assuming a tablet core of pure drug, the fraction of drug released with zero-order kinetics is given by Eq. 10:
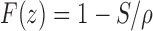 |
10 |
where F(z) is the fraction released by zero-order kinetics, S is the drug’s solubility (grams per cubic centimeter), and ρ the density (grams per cubic centimeter) of core tablet. Drug with the high solubility (<50 mg/ml) would be released (>95%) with zero-order kinetics according to Eq. 10 (18). The rate of drug release from an osmotic system is directly proportional to aqueous solubility of drug.
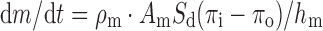 |
11 |
where dm/dt is the rate of drug release per unit time, ρm is the permeability of membrane, Sd is the solubility of solute (drug), Am is the area of membrane, πi is the osmotic pressure inside the tablet, πo is the osmotic pressure outside the tablet, and hm is the membrane thickness (4). Equation 11 gives an idea for drug release from osmotic pump tablet and rate-limiting factors (permeability of membrane, membrane thickness, and osmotic pressure) which could control the release of water-soluble drug.
However, incorporation of hydrophilic polymers could alter the solubility of drug, depending upon nature of hydrophilic gel matrix formation and by increasing the viscosity within core (Fig. 5). In order to modulate the movement of drug inside the tablet core, hydrophilic polymers like PEO WSR Coagulant and HPMC K4M were used in combination that could extend drug release (24). Selection of excipients was based on viscosity of hydrophilic polymers and compressibility characteristics.
Effect of Hydrophilic Polymers
It is clearly evident in batches I and II that higher concentration of hydrophilic polymers (Table I) significantly retards the release rate of drug. This could be due to the increase of viscosity, and dense gel matrix may resist the easy movements of drug molecules (Fig. 5)
Different Grades of Polyethylene Oxide
DEC is a highly water-soluble molecule which would generate very high osmotic pressure inside the tablet core and will release the drug immediately; thus, to control its release, the core needed to have sufficient viscosity to withstand up to longer period. Hydrophilic polymers would be expected to produce greater viscosity in core. Thus, two different viscosity grades of PEO (WSR Coagulant and WSR N60K) were studied. It was noticed that WSR Coagulant behaved more promisingly to control the release of DEC compared to WSR N60K. Batch V containing PEO WSR Coagulant and batch VI containing PEO WSR N-60K was compressed and coated. The in vitro release profile of batch VI after 6 h showed more than 60% drug release as compared to 30% of drug release in batch V (Fig. 6). This could be due to the fact that release of drug was dependent upon the viscosity of hydrophilic polymers. Increment in the viscosity impedes the movement of drug molecules within the system as well as lesser permeation power of water generates weak osmotic pressure in the EOPT. Viscosity of 1% aqueous solution of WSR Coagulant is almost equivalent to 5% solution of WSR N-60K at 25°C (25). However, PEO is highly swellable than HPMC; thus, it could not be used beyond a threshold limit. So the excess use of PEO may affect the integrity of EOPT by two possible ways: either breakage of SPM leading to burst release or chocking of orifice by leaching of swollen PEO which could block the passage for release of drug.
Fig. 6.

Effect of different formulation variables on drug delivered from an elementary osmotic pump tablet. Each dissolution tests were performed in triplicate
Different Grades of HPMC
The effect of two different viscosity grades of HPMC (K4M in batch V and K100 Premium in batch VII) was studied. The in vitro release profile of batch VII was found higher compared to batch V. The batch VII showed 40% and 80% release compared to 20% and 50% release of batch V after 4 and 12 h, respectively (Fig. 6). This decrease in drug release in batch V is due to the fact that K4M possessed higher viscosity than K100 Premium of batch VII. As reported, viscosity of 2% (w/v) aqueous solution of K4M is four times greater than 2% K100 Premium. Thus, K4M retarded the drug release more than K100 Premium. However, HPMC K4M possesses less viscosity than PEO WSR Coagulant (25). Again there was certain threshold limit for the use of HPMC due to similar reasons described for PEO as both are hydrophilic in nature. So the desired quantities of hydrophilic polymers must be within the range without altering the integrity of EOPT.
Effect of Diluents
Diluents were required to improve the hardness of tablets. Preliminary observation suggested that the batches without diluents exhibited very poor hardness value (only 3–4 kg/cm2) which were not acceptable for robustness of tablets. MCC (hydrophilic) and DCP (hydrophobic) compressed in batches VIII and V, respectively, and were compared in terms of their effects on drug release. The drug release rate of batch VIII was found to be higher than batch V. This measurable improvement in release rate can be seen in Fig. 6. It is interesting to note that there may be two reasons behind increase in release behavior: Firstly, hydrophilic nature of MCC increases the solubility of the DEC which further increases the rate of drug release (21), and secondly, MCC possesses disintegrating nature, which may enhance the porosity of core tablet material thereby increasing the penetration of solvent in tablet. The increase of porosity makes easy entry of solvent inside the core tablet, building up the osmotic pressure owing to increase in solubilization of drug (26). In contrast, hydrophobic nature of DCP neither increases the solubility of drug nor alters the disintegration of core tablet. Thus, DCP shows no effect on solubility and release behavior of drug from tablet.
Characteristics of SPM
Types of Plasticizer
SPM coating maintains the integrity of osmotic system by allowing the passage of SGF through it and resists the movement of drug. Combination of water-insoluble coating polymer (e.g., CA) with hydrophilic (e.g., PEG 400) or hydrophobic (e.g., Castor oil) plasticizer has been studied. Plasticizers were added in coating solution to increase the flexibility of SPM to maintain the pressure inside core tablet during exposure to dissolution media. In order to compare the effect of different plasticizers, batch V containing CA/castor oil (0.2:0.004) and batch IX containing CA/PEG (0.2:0.004) were coated separately. Figure 6 shows that the presence of water-soluble plasticizer in batch IX promotes in situ micropore formation within SPM and enhances drug release by two probable mechanisms: increased imbibition of solvent and/or leaching of drug through micropores of SPM. However, hydrophobic plasticizer (castor oil) had shown slower drug release in batch V in contrast to batch IX; thus, castor oil best suits to control delivery of water-soluble drug compared to PEG 400.
Thickness of SPM
To study the effect of coat thickness on release profile, batch V with coat thickness of 105 μm was compared with batch X with coat thickness of 200 μm. Interestingly, the difference of 105 μm coat thickness between two batches produced no significant difference in the drug release profiles. This indicated that the developed FEOPT system was robust with respect to variations in the SPM coating thickness. However, an increase in lag time of 30 min was observed in case of batch X as compared to batch V (Fig. 6). Coating thickness increases the time for SGF penetration through SPM which might increase the lag time. Moreover, the amount of drug release from EOPT was solely governed through orifice not from SPM (impermeable to drug). As the water enters via continuous imbibition through SPM and creates osmotic pressure gradient inside the tablet, drug is released from orifice to the external medium till equilibrium of osmotic pressure is attained on both sides (12).
Formulation Aspects of FEOPT
To improve gastric residence of EOPT, floating system was designed (27). The optimized batch of EOPT (batch V) was further subjected for development of floating system. In the present study, we have combined the property of gas-generating system (gelling agent and sodium bicarbonate) and polymeric film to obtain FEOPT tablets which can float within the stomach. Gas was produced in gas-generating layer as a consequence of imbibition of SGF through polymeric film and reacts with sodium bicarbonate. The floating tablets composed of drug-loaded core were coated with a gas-generating layer and polymeric film, respectively (Figs. 1 and 7). Since sodium bicarbonate itself could not adhere onto the core tablets, HPMC was used as a gelling agent to entrap the CO2 gas bubbles within gas-generating layer. The ideal polymeric film should be highly water permeable in order to facilitate the effervescent reaction. Wet or hydrated coatings should be impermeable to the generated CO2 to maintain floatation. The polymeric film should be sufficiently flexible in wet state to withstand the pressure of generated gas and avoid rupturing (16).
Fig. 7.

Schematic representation of mechanism of floatation and drug release: A floating elementary osmotic pump tablet (FEOPT) containing elementary osmotic pump tablet (EOPT), gas-generating layer, and polymeric film; B CO2 gas bubbles formation due to chemical reaction between sodium bicarbonate and SGF (within gas-generating layer) caused floatation of FEOPT; and C movement of gas bubbles in upward direction and descending of EOPT within FEOPT
Effect of Gelling Agent
Mixture of HPMC K4M and PVP swells and forms a gel matrix on contact with water. This gel matrix entraps CO2 gas and helps on the floatation of FEOPT (28). To obtain optimal ratio of HPMC and PVP for gel matrix, amount of sodium bicarbonate was kept constant. Varying PVP from 1% to 2% by keeping constant amount of HPMC, floating duration was increased from 10 to 12 h. Now, with the increase of HPMC from 6% to 7% by keeping constant amount of PVP, floating duration increased from 12 to 15 h (Table II). Further, with the increase of HPMC from 7% to 9%, the floating duration did not increase significantly because major role for buoyancy was played by sodium bicarbonate instead of HPMC (Table II). Gel matrix acts as a net to entrap CO2 gas, also loses its density with swelling, and provides synergistic effect in floatation. CO2 gas acts as a disintegrating agent for net which dissolves gel matrix rapidly. Thus, to prevent rapid disintegration of gel matrix, PVP incorporated as a binder. It holds the gel net more firmly and aids in the entrapment of CO2 gas.
Effect of Gas-Generating Agent
Floatation of tablet is purely dependent upon the principle of buoyancy. Table II shows that batch V/FT6 was optimized in terms of duration of floatation. To obtain optimal amount of sodium bicarbonate, the amount of HPMC and PVP kept constant. Subsequent increase of sodium bicarbonate from 4% to 6% showed increased floating duration from 15 to 24 h and decreased floating lag time. Further increase of sodium bicarbonate decreases floating lag time as well as duration of floatation. Interesting two explanations for this are: Firstly, the excess gas could not be counter balanced by polymeric film which may cause breakage of film, and secondly, CO2 gas could disintegrate gel matrix; hence, the gas can escape immediately leading to sedimentation of tablet.
Role of Polymeric Film
In our earlier batches (data has not shown) despite of gas formation, the gas-generating tablets could not float for significant period due to absence of any barrier which could prevent the escape of CO2 gas. Absence of any barrier leads the tablet to sink at the bottom. We tried polymeric film coating of Eudragit RL 30D on gelling layer, which acted as flexible but non-swellable barrier and can withstand the pressure of CO2 gas (16). It has been found that with the increase on coating thickness (5%, 6%, and 7%) of polymeric film in different batches (batch V/FT7, V/FT8, and V/FT9), floating lag time and floating duration increased (Table II). This phenomenon can be attributed to the fact that polymeric film provides a shielding layer thus reduces instant penetration of SGF through film, which in turns delays conversion of sodium bicarbonate into CO2 and hence increases the floating lag time. Although SGF rapidly enters in the thinner polymeric films (<5%) thereby decreasing the floating lag time, thinner polymeric films cannot withstand to the pressure of gas formed inside the gel, resulting in coat burst.
Mechanism of Drug Release
The sequence of events observed when a FEOPT tablet comes in contact with SGF is described in Fig. 8. In general, reservoir systems containing drug and polymer can have three different stages of tablet dissolution: a dry or non-hydrated part (material that has not seen any water molecule), another part which is highly viscous, and lastly a part which is in solution state (less viscous). Within 3 min, the CO2 gas bubbles are seen in the surface of FEOPT system, and it becomes buoyant in 5 min as shown in Fig. 8b, c. The CO2 has the tendency to move against gravity which brought over all tablets to move upward as the resultant density became lesser. SGF influx creates osmotic pressure inside FEOPT, and to counterbalance this pressure, less viscous part flows out from the hole at very first, followed by more viscous, and at last the dry part with their subsequent conversion. The EOPT resided inside the FEOPT has relatively higher density, which brings it toward gravity within FEOPT as shown in Fig. 7c. Inversion of tablet takes place in SGF and orifice turned to down face or toward the gravity (Fig. 8c, d). This inversion of tablet also revealed the differences in density within FEOPT. Once the drug is released through orifice of EOPT, some amount of drug get diffused within swollen gas-generating layer of FEOPT, which would be further diffused out through polymeric film of FEOPT (16).
Fig. 8.

Floating sequences of FEOPT in SGF (pH = 1.2) at different time intervals. a 0 min, FEOPT at the bottom; b 3 min, gas bubbles formation; c 5 min, initialization of floatation and drug release through orifice; d floatation lasted up to 24 h
To observe the effect of gas-generating gelling layer and polymeric film, the optimized batch of EOPT and FEOPT was subjected for release study. Figure 9 shows that the optimized batch V of EOPT and batch V/FT6 of FEOPT did not show significant difference in release profiles. Eventually, this revealed that the polymeric film and gelling layer of FEOPT provide only floatation without altering drug release significantly. The schematic representation of mechanism of floatation (Video 1: Clip of floating lag time and floating duration of FEOPT) and drug release (Video 2: Clip showing drug release through orifice of FEOPT) is represented in Fig. 7.
Fig. 9.

Drug delivered from an elementary osmotic pump tablet, floating elementary osmotic pump tablet, and after 3 months storage. Each dissolution tests were performed in triplicate
Effect of pH
The effect of pH on release profile was studied on optimized batch of FEOPT (batch V/FT6) under different pH conditions: pH 1.2 (SGF) and pH 4.5 (phosphate buffer) for 24 h. From batch V/FT6, 15%, 56%, and 96% drug was released in 4, 12, and 24 h at pH 1.2, respectively, whereas at pH 4.5, the cumulative percent release was almost 16%, 55%, and 98% after 4, 12, and 24 h, respectively. Linear and similar pattern of drug release under two different pH conditions shows that the drug release from FEOPT was found to be independent of the pH of surrounding environment (Fig. 10) (4). Thus, FEOPT system follows zero-order release kinetic at all pH condition.
Fig. 10.

Different pH conditions of dissolution media on drug delivered from batch V/FT6. Dissolution was performed for first 2 h in pH 1.2, next 2 h in pH 4.5 and pH 7.4 for the remaining h. Each dissolution tests were performed in triplicate
Effect of Agitation Intensity
To assess the effect of agitation intensity on release profile, three agitation intensities (50, 100, and 150 rpm) were selected for batch V/FT6. The difference (f1) and similarity (f2) factor for FEOPT (batch V/FT6) at three different rotation speeds are presented in Table IV. The difference factor (f1) and similarity factor (f2) values were found to be 3.21 and 81.20 (50 rpm), 5.1 and 79.26 (100 rpm), and 10.19 and 66. 52 (150 rpm). Release profile of DEC from FEOPT at different agitation intensities is shown in Fig. 11. It has been found that different rotation speeds could not significantly affect drug release. Thus, it can be expected that to a large extent drug release from FEOPT is dependent upon osmotic pressure built inside the FEOPT and independent of the hydrodynamic conditions of the absorption site (12).
Fig. 11.

Effect of agitation intensity (50, 100, and 150 rpm) on drug delivered from floating elementary osmotic pump tablet of batch V/FT6. Each dissolution tests were performed in triplicate
Mathematical Modeling of In Vitro Release Kinetics
In vitro release data of different batches were fitted to various mathematical models in order to ascertain the kinetics of drug release. Based on Korsmeyer–Peppas power model, drug release data were analyzed for curve fitting and drug release exponents (n). Results confirmed that batches I and II showed super-case II type of release, batch III showed non-Fickian diffusion kinetics, whereas batches IV and V showed zero-order kinetics. The linear nature of plots between percent cumulative drug release and time suggests that batch V followed very close to the zero-order kinetic, which was further confirmed by the higher sum of correlation coefficient (R2 = 0.9533, n = 0.959, almost equivalent to 1) among all batches (Table V).
Table V.
Fitting In Vitro Drug Release Data of the Batch V/FT6 According to Various Mathematical Models
| Models | Zero order | First order | Higuchi | Korsmeyer–Peppas | |||||
|---|---|---|---|---|---|---|---|---|---|
| Batches | R 2 | K o | R 2 | K F | R 2 | K H | n | R 2 | K KP |
| I | 0.9668 | 2.44 | 0.9274 | 0.0155 | 0.8233 | 0.1178 | 1.0194 | 0.9444 | −1.7709 |
| II | 0.9887 | 3.2952 | 0.9884 | 0.0246 | 0.9295 | 0.1672 | 1.4547 | 0.9659 | −1.961 |
| III | 0.91 | 4.4037 | 0.9868 | 0.0638 | 0.9655 | 0.2373 | 0.8959 | 0.9765 | −1.1114 |
| IV | 0.9732 | 4.3117 | 0.9224 | 0.0616 | 0.9669 | 0.2252 | 0.9533 | 0.9922 | −1.2382 |
| V | 0.9949 | 4.1945 | 0.8034 | 0.0631 | 0.9472 | 0.2141 | 0.959 | 0.9982 | −1.2995 |
| VI | 0.972 | 10.233 | 0.9941 | 0.0887 | 0.9712 | 0.3055 | 0.7775 | 0.9957 | −0.7675 |
| VII | 0.9351 | 5.761 | 0.9546 | 0.0729 | 0.9928 | 0.2421 | 0.3635 | 0.3230 | −0.5058 |
| VIII | 0.9265 | 6.1325 | 0.9717 | 0.0762 | 0.9764 | 0.2506 | 0.5095 | 0.3681 | −0.6276 |
| IX | 0.9503 | 5.7614 | 0.9879 | 0.0601 | 0.9774 | 0.2319 | 0.4763 | 0.3333 | −0.6384 |
| X | 0.9965 | 4.4635 | 0.9888 | 0.0312 | 0.902 | 0.1602 | 0.0848 | 0.0066 | −0.5824 |
n drug release exponents, K KP Korsmeyer–Peppas release constant, R 2 correlation coefficient, K 0 zero-order release rate constants, K F first-order release rate constant of different models
Accelerated Stability Study
Stability study provides the evidence on the quality of drug product, and FEOPT (batch V/FT6) formulation was charged for 3 months under ICH guidelines. Samples withdrawn after 3 months showed no significant difference in the characteristic peaks of FT-IR and XRD and also in terms of physical properties, drug content, hardness, floating lag time, and duration of floatation. The in vitro release of before and after 3 month storage was found to be similar as shown in Fig. 9.
CONCLUSION
From the present study, an optimized formulation of FEOPT for DEC, a water-soluble drug, was successfully prepared. The FEOPT tablet consists of an EOPT surrounded by a gas-generating gelling layer and a polymeric layer with an orifice. Developed FEOPT showed excellent floating properties (floating lag time <8 min and floating duration ≥24 h) with zero-order drug release up to 24 h. The data on physical parameters provided an insight for selection of excipients for the development of FEOPT. Drug release from FEOPT system was found to be independent to pH and hydrodynamic conditions of the dissolution medium. Other variables like role of hydrophilic polymers, thickness of coating, and level of gas-generating agent also have been investigated individually, which provided deeper understanding of floating and drug release mechanism. Mathematical modeling of drug release data suggested that FEOPT could follow zero-order kinetic and provide required controlled release up to 24 h. Accelerated stability data confirmed that the developed FEOPT are stable and complied with reproducible release performance. In view of overall results reported in the present study, it may be proposed that FEOPT can be a newer osmotic drug delivery platform for controlled delivery of water-soluble class of drugs.
Electronic supplementary materials
Below is the link to the electronic supplementary material.
Clip showing floating lag time and floating duration of FEOPT. (MPG 48050 kb)
Clip showing release of drug through orifice of FEOPT (MPG 1014 kb)
ACKNOWLEDGMENTS
The authors are thankful to Dr. A. Patani (Director, Inga Laboratories, Mumbai) for providing gift sample of DEC and Prof. Dhananjay Pandey, School of Material Science, IT-BHU for providing XRD facility.
Conflict of Interest
There is no conflict of interest between authors. The authors have not received any financial assistance from any funding agency for this research work.
REFERENCES
- 1.Li X, Pan W, Nie S, Wu L. Studies on controlled release effervescent osmotic pump tablets from Traditional Chinese Medicine Compound Recipe. J Control Release. 2004;96:359–367. doi: 10.1016/j.jconrel.2004.01.026. [DOI] [PubMed] [Google Scholar]
- 2.Verma RK, Mishra B, Garg S. Osmotically controlled oral drug delivery. Drug Dev Ind Pharm. 2000;26:695–708. doi: 10.1081/DDC-100101287. [DOI] [PubMed] [Google Scholar]
- 3.Kumar P, Singh S, Mishra B. Colon targeted delivery systems of metronidazole based on osmotic technology: development and evaluation. Chem Pharm Bull. 2008;56:1234–1242. doi: 10.1248/cpb.56.1234. [DOI] [PubMed] [Google Scholar]
- 4.Kumar P, Singh S, Rajinikanth PS, Mishra B. An overview of osmotic pressure controlled release formulation. J Pharm Res. 2006;5:34–45. [Google Scholar]
- 5.Kasim NA, Whitehouse M, Ramachandran C, Hussain AS. Molecular properties of WHO essential drugs and provisional biopharmaceutical classification. Mol Pharm. 2003;1:85–96. doi: 10.1021/mp034006h. [DOI] [PubMed] [Google Scholar]
- 6.Peixoto CA, Alves LC, Brayner FA, Florencio MS. Diethylcarbamazine induces loss of microfilarial sheath of Wuchereria bancrofti. Micron. 2003;34:381–385. doi: 10.1016/S0968-4328(03)00099-4. [DOI] [PubMed] [Google Scholar]
- 7.Hakim SL, Vythilingam I, Marzukhi MI, Mak JW. Single-dose diethylcarbamazine in the control of periodic brugian filariasis in Peninsular Malaysia. Trans R Soc Trop Med Hyg. 1995;89:686–689. doi: 10.1016/0035-9203(95)90445-X. [DOI] [PubMed] [Google Scholar]
- 8.Baveja SK, Ranga RKV, Devi KP. Sustained release tablet formulation of diethylcarbamazine part III. Int J Pharm. 1985;27:157–162. doi: 10.1016/0378-5173(85)90065-1. [DOI] [Google Scholar]
- 9.Edwards G, Breckenridge AM, Adjepon-Yamoah KK, Le M, Ward SA. The effect of variations in urinary pH on the pharmacokinetics of diethylcarbamazine. Br J Clin Pharm. 1981;12:807–812. doi: 10.1111/j.1365-2125.1981.tb01311.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davis SS, Hardy JG, Far JW. Transit of pharmaceutical dosage forms through the small intestine. Gut. 1986;27:886–892. doi: 10.1136/gut.27.8.886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bardonnet PL, Faivre V, Pugh WJ, Piffaretti JC, Falson F. Gastroretentive dosage forms: overview and special case of Helicobacter pylori. J Control Release. 2006;111:1–18. doi: 10.1016/j.jconrel.2005.10.031. [DOI] [PubMed] [Google Scholar]
- 12.Mishra M, Mishra B. Design and evaluation of microporous membrane coated matrix tablets for a highly water soluble drug. Chem Pharm Bull. 2010;58:995–1000. doi: 10.1248/cpb.58.995. [DOI] [PubMed] [Google Scholar]
- 13.Seo DH, Jeong Y, Kim DG, Jang MJ, Jang MK, Nah JW. Methotrexate-incorporated polymeric nanoparticles of methoxy poly(ethylene glycol)-grafted chitosan. Colloids Surf B Biointerfaces. 2009;69:157–163. doi: 10.1016/j.colsurfb.2008.10.020. [DOI] [PubMed] [Google Scholar]
- 14.Waterman KC, MacDonald BC, Roy MC. Extrudable core system: development of a single-layer osmotic controlled-release tablet. J Control Release. 2009;134:201–206. doi: 10.1016/j.jconrel.2008.11.017. [DOI] [PubMed] [Google Scholar]
- 15.Ramakrishna N, Mishra B. Plasticizer effects and comparative evaluation of cellulose acetate and ethyl cellulose–HPMC combination coating as semi permeable membrane for oral osmotic pump of naproxen sodium. Drug Dev Ind Pharm. 2002;28:403–412. doi: 10.1081/DDC-120003001. [DOI] [PubMed] [Google Scholar]
- 16.Sungthongjeen S, Sriamornsak P, Puttipipatkhachorn S. Design and evaluation of floating multi-layer coated tablets based on gas formation. Eur J Pharm Biopharm. 2008;69:255–263. doi: 10.1016/j.ejpb.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 17.Ileleji KE, Zhou B. The angle of repose of bulk corn stover particles. Powder Technol. 2008;187:110–118. doi: 10.1016/j.powtec.2008.01.029. [DOI] [Google Scholar]
- 18.Kumar P, Singh S, Mishra B. Gastroretentive drug delivery system of ranitidine hydrochloride based on osmotic technology: development and evaluation. Curr Drug Deliv. 2008;5:332–342. doi: 10.2174/156720108785914943. [DOI] [PubMed] [Google Scholar]
- 19.Swamy PV, Gada SN, Shirsand SB, Kinagi MB, Shilpa H. Design and evaluation of cost effective orodispersible tablets of diethylcarbamazine citrate by effervescent method. Int J Pharm Sci Res. 2010;1:258–264. [Google Scholar]
- 20.Srivastava AK, Wadhwa D, Ridhurkar D, Mishra B. Oral sustained delivery of atenolol from floating matrix tablets—formulation and in vitro evaluation. Drug Dev Ind Pharm. 2005;31:367–374. doi: 10.1081/ddc-54313. [DOI] [PubMed] [Google Scholar]
- 21.Maulvi FA, Dalwadi SJ, Thakkar VT, Soni TG, Gohel MC, Gandhi TR. Improvement of dissolution rate of aceclofenac by solid dispersion technique. Powder Technol. 2011;207:47–54. doi: 10.1016/j.powtec.2010.10.009. [DOI] [Google Scholar]
- 22.Waterman KC, Adami RC. Accelerated aging: prediction of chemical stability of pharmaceuticals. Int J Pharm. 2005;293:101–125. doi: 10.1016/j.ijpharm.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 23.Costa P, Lobo JMS. Modeling and comparison of dissolution profiles. Eur J Pharm Sci. 2001;13:123–133. doi: 10.1016/S0928-0987(01)00095-1. [DOI] [PubMed] [Google Scholar]
- 24.Prabakaran D, Singh P, Kanaujia P, Vyas SP. Effects of hydrophilic polymers on the release of diltiazem hydrochloride from elementary osmotic pumps. Int J Pharm. 2003;259:173–179. doi: 10.1016/S0378-5173(03)00230-8. [DOI] [PubMed] [Google Scholar]
- 25.Rowe RC, Sheskey PJ, Quinn ME. Handbook of pharmaceutical excipients. 6. London: Pharmaceutical; 2009. [Google Scholar]
- 26.Pesonen T, Paronen P, Ketolainen J. Disintegrant properties of an agglomerated cellulose powder. Int J Pharm. 1989;57:139–147. doi: 10.1016/0378-5173(89)90302-5. [DOI] [Google Scholar]
- 27.Iannuccelli V, Coppi G, Bernabei MT, Cameroni R. Air compartment multiple-unit system for prolonged gastric residence. Part I. Formulation study. Int J Pharm. 1998;174:47–54. doi: 10.1016/S0378-5173(98)00229-4. [DOI] [Google Scholar]
- 28.Chena RN, Ho HO, Yu CY, Sheu MT. Development of swelling/floating gastroretentive drug delivery system based on a combination of hydroxyethyl cellulose and sodium carboxymethyl cellulose for losartan and its clinical relevance in healthy volunteers with CYP2C9 polymorphism. Eur J Pharm Sci. 2010;39:82–89. doi: 10.1016/j.ejps.2009.10.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Clip showing floating lag time and floating duration of FEOPT. (MPG 48050 kb)
Clip showing release of drug through orifice of FEOPT (MPG 1014 kb)