

Abstract
Reactive impurities in pharmaceutical excipients could cause drug product instability, leading to decreased product performance, loss in potency, and/or formation of potentially toxic degradants. The levels of reactive impurities in excipients may vary between lots and vendors. Screening of excipients for these impurities and a thorough understanding of their potential interaction with drug candidates during early formulation development ensure robust drug product development. In this review paper, excipient impurities are categorized into six major classes, including reducing sugars, aldehydes, peroxides, metals, nitrate/nitrite, and organic acids. The sources of generation, the analytical method for detection, the stability of impurities upon storage and processing, and the potential reactions with drug candidates of these impurities are reviewed. Specific examples of drug–excipient impurity interaction from internal research and literature are provided. Mitigation strategies and corrective measures are also discussed.
Key words: excipients, impurities, interaction, mitigation, variability
INTRODUCTION
Degradation products in drug product formulations are sought to be tightly controlled by the industry. Depending upon the drug’s daily dose, regulatory guidelines require the characterization and toxicological evaluation of degradates if they accumulate in the drug product above a threshold concentration. Drug degradation in products often results from the reaction of active pharmaceutical ingredients (API) with excipients used in the formulation. Many of the reported drug–excipient reactions involve hydrolysis, oxidation, or specific interaction of drugs with reactive impurities in excipients. Excipients are multicomponent materials, and the appropriateness of the term “impurity” has been questioned. Although some of the components may be innocuous or even desirable, this review is focused on those reactive “components” that are detrimental to the drug products and are hence referred to as “reactive impurities” throughout the manuscript.
Depending on the application of a specific excipient as well as other formulation and processing factors, the presence of these reactive impurities in excipients even in trace amounts could influence the safety and efficacy of the drug products, especially for highly potent APIs which have low dose and high excipient/API ratio in the drug product. Some examples of drug incompatibility with reactive impurities in excipients in drug products are provided in Table I.
Table I.
A Sample of Drug Incompatibility with Excipient Impurities
| Drug | Impurity | Excipient | Drug loading (w/w) |
|---|---|---|---|
| BMS-203452 (18) | Formaldehyde | PEG 300 or Tween 80 | 1% |
| Fluoxetine HCl (6) | Reducing sugars | Lactose | 10% |
| Org-30659 (94) | Lactose phosphate | Lactose | 0.10% |
| Compound A (39) | Peroxides | Povidone/Copovidone | 2–3% |
| Compound B (39) | Peroxides | Povidone/Copovidone | 2–3% |
| Raloxifene (37) | Peroxides | Povidone/Copovidone | 12.50% |
| CP448187 (82) | Free radicals/peroxides | Microcrystalline Cellulose | 0.50% |
| BMS-A (9) | Free radicals/peroxide/reducing sugars | Microcrystalline Cellulose | 0.83% |
| Vigabatrin (8) | Reducing sugars, aldehydes | Microcrystalline cellulose | – |
| Irbesartan (19) | Formaldehyde | PEG in film coating | Low strength |
| Haloperidol (27) | Furfuraldehyde | Lactose | 0.575% |
| Varenicline (16) | Formic acid/formaldehyde | PEG | 0.68% |
| Hydralizine (28) | Aldose | Starch | 10% |
A robust formulation is one that is able to accommodate the typical variability in API, excipients, and processes (1). The choice of excipients in the design of formulation is made based on their function as well as chemical compatibility with the drug substance. In a typical drug–excipient compatibility experiment, drug stability at accelerated temperature is assessed in the presence of single or multiple excipients (either as powder blend or compact) with or without humidity/water. Although these experiments are useful as a first step to eliminate incompatible excipient(s), the chemical compatibility of drugs with different lots of excipients or excipients from different vendors is usually never studied prospectively (2). The excipient lot-to-lot variability in drug compatibility might arise from the variability in the levels of reactive impurities in excipients. Additionally, the drug product stability may also be influenced by formulation processes, environmental conditions (microenvironmental pH, temperature, water content, and/or water activity (RH), and oxygen), and any changes in the drug/excipient ratio or API particle size.
Understanding the excipient (i.e., reactive impurity sources, variability, etc.) and its impact on the drug product stability is essential in designing and developing a robust drug product. Some of the common reactive excipient impurities that have been reported (e.g., Table I) to cause drug degradation are reducing sugars, aldehydes, hydroperoxides, organic acids and esters, heavy metals and trace metals, nitrates/nitrites, and free radicals. It is unrealistic to expect compendial limits, if available, for the reactive impurities in excipients to provide proper control as the tolerance of the reactive impurities varies widely between drug products. A reactive impurity that may be problematic for a particular drug product may not be a problem for many other drugs. The reactive impurities may either be residues from excipient manufacturing processes or degradation products of the excipients.
This review paper focuses on profiling these reactive impurities in ten commonly used solid dosage form excipients, namely, lactose, microcrystalline cellulose, povidone, crospovidone, hydroxypropyl cellulose, sodium starch glycolate, sodium croscarmellose, pre-gelatinized starch, stearic acid, magnesium stearate and silicon dioxide, and the chemical interaction between the API and the reactive impurity in excipients in the drug products. The origin of these reactive impurities, their fate upon processing and storage, and the analytical detection and quantitation methods are provided. Potential chemical reactions between the reactive impurities and susceptible APIs are identified. Finally, strategies to mitigate the potential incompatibilities are also reviewed.
NATURE OF REACTIVE IMPURITIES IN EXCIPIENTS
Reducing Sugars
Glucose and lactose are reducing sugar excipients. Incompatibility of amine drugs with these sugars is well known. Trace level reducing sugars are also found in non-reducing excipients such as microcrystalline cellulose (MCC), starch, mannitol, maltitol, and sucrose. Some of these excipients also contain aldehyde impurities and will be discussed in the next section.
Sources of Reducing Sugar Impurities
The manufacturing processes utilized by excipient vendors are often trade secrets, but it is conceivable that reducing sugar impurities could be generated during the manufacturing processes of the excipients (e.g., microcrystalline cellulose) where acid hydrolysis and milling are commonly used. They can also be generated as degradation products of these polysaccharide excipients during long-term exposure to heat and moisture. For example, MCC may be manufactured by controlled hydrolysis of α-cellulose from plant with dilute mineral acid (3). Trace levels of glucose (40–80 ppm) were reported in some lots of MCC (Table II). Starch is a mixture of amylose and amylopectin, the ratio of which differs depending on the source of plants. It is prepared from plant seeds or roots like corn, wheat, potato, and tapioca through coarse milling, water washing, wet sieving, and centrifugal separations. Degradation of starch into smaller chains during the isolation and fractionation steps can hardly be avoided. Mannitol is produced by catalytic or electrolytic reduction of monosaccharides such as mannose and glucose. Trace levels of reducing sugar from mannitol was reported to cause oxidative degradation of a cyclic heptapeptide from a lyophilized formulation (4). Lactose is a natural disaccharide consisting of galactose and glucose. Lactose and its monosaccharide components can undergo Maillard reaction with primary amines (5) and secondary amine drugs such as Fluoxetine Hydrochloride (6).
Table II.
Profiling of Reactive Impurities in Selected Lots of Pharmaceutical Excipients
| Excipients | Sources/lot | Impurity (ppm) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Glucose | HCHO | Hydrogen peroxide | NO2 | NO3 | Monochloroacetate | Heavy metals | Trace metals | ||
| Microcrystalline cellulose, PH102 | FMC/1 | 79.6 | 4.8 | <2 | N/A | N/A | N/A | <10 | <5 Mg, Mn; <10 Al, Cr, Cu, Fe, Ni, Zn; 10 Ca |
| FMC/2 | 59.5 | 5.1 | <2 | 9.4 | 23.0 | 0.9 | N/A | N/A | |
| FMC/3 | 40.7 | 4.1 | ND | N/A | N/A | N/A | N/A | N/A | |
| Lactose Fast Flo | Foremost | ND | N/A | <2 | 10.4 | 12.4 | 12.0 | <10 | <5 Cr, Cu, Fe, Mg, Mn, Ni, Zn; <10 Al; 15 Ca |
| Lactose monohydrate | Foremost/1 | ND | 1.4 | <2 | 5.1 | 9.1 | 1.0 | <10 | <5 Cr, Cu, Fe, Mg, Mn, Ni, Zn; <10 Al, Ca |
| Foremost/2 | ND | ND | <2 | 5.5 | 8.0 | 0.9 | <10 | <5 Cr, Cu, Fe, Mg, Mn, Ni, Zn; <10 Al, Ca | |
| Lactose anhydrous | Quest/1 | ND | 7.4 | <2 | 5.4 | 4.3 | 0.6 | <10 | <5 Cr, Cu, Fe, Mg, Mn, Ni, Zn; <10 Al; 37 Ca |
| Quest/2 | ND | 3.6 | <2 | 3.7 | 6.0 | 0.6 | <10 | <5 Cr, Cu, Fe, Mg, Mn, Ni, Zn; <10 Al; 32 Ca | |
| Pre-gelatinized starch | Colorcon/1 | ND | 14.7 | <2 | 14.5 | 29.2 | 4.4 | <10 | <10 Cr, Cu, Fe, Mg, Mn, Ni, Zn; <20 Al, Ca |
| Colorcon/2 | ND | 10.9 | <2 | 11.8 | 22.9 | 2.3 | <10 | <10 Cr, Cu, Fe, Mg, Mn, Ni, Zn; <20 Al, 21 Ca | |
| Colorcon/3 | ND | 11.1 | N/A | N/A | N/A | N/A | N/A | N/A | |
| Povidone | ISP/1 | INC | INC | 37 | 2.2 | 13.6 | ND | <10 | <5 Cr, Cu, Fe, Mg, Mn, Ni, Zn; <10 Al, Ca |
| ISP/2 | INC | INC | 72 | 1.6 | 13.1 | ND | <10 | <5 Cr, Cu, Fe, Mg, Mn, Ni, Zn; <10 Al, Ca | |
| Crospovidone | ISP/1 | ND | 40.8 | 66 | 17.2 | 52.4 | ND | N/A | <5 Mn; <10 Al, Cr, Cu, Fe, Ni, Zn; 5 Mg; 10 Ca |
| ISP/2 | ND | 8.5 | 69 | 10.5 | 30.4 | ND | N/A | <5 Mg, Mn; <10 Al, Ca, Cr, Cu, Fe, Ni, Zn | |
| Sodium starch glycolate | Roquette/1 | – | 4.6 | <2 | 279.2 | 183.1 | ND | <10 | <5 Cr, Cu, Fe, Mn, Ni, Zn; <10 Al, 79 Ca; 9 Mg |
| Roquette/2 | – | 1.5 | <2 | 285.6 | 117.3 | 135.8 | <10 | <5 Cr, Cu, Fe, Mn, Ni, Zn; <10 Al, 75 Ca; 8 Mg | |
| Croscarmellose Na | FMC/1 | ND | 6.5 | <2 | 2.4 | 23.8 | 52.2 | N/A | N/A |
| FMC/2 | ND | 6.6 | <2 | 1.4 | 10.3 | 21.6 | <10 | <10 Cr, Cu, Fe, Mg, Mn, Ni, Zn; <20 Al, 42 Ca | |
| Magnesium stearate | Mallincrodt/1 | ND | 3.8 | <2 | 2.1 | 6.0 | ND | <10 | <5 Mn; <10 Al, Ca, Cr, Cu, Fe, Ni, Zn; |
| Mallincrodt/2 | ND | 3.7 | <2 | 5.3 | 12.5 | 0.7 | N/A | N/A | |
| Stearic acid | Crompton | ND | 3.1 | <2 | 3.5 | 6.6 | ND | ND | <5 Mn; <10 Al, Ca, Cr, Cu, Fe, Ni, Zn; 30 Mg |
| Hydroxypropyl cellulose | Hercules/1 | ND | 11.4 | 13 | N/A | N/A | N/A | N/A | N/A |
| Hercules/2 | ND | 9.4 | 13 | 0.9 | 3.5 | ND | <10 | <5 Cr, Cu, Fe, Mg, Mn, Ni, Zn; <10 Al, 23 Ca | |
| Silicone dioxide | Degussa/1 | ND | 6.1 | <2 | 5.8 | 12.5 | ND | N/A | 7 Mg; <5 Mn; <10 Al, Ca, Cr, Cu, Fe, Ni, Zn |
| Degussa/2 | N/A | N/A | <2 | 1.5 | 8.7 | ND | N/A | 200 Al; 480 Ca; 30 Fe; 130 Mg; <5 Mn, <10 Cr, Cu, Ni, Zn | |
ND not detectable, N/A not available, INC incompatible
Heavy metals and trace metals analysis conducted using Inductively Coupled Plasma Atomic Emission Spectroscopy (ICP-AES) - Microwave digestion in acid was used for treatment of insoluble excipients
Analytical Method and Levels of Detection of Reducing Sugar
High-performance liquid chromatography (HPLC) determination of trace levels of reducing sugars, such as glucose, is challenging due to the absence of a chromophore for UV detection and the difficulty of achieving adequate retention in an HPLC column. Conventional glucose assays, such as enzymatic and colorimetric assays, typically lack specificity and are not sufficiently sensitive to detect glucose at parts per million concentrations. A reversed-phase HPLC method to determine trace levels of glucose and formaldehyde in pharmaceutical excipients was reported by our lab (7). The method utilizes pre-column derivatization of analytes with 2,4-dinitrophenylhydrazine (DNPH) to enable UV detection of trace glucose and formaldehyde. Reactive impurities in excipient samples were extracted in 1:1 acetonitrile/water solution prior to the derivatization reaction. The reaction reagent is composed of 0.2 mg/mL DNPH and 96 mM HCl in 20% (v/v) acetonitrile. Derivatization is carried out at room temperature by mixing 1 mL of the DNPH reagent and 1 mL of the extracted sample solution and stirred for at least 2 h before HPLC analysis. HPLC analysis was performed on a YMC ODS-AQ column with a gradient of acetonitrile (0.1% TFA). The wavelength for UV detection is 354 nm. Kinetic studies were conducted to determine the minimum amount of DNPH and the length of time required for the derivatization reaction. The detection limit for glucose is as low as 1 ppm, and the limit for formaldehyde is 0.3 ppm. Several batches of excipients were analyzed for glucose, formaldehyde, and other reactive impurities; the results are presented in Table II.
Chemical Interactions with Reducing Sugar Impurities
The major chemical interaction associated with reducing sugar impurities is the Maillard reaction with amine drugs. George et al. (8) reported a drug incompatibility associated with Avicel® (microcrystalline cellulose) in Vigabatrin tablets. Products of the Maillard reaction between the primary amine drug, Vigabatrin, and the monosaccharide component of MCC, i.e., glucose, were implicated in the browning of Vigabatrin tablets on aging. In another study, MCC was found to be responsible for granulation discoloration in a capsule formulation of BMS compound A (9). Approximately 40 ppm of glucose was detected in the particular lot of MCC used in the formulation. The discoloration was indicative of Maillard reaction occurring between glucose impurity from MCC and l-phenylalanine in the drug complex. The chemistry of Maillard reaction was reviewed by Njoroge and Monnier (10), and a four-step reaction scheme was provided. Glycosylamine is usually formed as the first step in the reaction in which the glycosidic hydroxyl group in the reducing sugar is replaced by the amine group. Glycosylamine could undergo Amadori rearrangement (11) to produce ketose-amines and react further with additional amines to produce advanced Maillard reaction products, such as melanoidin, leading to discoloration. The degradation levels are usually dependent on drug loading, microenvironmental pH, and moisture content in the formulation. Low drug loading, high moisture content, and alkaline microenvironmental pH will contribute to a faster reaction rate. Lactose, as a reducing sugar excipient, is usually avoided in formulations containing amine drugs. It could contain many other reactive impurities such as formic acid, acetic acid, furfuraldehyde, and other aldehydes. The potential chemical interaction of API with the aldehydes and organic acids will be discussed in the following sections of the review.
Aldehydes
Formaldehyde, acetaldehyde, and furfuraldehyde (and possibly other aldehydes) are common aldehyde impurities which exist in MCC, starch, pre-gelatinized starch, crospovidone, hydroxypropyl cellulose, polyethylene glycol, and lactose (Table II).
Analytical Methods and Levels of Detection of Aldehydes
Headspace gas chromatography is the most commonly used method to determine trace volatile impurities in pharmaceutical excipients. Li et al. (12) developed an analytical method to detect and quantify trace amounts of C1–C8 aliphatic aldehydes and benzaldehyde in excipients by headspace gas chromatography (GC). The method involves the derivatization of aldehydes with O-2,3,4,5,6-(penta fluorobenzyl) hydroxylamine hydrochloride (PFBHA), followed by static headspace GC of PFBHA aldehyde oximes with MS detection. Thirty typical excipients were screened for formaldehyde and acetaldehyde using this method. Among the excipients tested, polyethylene glycol (PEG) 200, 400, and 600 exhibited significantly high levels of formaldehyde (65.2–107.0 ppm) and acetaldehyde (2.7–12.5 ppm). Another gas chromatography/mass spectrometry (GC/MS) method developed by del Barrio et al. (13) determines formic acid and formaldehyde in excipients simultaneously. The method utilized a one-step procedure requiring the dissolution or dispersion of samples in acidified ethanol to convert formic acid and its esters to ethyl formate and formaldehyde to diethoxymethane. Identification and quantification of the derivatized analytes were conducted by GC/MS. The authors claimed the detection limit for formic acid to be 0.5 ppm (range, 0.5–10,000 ppm) and 0.2 ppm (range, 0.2–10,000 ppm) for formaldehyde. There are several other methods for the measurement of formaldehyde in the literature, such as the colorimetric method using chromotropic acid (CTA) or acetylacetone and the Purpald (4-amino-3-hydrazino-5-mercapto-1,2,4-triazole) method which requires the oxidation of the formaldehyde–Purpald adduct for color development. The CTA (4, 5-dihydroxynaphthalene-2,7-disulfonic acid) assay is a popular method for the detection of formaldehyde as it is highly specific. This colorimetric assay, however, requires lengthy heating of the sample under strong acidic conditions (e.g., 100°C, 30 min). The CTA assay is not suitable for the determination of formaldehyde in starch- and cellulose-based excipients as the strong acid may hydrolyze the end glucose on the polymeric chain of these excipients and release intrinsic aldehydes, causing false high levels of aldehyde readings.
The DNPH methods described under “Reducing Sugars” was developed in our labs and was shown to be a simple and sensitive method to determine trace formaldehyde, acetaldehyde, and other aldehydes in excipients. It works at room temperature and the reaction time is 1 h. DNPH derivatization is an acid-catalyzed reaction at pH 1.5–2; however, starch and cellulose excipients (and their derivatives) will not be degraded as they are not in contact with the acidic reaction solution. The reactive impurities from these excipients were extracted out in 1:1 acetonitrile/water in the sample preparation step. The extracted solution was filtered through 0.45-μm membranes before adding to the DNPH solution for the derivatization reaction. The method utilized HPLC for separation and quantification and could be beneficial for labs without GC/MS capability.
Sources of Aldehyde Impurities and Chemical Interactions with API
Formaldehyde could be formed from the breakdown of the polymeric chain of polyethylene glycol and polysorbates (14–17) (Fig. 1). Nassar et al. (18) reported 2–165 ppm of formaldehyde in various lots of PEG 300 and polysorbate 80. Formaldehyde in these excipients reacted with the drug substance BMS-204352 and formed a hydroxymethyl derivative in the parenteral formulation (Fig. 2). Trace level of formaldehyde (8 ppm) in BMS-203452 formulation (10 mg/mL) was sufficient to generate 1% degradation product. Similarly, film-coated tablets of Avapro™ (Irbesartan) were found to degrade to a hydroxymethyl derivative of the drug substance during long-term stability studies of the low-dose tablet (19) (Fig. 3). The formaldehyde adduct formation was attributed to the formaldehyde impurity from PEG used in the tablet coating material—Opadry™ II white. Eliminating PEG from the blend of Opadry™ prevented the formation of the degradates. Another example of API–aldehyde interaction is Fenfluramine, where formaldehyde reacts with the amine group in the API to form an N-methyl derivative (Fig. 4) (20). Formaldehyde could also be formed as a degradation product of the drug. In the case of hydrochlorothiazide (HCTZ) bead formulation, formation of a trace amount of formaldehyde was observed due to the hydrolysis of HCTZ under high humidity conditions. It subsequently reacted with sodium starch glycolate, decreasing its functionality as a disintegrant and therefore retarding dissolution of the formulation (21). In addition, formaldehyde is known to cross-link gelatin capsule shells, leading to dissolution slowdown and incomplete drug release. The cross-linking is a result of formaldehyde interaction with amino groups in gelatin to form an insoluble protein. Formaldehyde is susceptible to air oxidation and could be partially converted to formic acid. Therefore, excipients having residual formaldehyde are expected to contain some formic acid impurity as well.
Fig. 1.

Formaldehyde/formic acid formation from oxidation and breakdown of polyethylene glycol and polysorbates (14,15)
Fig. 2.

Chemical interaction between BMS-204352 and formaldehyde (18)
Fig. 3.

Proposed mechanism of the degradation of irbesartan by formaldehyde (19)
Fig. 4.

Reaction mechanism for the methylation of amines (20)
Furfuraldehyde, an aromatic aldehyde, can be formed during the manufacturing process of those excipients which are sourced from plants. Many plant materials contain hemicellulose, a polymer of sugars containing five carbon atoms. When heated with sulfuric acid, hemicellulose undergoes hydrolysis to yield xylose and other five carbon sugars, which may undergo dehydration to form furfuraldehyde (22). 5-Hydroxymethyl-2-furfuraldehyde (HMF) can also be formed as a result of heat sterilization of parenteral solutions containing hexoses. It is reported that spray-dried lactose contains furfuraldehyde (23,24). The presence of HMF was found to correlate with the discoloration of lactose. The reaction between HMF and primary amine drugs could lead to the formation of Schiff bases (25,26). The “browning reaction” is base-catalyzed and may therefore be accelerated in solutions with high pH or in solid dosage forms if alkaline lubricants are used. In the case of Vigabatrin, direct reaction of the drug with HMF resulted in the formation of a colored compound (II) which was isolated from the pigment of aged Vigabatrin tablets (Fig. 5). The amorphous content of lactose, equilibrium moisture content, microenvironmental pH, and salt/free base form of the drug can contribute to the extent of the reaction. The impurity, 5-HMF, can also react with a carbonyl (ketone) compound with an α-hydrogen to form a condensation product. Janicki and Almond (27) showed that Haloperidol reacted with HMF to form the condensation product as shown in Fig. 6.
Fig. 5.

Reaction of vigabatrin with HMF to form a colored compound II (8)
Fig. 6.

Reaction of haloperidol with HMF to form a condensation product (27)
Starch is a polymer of α-d-glucose units. The terminal glucose unit at the end of the chain contains an aldehyde functionality in equilibrium with the cyclic hemiacetal. The terminal aldehydes in starch have been shown to react with the hydrazine moieties of hydralazine HCl and form phthalazine hydrazone and/or triazolophthalazine derivative (28) (Fig.7). Starch also has the potential to physically adsorb drug substance (29), potentially leading to drug retention during extraction or dissolution that can translate to incomplete physiological absorption (30).
Fig. 7.

Reaction of starch with hydrazine moieties of hydralazine HCl (28)
Hydrogen Peroxide and Hydroperoxide
The sources of reactive peroxides in drug product formulations are from polymeric excipients such as povidone, hydroxypropylcellulose, crospovidone, polyethylene glycol, polyethylene oxide, and polysorbate. Many pharmaceutical excipients contain trace levels of hydroperoxide (HPO) impurities, especially polymeric excipients which are commonly generated through radical reactions leaving trace peroxides as a by-product.
Peroxides can, in general, be either organoperoxides (ROOR′) or hydroperoxides (ROOH) (31). A general free radical mechanism of peroxide generation involves homolytic cleavage of the C–H bond next to a heteroatom, followed by the addition of oxygen which leads to peroxy radical formation. The peroxy radical can then participate in an autocatalytic cycle by the abstraction of a hydrogen radical from another reactant to form a hydroperoxide while generating another carbon free radical (32). The O–O bond in peroxides is particularly weak and can cleave to form alkoxy (RO⋅) and hydroxy (⋅OH) free radicals. Peroxides and free radicals can also lead to the formation of other reactive oxygen species, such as superoxide anion (O2−⋅), hydrogen peroxide (H2O2), and organic hydroperoxides (ROOH). The organoperoxides, such as benzoyl peroxide and methyl ethyl ketone peroxide, are typically used as initiators of polymerization reactions, bleaching agents, and free radical generators in experimental systems. Hydroperoxides, on the other hand, are the common peroxide impurities in pharmaceutical excipients and play a major role in drug degradation (33).
Analytical Methods for Detection of Hydrogen Peroxide and Hydroperoxide
Measurement of trace HPO in pharmaceutical excipients has been reported in the literature (34,35). One of the methods is the HPLC-based HPO assay, involving triphenylphosphine (TPP) for total HPO content (ROOH and H2O2) (35). TPP reacts with ROOH or H2O2 to give 1 mol of triphenylphosphine oxide (TPPO) for each mole of HPO. The detection limit of TPPO at 220 nm by HPLC is <10 pmol, corresponding to less than one peroxide molecule in a 10-mg sample. Some other techniques for HPO measurement include the ferrous oxidation–xylenol orange (FOX2) method for total HPO, the liquid chromatography-based electrochemical determination of hydrogen peroxide using platinum and enzyme electrodes (36) (detection limit, >1 ng/mL), and the enzyme-based Reflectoquant® colorimetric test for inorganic peroxide (0.2–20 ppm). Other analytical methods, such as the titanium sulfate method or the iodide titration methods reported in US Pharmacopoeia, British Pharmacopoeia, and European Pharmacopoeia, are for the determination of more concentrated hydrogen peroxide solutions.
Sources of Hydrogen Peroxide and Hydroperoxide Impurities
Peroxides may be introduced into an excipient during the manufacturing process. Polymeric excipients such as polyethylene oxide derivatives and polyvinylpyrrolidones (Povidone, PVP) often use peroxides to initiate the polymerization reaction, and it is generally thought that this is the primary source of oxidants in polymeric excipients as it is difficult to completely eliminate them from the final product (36). As an example of such a process, the manufacture of polyvinylpyrrolidone was reported to involve a series of reactions. First, butyrolactone is synthesized through the Reppe process. Butyrolactone then reacts with ammonia to produce pyrrolidone, followed by a vinylation reaction of pyrrolidone and acetylene under pressure. The product, vinylpyrrolidone, is subsequently polymerized in the presence of catalysts to produce povidone. More recently, it was reported that commercial solutions of PVP contain non-detectable levels of peroxides, suggesting that the introduction of peroxides may occur after the synthesis during the drying process. PVP freeze-dried from commercial solutions does not increase in peroxides on storage; however, peroxides in spray-dried, drum-dried, or belt-dried PVP increases with time upon storage, implicating the high temperatures used in these processes as a source of peroxide formation in these materials (32). Peroxide content can increase on storage due to reactions with the excipient, and the rate of increase can depend on how the excipient is manufactured. For PVP, the drum-drying process is conducted at a higher temperature and with longer residence time than the other drying processes. This process builds up peroxide more rapidly than spray-dried PVP (37). Our studies indicate that peroxide levels in PVP decrease as humidity increases (Fig. 8). At 40°C/60%RH, the hydroperoxide level in PVP decreased significantly compared with the initial level (about tenfold reduction).
Fig. 8.

Hydroperoxide levels in povidone (PVP K-30) as a function of temperature and humidity at 10 weeks
Water-insoluble cross-linked PVP (crospovidone) is prepared from the polymerization of PVP in the presence of catalysts such as alkaline metals, their oxides, or hydroxides at 140°C. This procedure often generates colored polymer products. A new pathway of synthesis for crospovidone was developed by Hofman and Herrle (36), where vinylpyrrolidone polymerization was carried out in the presence of cross-linking reagents N,N-divinyl imidazolidon and radical initiators (AIBN, organic peroxides). Tallon et al. reported that cross-linker type B, N,N-divinyl imidazolidinone, has twice the number of oxidation sites compared with cross-linker type A, which is ethylidene vinylpyrrolidinone. Peroxide accumulation on storage is much greater in type B crospovidone (38).
Cellulosic excipients can utilize either peroxide or a hypochlorite bleaching agent which can remain in the excipients at trace levels and become a source of undesirable oxidation of drug substance. Polyethylene oxides are formed by generating high-molecular-weight material and oxidizing the material to the desired molecular weight range, leaving residual peroxides as a potential trace impurity.
Chemical Interactions of Hydrogen Peroxide and Hydroperoxide Impurities with API
Any drug substance that is prone to oxidation can be susceptible to interactions with hydroperoxide containing excipients such as PVP, crospovidone, and HPC. The APIs can exhibit two-electron reactions such as the formation of N-oxide (38,39) and oxidation of thiols (40) or one-electron oxidation such as the abstraction of benzylic hydrogen atoms (41), etc. (Fig. 9).
Fig. 9.

Piperazine reaction with hydrogen peroxide from crospovidone (38,39)
Trace Heavy Metals
Metals are ubiquitous in pharmaceutical excipients at very trace levels and can catalyze the oxidation of pharmaceuticals. Inductively coupled plasma–atomic emission spectroscopy is the common analytical method to determine trace metals in excipients. The sensitivity of the method can be as low as parts per billion to parts per million levels. The reaction of molecular oxygen with most organic molecules is thermodynamically favored; however, the triplet state of molecular oxygen (the ground state) represents a kinetic barrier. Trace metal impurities can react with triplet oxygen, reducing the molecule to more kinetically favored oxidizing agents such as superoxide (42).
Another common mode of transition metal-mediated oxidation involves the Fenton-like reactions where the oxidized or reduced form of a catalytic transition metal such as Fe(III) or Fe(II) react with hydrogen peroxide to produce several more reactive species. Hydrogen peroxide can be reduced to a hydroxyl radical and hydroxide or oxidized to a peroxy radical and a proton (43).
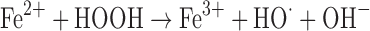 |
1 |
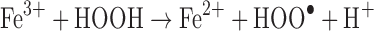 |
2 |
Metal chelators are often employed to scavenge reactive transition metals from formulations; however, hydrogen peroxide can often react with the chelator–metal complex, generating reactive species as well (31).
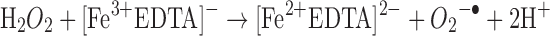 |
3 |
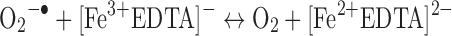 |
4 |
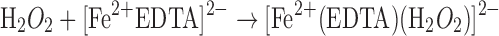 |
5 |
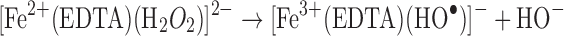 |
6 |
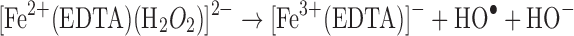 |
7 |
Nitrite and Nitrate
Sources of Nitrite and Nitrate Impurities
Nitrates and nitrites are common nitrosating impurities that can be found in most excipients at parts per million levels. Sodium starch glycolate, croscarmellose sodium, pre-gelatinized starch, PVP, cPVP, and Lactose Fast Flo® are excipients that carry trace levels of nitrate or nitrite impurities. To screen trace levels of nitrate and nitrite in excipients, an ion exchange chromatography method was developed where an AS-11 Dionex was used as the analytical column and an AG-11 Dionex used as the guard column. Results from the impurity profiling of 26 excipient samples are presented in Table II.
Chemical Interactions of Nitrite and Nitrate with API
Nitrogen-containing pharmaceutical compounds have the potential to form N-nitroso compounds (NOC) as degradates in the drug product due to interactions with nitrite or nitrate impurities in the excipients. Degradation of the drug substance of any kind can cause a loss of potency, but nitrosation at trace levels can also be carcinogenic (44). The exact source of nitrates and nitrites in excipients has not been investigated, but it is possible that these trace impurities could come from processing water, processing steps requiring acid titration, bleaching, and potentially from oxidation of air as the excipient is being heated in a drying process (45). Functional groups that can potentially form NOCs include dialkyl, alkylaryl, diaryl, cyclic secondary amines, N-alkylureas, N-alkylcarbamates, and N-alkylamides to form nitrosamines or nitrosamides. To a much lesser extent, tertiary amines, cyanamides, guanidines, amidines, hydroxylamines, hydrazines, hydrazones, and hydrazides may also form NOCs. Brambilla and Martelli (46) reviewed 182 drugs that react with nitrite. Nitrite and nitric acid are not effective nitrosating agents directly, but rather form more reactive NO+ carrier species (specifically, N2O3 in the case of nitrite) under mildly acidic conditions (47).
Nitrosation of this variety depends on the balancing of two opposing pH effects—the reaction of two molecules of nitric acid to form N2O3 at a low pH and the deprotonation of the amine at higher pH, leading to an optimum pH range of 2.5–3.4 for this reaction to occur. The reaction rate is proportional to the concentration of the amine and the square of the concentration of nitrite. Many less reactive amino compounds are not nitrosated by N2O3, but can react with the nitrosonium ion (NO+) formed from nitric acid at low pH. The reaction rate for these reactions is proportional to the concentration of the amine, the concentration of nitrite, and the concentration of hydronium ion. Figure 10 illustrates the nitrosation of Bromhexine by N2O3 (48).
Fig. 10.

Nitrosation of bromhexine (48)
Contrasting the concern for nitrosating species in various excipients, nitrates and nitrites are found at reactive levels in human saliva and stomach milieu. There is an endogenous pathway for the production of nitrates and nitrites as well as the availability of dietary nitrates (which are partially converted to nitrite endogenously) from fruits and vegetables, animal-based products such as processed meats, as well as water (49,50). Typical formulator concern for the production of potentially toxic drug-related NOC is limited to the generation of these species in the drug product; however, substantial formation can occur after ingestion as well, questioning the significance of the mitigation strategies employed for the drug product. Work comparing drug product formation of drug-related NOC species with endogenous formation would be valuable in guiding development scientists in understanding what level of NOC formation mitigation in the drug product is meaningful.
As many of the foods we eat also contain compounds with functional groups prone to nitrosation (e.g., amino acids), a substantial amount of endogenous NOC formation (i.e., not drug-related) is observed through dietary sources, suggesting that the impact of exposure to pharmaceutical NOCs compared with dietary NOCs may need to be investigated to understand whether a substantial difference exists.
Organic Acids
Formic acid and its esters, acetic acid and monochloro acetic acid, are trace organic acid impurities that may be present in pharmaceutical excipients (Table III) (13). Residual organic solvents from the synthesis and purification of excipients may go through further degradation to form organic acids.
Table III.
Reported Trace Organic Acids Impurities in Pharmaceutical Excipients
| Impurity | Excipient | Reported levels (ppm) |
|---|---|---|
| Formic acid (13) (including formyl esters) | Polyethylene glycol | 10–1,000 s (MW-dependent) |
| Hydroxypropyl methylcellulose | 10–100 | |
| Povidone | 1000 s | |
| Polyvinyl alcohol | 30–40 | |
| Acetic acid (51,95) | Polyvinyl alcohol cellulose acetate butyrate | 100 s |
| Monochloro acetic acid | Sodium starch glycolate | 0–140 |
| Croscarmellose sodium | 22–53 |
Sources of Organic Acid Impurities
Oxidation of PEGs at high temperature at a central carbon followed by chain scission could generate formaldehyde. Air oxidation of formaldehyde at temperatures used for accelerated stability testing could lead to the formation of formic acid, which would then react with alcohols to form esters. Formic acid could also be formed through the oxidation of terminal groups in PEG (14) and polysorbates (15–17) (Fig. 1).
Polyvinyl alcohol (PVA) is frequently used as a film-forming polymer in tablet coating systems. Hydrolysis of polyvinyl acetate is a common method for the manufacture of PVA, which leads to the presence of polyvinyl acetate, acetic acid, and also some methyl acetate in the commercially available PVA. Also, relatively high levels (20–60 ppm) of formic acid can be present in the PVA as an impurity. We observed that the formic acid level does not increase in PVA (solid powder) on storage, while it increases significantly in a mixture of PVA with PEG (both as solid powder). The rate of increase of formic acid in the mixture of PVA and PEG was greater than that in PEG alone (51).
Monochloroacetate is a potentially reactive impurity in croscarmellose sodium and sodium starch glycolate. Croscarmellose sodium is produced from the reaction of sodium monochloroacetate with primary alcohols on the cellulose backbone. The levels of monochloroacetate in selected batches of croscarmellose sodium and sodium starch glycolate is presented in Table II.
Some weakly basic drugs can compete with the sodium counterion in croscarmellose sodium and sodium starch glycolate, adsorbing onto the surface of the disintegrant particles. Adsorption of weakly basic drugs (52–54) and salts of weakly basic drugs to croscarmellose sodium has been observed to cause incomplete in vitro dissolution and/or incomplete extraction. Sodium starch glycolate is a substituted and cross-linked derivative of potato starch. Starch is carboxymethylated by reacting it with sodium chloroacetate in an alkaline medium followed by neutralization with citric acid, or some other acids. Cross-linking may be achieved by either physical methods or chemically by using reagents such as phosphorus oxytrichloride or sodium trimetaphosphate. Solid-state ion transfer reaction between a salt of a weak base and carboxymethylcellulose sodium (NaCMC) is shown below.
| 8 |
Analytical Methods for Organic Acids
The levels of organic acids in excipients are not usually tested by the excipient manufacturer due to their low toxicity as class III solvents. The development of analytical methods for the quantitation of trace organic acid impurities is challenging (13). The GC/MS method is the most commonly used method to determine these impurities (18). For example, the formyl species detection methods usually require derivatization with an alcohol, such as ethanol, to form an ester, followed by HPLC or GC separation and detection. These methods are nonspecific with respect to the type (free acid versus ester) and relative proportion of formyl species present in the starting materials. The ion exchange HPLC method described under “Nitrate and Nitrite” could be used to determine trace monochloroacetate in excipients. Ion exchange chromatography could also be used to detect other organic acids.
Chemical Interactions of Organic Acids with API
These trace impurities can potentially react with the amino and/or hydroxyl groups in drugs to form significant levels of degradants. Drugs with alcohol groups can form esters with organic acids or undergo transesterification with esters (e.g., parabens). Similarly, acidic drugs can esterify with excipients containing alcohol groups. For example, ester formation was reported between the carboxylic acid moiety of cetirizine and polyols such as sorbitol and glycerol in oral solution and oral drop formulation (55).
Formic acid can cause drug instability in the formulations. For example, Waterman et al. (16) observed N-formylation and N-methylation of the secondary amine group in varenicline. These were attributed to the presence of formaldehyde and formic acid in the formulation. N-methylvarenicline (NMV) and N-formylvarenicline (NFV) were formed in the osmotic tablets of varenicline, a secondary amine (52). NMV was produced by the reaction of the amine moiety with both formaldehyde and formic acid impurities in an Eschweiler–Clarke reaction, while NFV was formed by the reaction of formic acid alone with varenicline (52).
In another study, optical isomerization of an experimental compound was observed at an asymmetric carbon atom that linked the pyrrolobenzodiazepine ring to a heterocyclic ring through an amide bond (56). This was in a soft gelatin capsule dosage form which contained a mixture of PEG 400 and glycerol. The degradation of the active was accelerated by the formic acid in the formulation. In yet another study, nonspecific degradation of miconazole, which is a tertiary amine compound and commonly used as an antifungal agent, was observed in a liquid formulation of hydrogenated castor oil and lactic acid (57). The hydrogen peroxide, formaldehyde, and formic acid from the hydrogenated castor oil on stability were identified as the cause of this instability.
The rate of N-formamide adduct formation in the drug product could be highly variable. It is affected by temperature, humidity, formyl species (free formic acid and formyl esters) level in the drug product, and microenvironment effects in solid-state reactivity (2). In the liquid state, a bimolecular reaction involving an amine drug and formic acid to form the N-formamide is directly proportional to the concentrations of the reacting species. In the solid state, temperature and moisture play a role by increasing the plasticity and molecular mobility of the reacting species (58). However, the inherent characteristics of solid-state reactions, such as the type, amount, and distribution of formyl species in the microenvironment, increase the variability in N-formamide formation in the drug product. Also, the presence of formyl species in the drug product could be attributed not only to the initial content of formyl species in the excipients but also to their generation over storage of the drug product, which can be highly variable.
Displacement of chlorine in monochloroacetate by an amine or alcohol could lead to an impurity with MW + 58 with respect to the parent compound (Fig. 11).
Fig. 11.
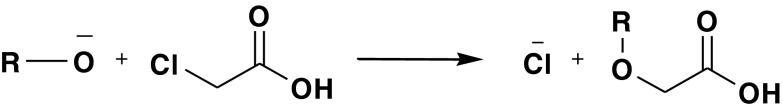
Displacement of chlorine in monochloroacetate
Mitigation Strategies
Early identification of the incompatibilities between an excipient or its impurities and the drug is the best approach to avoid undesirable surprises during late-stage development or commercialization. Excipients that show incompatibilities in compatibility and/or stability studies should preferably be avoided unless an appropriate substitute is unavailable. Various mitigation strategies and controls are discussed in the following sections. While some of the mitigation strategies such as modification of API crystal properties, formulation composition, manufacturing process, and packaging/storage conditions can be implemented by the formulation scientists, the others require a great deal of communication and cooperation not only between various internal groups such as R&D, sourcing, purchasing, and manufacturing/technical operation within a pharmaceutical company but also between the excipient vendor and the pharmaceutical company.
Change in the API Properties and API/Excipient Ratio
Chemical degradation of drugs in the products can be influenced by the API form and powder characteristics. For example, Badawy (59) showed that the mesylate salt form of DMP 754 had better stability than the acetate salt in the tablet dosage forms. An additional way of mitigating the hydrolytic instability of DMP 754 (60) was the incorporation of pH modifiers. In addition to hydrolysis, oxidation and addition reactions can also be mitigated by pH modification. For instance, lowering the pH in an oral solution reduced the reactivity of the piperazine nitrogen, thus mitigating the reaction of the drug with hydrogen peroxide impurity to form the N-oxide degradant (39). Also, Gold et al. (61) showed that the cross-linking between gelatin and formaldehyde impurity is pH-dependent, and lowering the pH reduces the interaction between the amine and formaldehyde.
The influence of powder characteristics on drug product stability is evident in solid-state reactions that depend on the exposed surface of drug particles (3). Thus, milling increased the reactivity of metoclopramide hydrochloride with amorphous lactose in physical mixtures (62). The authors attributed this to the increase in the surface area of drug particles in addition to the increase in amorphous content and crystal defects in drug particles as a result of milling. The authors also observed that spatial proximity of reacting solid particles can increase reactivity. Thus, tablets of metoclopramide hydrochloride with all three kinds of lactose (spray-dried anhydrous, spray-dried monohydrate, and amorphous) had higher Maillard reaction rates than physical mixtures of respective powders. Drug reactivity further depended on the compression force used for tableting in the case of spray-dried anhydrous lactose. Therefore, selection of appropriate drug particle size and particle size reduction technique can influence drug product stability. Also, in the case of compressed tablet dosage forms, the selection of appropriate solid fraction and hardness range of tablets can help minimize reactivity in the solid state.
In addition, solid-state surface reactions that depend on the surface exposure of reactive functional groups can be influenced by the polymorphic form of the API. Thus, Chen et al. (63) hypothesized that the different reactivities of two polymorphs of flufenamic acid with magnesium oxide, an acid–base reaction, in physical mixtures at high-temperature and humidity conditions were due to differences in the surface exposure of reactive functional groups. In cases where one or more excipients are involved in drug instability, drug loading in the formulation can also play a significant role. Thus, Desai et al. (64) observed greater instability of peliglitazar to acid- and/or base-catalyzed degradation in conventional tablet formulations with low drug-to-excipient ratio compared with an active coated formulation with high drug-to-excipient ratio in the film coat.
Antioxidants and Other Stabilizers in Excipients and Drug Product Formulations
Oxidation during storage and processing frequently generates reactive excipient impurities. For example, generation of formic acid from PEG is hypothesized to follow a free radical oxidation mechanism. The use of antioxidants in excipients (such as BHT in PEG) and drug products is a common stabilization strategy.
Free radical oxidation reactions tend to propagate themselves until the substrate is depleted. The initiation of a free radical reaction could involve heavy metal(s), peroxides, or oxygen acting together with environmental stresses such as heat or light. These reactions could be terminated by bimolecular reactions of one free radical with another free radical or a stabilizing conjugated system, which can produce a relatively nonreactive product (31).
Free radical-mediated oxidative degradation reactions could be stabilized by inhibiting the initiation and/or propagation phases and/or promoting chain termination. One of the stabilizing approaches commonly utilized in drug product formulations of susceptible compounds is to use an antioxidant. Based on their mechanism of action (65), antioxidants could act as:
Initiation inhibitors. Antioxidants could react with or remove reactive impurities that may act as catalytic initiators of free radical reactions, such as peroxides. Antioxidants may also react with initiating free radicals and terminate the reaction, thus preventing a reaction cascade pathway. Such antioxidants could include ethylenediaminetetraacetic acid (EDTA), which acts as a heavy metal chelating agent.
Terminators of free radicals. Some antioxidants, such as BHA and BHT, can react with free radicals. This inhibits the propagation phase of the free radical chain reaction.
Antioxidants as reducing agents. Antioxidants may possess lower redox potential than the substrate undergoing oxidation in a formulation. In such cases, antioxidants may act as a reducing agent by getting preferentially oxidized, thus protecting the substrate by competitive reactivity. Ascorbic acid, thiols (such as thioglycerol and thioglycollic acid), and polyphenols (such as propyl gallate) can act as reducing agents.
The choice of an antioxidant for a given formulation is usually empirical. There is a general lack of a consensus approach on the relative prioritization and basis for the selection of antioxidants. Few investigations have reported the relative efficacy of some antioxidants under given experimental conditions. For example, oxidative degradation of an experimental compound, RG 12915, at the benzofuran and quinuclidine moieties to produce a hydroperoxide and an N-oxide, respectively, was metal (Cu3+)-catalyzed and was prevented by EDTA. Metal chelation by EDTA is presumed to be the mechanism in this case. Also, propyl gallate as an antioxidant inhibited the oxidation of the benzofuran (cyclic oxygen). However, it did not inhibit the oxidation at the quinuclidine (cyclic nitrogen) moiety (66). Another study found that α-tocopherol was more effective than BHA, which, in turn, was more effective than propyl gallate in preventing the oxidative degradation of lovastatin in an aqueous solution (67).
In addition to antioxidants, noncovalent complex formation between drug and excipients can stabilize a drug. For example, hydrolysis of benzocaine (68), procaine (69), tetracaine (70), riboflavin (71), and phenyl benzoates (72) in aqueous solution is inhibited by complexation with caffeine. The formation of a stacking complex structure where the drug is sandwiched between caffeine molecules, thus hindering solvent access to the reaction site on the drug molecule, was hypothesized as the protective mechanism.
A similar protective effect is often obtained with the use of cyclodextrins in cases where cyclodextrins form inclusion complexes with drugs and hinder solvent access to the reactive site on the molecule. Thus, prostaglandins and prostacyclins can be stabilized in solution (73,74) and solid (75) states by complexation with cyclodextrins. The use of cyclodextrins for reduction of degradation has been reported for several drugs undergoing different degradation pathways such as hydrolysis and oxidation. However, cyclodextrin complexation may also enhance drug degradation in cases where the mechanism of drug degradation can proceed within the environment and the steric arrangement of the drug in the cyclodextrin complex. Thus, β-cyclodextrin increases the hydrolytic ester cleavage of aspirin (76) and γ-cyclodextrin destabilizes doxorubicin (77).
Other stabilizers that may be used in drug product formulations include pH modifiers that affect the microenvironmental pH in the immediate vicinity of the drug particles. For example, ester compounds are particularly sensitive to hydrolytic degradation in the solid dosage form, especially when manufactured by the wet granulation process. Therefore, the wet granulation process is generally not preferred for the formulation of an ester compound unless the rate of hydrolysis can be controlled by another mechanism—such as modification of microenvironmental pH. Thus, an ester prodrug which can be stabilized by pH modulation in the dosage form could actually be more stable when formulated by wet granulation than by dry granulation (60,61). This is attributed to a more uniform distribution of the pH modifier in the formulation prepared by wet granulation (78,79). For example, pH modification reduced the drug degradation rate of acetylsalicylic acid in a wet granulation formulation.
Sometimes, mitigation strategies can be effective even in cases where complete mechanism or degradation pathways are unknown. This is particularly the case when the impurities in the formulations and excipients are elusive, such as free radicals. For example, a capsule formulation of BMS compound A showed degradation and discoloration under stressed conditions (10). The degradation products were only observed when the form of the drug was a drug–phenylalanine complex. The authors hypothesized that Maillard reaction between phenylalanine and reducing sugar impurities in microcrystalline cellulose leads to the formation of free radicals that oxidize BMS compound A. Incorporation of radical-scavenging antioxidants stabilized compound A from oxidation.
Drug Product Processing
In addition to contribution from starting materials, API impurities in drug products that are formed by the reaction of API with excipient impurities could often be related to the processing of the drug product. Pharmaceutical manufacturing processes can sometimes expose the formulation to high-temperature (such as fluid bed drying), high-moisture (such as wet granulation), and high-pressure (such as high shear wet granulation, roller compaction, and tablet compression) conditions. In the case of sensitive APIs, these conditions can increase the formation of API impurities. For example, Harmon et al. (80) observed the formation of an oxidative degradation product of API in a fluid bed granulation process that was related to the content of peroxides in the formulation. The authors observed that the hydroperoxide content of the formulation increased during processing and depended more on the process parameters than the input material peroxide content. A control of process parameters was able to resolve the issue in the drug product. In another study, Reed et al. (81) observed the photochemical degradation of a phenyl-ether-based API that was related to the heavy metal content in the excipients used in the formulation. The authors observed a concurrent effect of light exposure and heavy metal content in the formulation and were able to control drug degradation by selecting an optimized processing condition.
In addition, drug product processing can influence the effectiveness of a stabilization strategy. Thus, the use of wet granulation allows better distribution of a pH modifier in a solid dosage form, leading to greater stabilization of a drug that degrades by pH-sensitive hydrolysis than dry granulation. Similarly, the use of milling can destabilize a drug in a solid dosage form by increasing the reactive surface (66). Also, oxidation of sensitive drugs that degrade by free radical mechanisms could be sensitive to the mechanically generated radicals. Thus, the use of high shear mixing increased the oxidative degradation rate of an oxidatively sensitive drug, which was attributed to the mechanoradicals generated from the excipient, microcrystalline cellulose (82).
Drug Product Packaging
Packaging of the drug product can be one of the ways to mitigate the degradation of drugs without modifying the formulation. A number of non-hydrolysis reactions are also influenced by the moisture activity/content in the dosage form. The desiccants such as silica gel can be co-packaged with the dosage form to mitigate the degradation of the drug. Several investigators have developed models to simulate the effect of desiccants on the moisture environment inside the package, which can be used to build predictive correlations with the drug product stability especially in the case of hydrolytically sensitive drugs (83–85). In addition, proprietary package inserts, such as oxygen scavengers, have been reported to improve the oxidation stability of drugs (86).
Change in Excipient Source
Although change in sourcing of excipients is an option, it is laden with obstacles and challenges (1). Responding to the growing industry need to control API reactivity with excipient impurities in drug product formulations, several excipients have recently become available commercially with lower levels of reactive impurities. For example, PEG 300, 400, and 600 with low levels of aldehydes and peroxides; polysorbate 80 with low levels of aldehydes and peroxides; low peroxide grade PVP; and low peroxide grade crospovidone are commercially available.
Excipient Packaging
An increase in the concentration of reactive impurities in excipients during excipient storage can compromise the benefit of using excipients with low initial levels of impurities. For example, in studies where peroxides in excipients were implicated in the oxidative degradation of drugs in their formulations, control of the initial peroxide concentration was recommended for drug product stabilization (87). Approaches for reducing peroxides from excipients include the use of enzymes (88), metals, or other additives (89); chemical modification of the cross-linker (32); supercritical fluid extraction (90); and vacuum drying (91). However, the peroxide impurities in excipients, such as povidone, tend to increase upon storage (92). This can limit the benefit of using an excipient with low starting peroxide concentration and is contingent on storage conditions.
In certain cases, packaging configuration of excipients that minimizes environmental exposure can help minimize the increase in reactive excipient impurities. For example, use of a coating on the polyethylene bags used as in-liners to make them more impermeable could reduce the increase in peroxide levels in an excipient upon storage at high-temperature and humidity conditions (93).
Raw Material Specifications
A frequently discussed approach involves setting acceptance criteria for the raw materials, i.e., excipients, when changes to the formulation or packaging cannot be made. Although global compendial excipient monographs are constantly being updated and harmonized, it is not reasonable to expect these monographs to capture the tolerance of a given reactive impurity for a specific product or application. The setting of specification, then, depends on the interface between the drug product manufacturer and the excipient vendor.
Setting incoming material specifications on excipients can be facilitated by an understanding of how the quantitative changes in reactive impurity level in an excipient used in the formulation impacts the drug product stability. There are several challenges to develop this relationship. To set meaningful specifications, one needs to obtain stability data of the dosage forms using excipients with a range of reactive impurities. Obtaining samples with different levels of impurities may not be feasible or practical for the vendor. In many cases, the typical batches provided by the vendor have a narrow range of reactive impurity levels. In addition, generating excipients with inherently different reactive impurity levels is challenging due to the large scale of excipient manufacture since excipients are utilized by several industries, (e.g., foods, cosmetics, etc.) where the demand is greater and often with reduced stringency in the requirements for certain impurities. Producing or spiking of reactive impurities into excipient or dosage form is problematic as many of these impurities are volatile.
Setting specifications beyond the requirements of compendial testing is often challenging. The application of a “use test” of every batch of incoming raw excipient for a given drug product is often difficult to develop and validate. In addition, a tight specification on reactive impurities of an excipient or a use test may only be specific for a given product and may not necessarily represent the general limits for other applications. Clearly, a strong technical, quality, and business partnership between the excipient vendor and the drug product manufacturer is essential for setting special specifications on the excipients in a particular dosage form.
CONCLUSIONS
Reactive impurities in pharmaceutical excipients could cause significant degradations in drug products. In this review, six major categories of reactive impurities from commonly used pharmaceutical excipients in solid dosage forms are profiled. The sources of generation, potential interactions with API, and the analytical methods of the excipient impurities are summarized. Knowledge of potential impurities in excipients along with an understanding of the stability “soft spots” of the drug will lead to better risk assessment due to excipient variability and implementation of a strategy for robust drug product to avoid any undesirable late-stage surprises. While some strategies involve changes in drug product design and process, other options involve greater cooperation and collaboration between excipient manufacturers and end users of the excipients.
Acknowledgments
The authors would like to thank Helen Fu, Seema Betigeri, Nancy Lewen, and Sandy Lee for their contributions in determining trace reactive impurities in excipient samples.
References
- 1.Moreton C. Functionality and performance of excipients in a quality-by-design world: part IV. Am Pharm Rev. 2010; Suppl. p. 18–21.
- 2.Narang AS, Rao VM, Raghavan K. Excipient compatibility. In: Qiu Y, Chen Y, Zhang GZ, Liu L, Porter W, editors. Developing solid oral dosage forms: pharmaceutical theory and practice. Burlington: Elsevier; 2009. pp. 125–46. [Google Scholar]
- 3.Kibbe A. Handbook of pharmaceutical excipients. 3. Washington: American Pharmaceutical Association; 2000. pp. 102–6. [Google Scholar]
- 4.Dubost DC, Kaufman MJ, Zimmerman JA, Bogusky MJ, Coddington AB, Pitzenberger SM. Characterization of a solid state reaction product from a lyophilized formulation of a cyclic heptapeptide. A novel example of an excipient-induced oxidation. Pharm Res. 1996;13(12):1811–4. doi: 10.1023/A:1016024923002. [DOI] [PubMed] [Google Scholar]
- 5.Katdare A, Chaubal M, editors. Excipient development for pharmaceutical, biotechnology, and drug delivery systems. New York: Informa Healthcare USA, Inc.; 2006. p. 100. [Google Scholar]
- 6.Wirth DD, Baertschi SW, Johnson RA, Maple SR, Miller MS, Hallenback DK, Gregg SM. Maillard reaction of lactose and Fluoxetine Hydrochloride, a secondary amine. J Pharm Sci. 1998;87:31–9. doi: 10.1021/js9702067. [DOI] [PubMed] [Google Scholar]
- 7.Huang G, Wu Y, Dali M. Determination of formaldehyde and glucose in pharmaceutical excipients by HPLC with pre-column derivatization. HPLC Conference 2007, San Francisco, CA. Poster presentation.
- 8.George RC, Barbuch RJ, Huber EW, Regg BT. Investigation into the yellowing on aging of Sabril tablet cores. Drug Dev Ind Pharm. 1994;20:3023–32. doi: 10.3109/03639049409041966. [DOI] [Google Scholar]
- 9.Wu Y, Dali M, Gupta A, Raghavan K. Understanding drug–excipient compatibility: oxidation of compound A in a solid dosage form. Pharmaceut Dev Tech. 2009;14(5):556–64. doi: 10.1080/10837450903182140. [DOI] [PubMed] [Google Scholar]
- 10.Njoroge FG, Monnier VM. The chemistry of the Maillard reaction under physiological conditions: a review. Prog Clin Biol Res. 1989;304:85–107. [PubMed] [Google Scholar]
- 11.Hodge JE. The Amadori rearrangement. Adv Carbohydr Chem. 1966;10:169–205. doi: 10.1016/S0096-5332(08)60392-6. [DOI] [PubMed] [Google Scholar]
- 12.Li Z, Jacobus LK, Wuelfing WP, Golden M, Martin GP, Reed RA. Detection and quantification of low-molecular-weight aldehydes in pharmaceutical excipients by headspace gas chromatography. J Chromatography A. 2006;1104:1–10. doi: 10.1016/j.chroma.2005.10.084. [DOI] [PubMed] [Google Scholar]
- 13.del Barrio MA, Hu J, Zhou P, Cauchon N. Simultaneous determination of formic acid and formaldehyde in pharmaceutical excipients using headspace GC/MS. J Pharm Biomed Anal. 2006;41:738–43. doi: 10.1016/j.jpba.2005.12.033. [DOI] [PubMed] [Google Scholar]
- 14.Glastrup J. Degradation of polyethylene glycol. A study of the reaction mechanism in a model molecule: tetraethylene glycol. Polym Degrad Stab. 1996;52:217–22. doi: 10.1016/0141-3910(95)00225-1. [DOI] [Google Scholar]
- 15.Hamburger R, Azaz E, Donbro M. Autoxidation of polyoxyethylenic nonionic surfactants and polyethylene glycols. Pharm Acta Helv. 1975;50:10–7. [PubMed] [Google Scholar]
- 16.Waterman K, Arikpo WB, Fergione MB, Graul TW, Johnson BA, MacDonald BC, Roy MC, Timpano RJ. N-methylation and N-formylation of a secondary amine drug (Varenicline) in an osmotic tablet. J Pharm Sci. 2008;97(4):1499–507. doi: 10.1002/jps.21119. [DOI] [PubMed] [Google Scholar]
- 17.Sakharov AM, Mazaletskaya LI, Skibida IP. Catalytic oxidative deformylation of polyethylene glycols with the participation of molecular oxygen. Kinet Catal. 2001;42:662–8. doi: 10.1023/A:1012371630951. [DOI] [Google Scholar]
- 18.Nassar MN, Nesarikar VN, Lozano R, Parker WL, Huang Y, Palaniswamy V, Xu W, Khaselev N. Influence of formaldehyde impurity in Polysorbate 80 and PEG 300 on the stability of a parenteral formulation of BMS-204352: identification and control of the degradation product. Pharmaceut Dev Tech. 2004;9(2):189–95. doi: 10.1081/PDT-120030249. [DOI] [PubMed] [Google Scholar]
- 19.Wang G, Fiske J, Jennings S, Tomasella F, Palaniswamy V, Ray K. Identification and control of a degradation product in Avapro film-coated tablet: low dose formulation. Pharm Dev Tech. 2008;13:393–9. doi: 10.1080/10837450802244918. [DOI] [PubMed] [Google Scholar]
- 20.Gannett P, Hailu S. In vitro reaction of formaldehyde with fenfluramine: conversion to N-methyl fenfluramine. J Anal Toxicol. 2001;25:88–92. doi: 10.1093/jat/25.2.88. [DOI] [PubMed] [Google Scholar]
- 21.Desai DS, Rubitski BA, Bergum JS, Varia SA. Effects of different types of lactose and disintegrant on dissolution stability of hydrochlorothiazide capsule formulations. Int J Pharm. 1994;110:257–65. doi: 10.1016/0378-5173(94)90248-8. [DOI] [Google Scholar]
- 22.Hoydonckx HE, VanRhijn WM, VanRhijn W, Devos DE, Jacobs PA. Furfural and derivatives. In: Ullmann’s encyclopedia of industrial chemistry. Weinheim: Wiley-VCH; 2007. doi:10.1002/14356007.
- 23.Brownley CA, Jr, Lachman L. Preliminary report on the comparative stability of certified colorants with lactose in aqueous solutions. J Pharm Sci. 1963;52(1):86–93. doi: 10.1002/jps.2600520121. [DOI] [PubMed] [Google Scholar]
- 24.Brownley CA, Jr, Lachman L. Browning of spray-processed lactose. J Pharm Sci. 1964;53(4):452–4. doi: 10.1002/jps.2600530428. [DOI] [PubMed] [Google Scholar]
- 25.Duvall RN, Koshy KT, Pyles JW. Comparison of reactivity of amphetamine, methamphetamine, and dimethylamphetamine with lactose and related compounds. J Pharm Sci. 1965;54(4):607–11. doi: 10.1002/jps.2600540426. [DOI] [PubMed] [Google Scholar]
- 26.Bisug SM, Huang WT. Interaction of dextroamphetamine sulfate with spray-dried lactose. J Pharm Sci. 1972;61:1770–5. doi: 10.1002/jps.2600611116. [DOI] [PubMed] [Google Scholar]
- 27.Janicki CA, Almond HR. Reaction of Haloperidol with 5-(hydroxymethyl)-2-furfuraldehyde, an impurity in anhydrous lactose. J Pharm Sci. 1974;63:41–3. doi: 10.1002/jps.2600630110. [DOI] [PubMed] [Google Scholar]
- 28.Lessen T, Da-Chuan Z. Interactions between drug substances and excipients. 1. Fluorescence and HPLC studies of triazolophtalazine derivatives from Hydralazine Hydrochloride and starch. J Pharm Sci. 1996;85:326–9. doi: 10.1021/js950218x. [DOI] [PubMed] [Google Scholar]
- 29.Al-Nimry SS, Assaf SM, Ibrahim MJ, Najib NM. Adsorption of Ketotifen onto some pharmaceutical excipients. Int J Pharm. 1997;149:115–21. doi: 10.1016/S0378-5173(96)04857-0. [DOI] [Google Scholar]
- 30.Aly SAS, Megwa SA. Drug excipient interaction: effect of adsorption of oxytetracycline hydrochloride by some tablet excipients on the physiological availability of the tablets. STP Pharma. 1987;3:652–7. [Google Scholar]
- 31.Hovorka S, Schoneich C. Oxidative degradation of pharmaceuticals: theory, mechanisms and inhibition. J Pharm Sci. 2001;90:253–69. doi: 10.1002/1520-6017(200103)90:3<253::AID-JPS1>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 32.Tallon MA, Malawer EG, Machnicki NI, Brush PJ, Wu CS, Cullen JP. The effect of crosslinker structure upon the rate of hydroperoxide formation in dried, crosslinked poly(vinylpyrrolidone) J Appl Polym Sci. 2008;107:2776–85. doi: 10.1002/app.27449. [DOI] [Google Scholar]
- 33.Wasylaschuk WR, Harmon PA, Wagner G, Harman AB, Templeton AC, Xu H, Reed RA. Evaluation of hydroperoxides in common pharmaceutical excipients. J Pharm Sci. 2007;96:106–16. doi: 10.1002/jps.20726. [DOI] [PubMed] [Google Scholar]
- 34.Huang T, Garceau ME, Gao P. Liquid chromatographic determination of residual hydrogen peroxide in pharmaceutical excipients using platinum and wired enzyme electrodes. J Pharm Biol Anal. 2003;31:1203–10. doi: 10.1016/S0731-7085(03)00022-0. [DOI] [PubMed] [Google Scholar]
- 35.Nakamura T, Maeda H. A simple assay for lipid hydroperoxides based on triphenylphosphine oxidation and high-performance liquid chromatography. Lipids. 1991;26:765–8. doi: 10.1007/BF02535628. [DOI] [PubMed] [Google Scholar]
- 36.Hoffmann E, Herrle K. U.S. Patent 3,759,880, 1973.
- 37.Hartauer K, Gordon A, Baertschi S, Ross J, Luke W, Pearson N, Richard E, Tingle C, Tsang P, Wiens R. Influence of peroxide impurities in povidone and crospovidone on the stability of raloxifene hydrochloride in tablets: identification and control of an oxidative degradation product. Pharm Dev Tech. 2000;5(3):303–10. doi: 10.1081/PDT-100100545. [DOI] [PubMed] [Google Scholar]
- 38.Kothari S, Paruchuri S, Rao VM, Desai D. Peroxide scavenging property of croscarmellose sodium and its potential to reduce N-oxidation of piperazine ring containing compounds. 2006; AAPS Poster, San Antonio, TX.
- 39.Freed AL, Strohmeyer HE, Mahjour M, Sadineni V, Reid DL, Kingsmill CA. pH Control of nucleophilic/electrophilic oxidation. Int J Pharm. 2008;357(1–2):180–8. doi: 10.1016/j.ijpharm.2008.01.061. [DOI] [PubMed] [Google Scholar]
- 40.Bahador K, Montazerozohori M, Habibi MH. Urea-hydrogen peroxide (UHP) oxidation of thiols to the corresponding disulfides promoted by maleic anhydride as mediator. Molecules. 2005;10(10):1358–63. doi: 10.3390/10101385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sims JL, Carreira JA, Carrier DJ, Crabtree SR, Easton L, Hancock SA, Simcox CE. A new approach to accelerated drug–excipient compatibility testing. Pharm Dev Tech. 2003;8(2):119–26. doi: 10.1081/PDT-120018476. [DOI] [PubMed] [Google Scholar]
- 42.Miller DM, Buettner GR, Aust SD. Transition metals as catalysts of “autoxidation” reactions. Free Radical Biol Med. 1990;8(1):95–108. doi: 10.1016/0891-5849(90)90148-C. [DOI] [PubMed] [Google Scholar]
- 43.Ohyashiki T, Kadoya A, Kushida K. The role of Fe3+ on Fe2+-dependent lipid peroxidation in phospholipid liposomes. Chem Pharm Bull. 2002;50(2):203–7. doi: 10.1248/cpb.50.203. [DOI] [PubMed] [Google Scholar]
- 44.Brambilla G, Martelli A. Genotoxic and carcinogenic risk to humans of drug–nitrite interaction products. Mutat Res. 2007;635:17–52. doi: 10.1016/j.mrrev.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 45.Wessel W, Schoog M, Winkler E. Polyvinylpyrrolidone (PVP), its diagnostic, therapeutic and technical application and consequences thereof. Drug Research. 1971;10:1469–82. [PubMed] [Google Scholar]
- 46.Bartsch H, Ohshima H, Pignatelli B. Inhibitors of endogenous nitrosation mechanisms and implications in human cancer prevention. Mutat Res. 1988;202:307–24. doi: 10.1016/0027-5107(88)90194-7. [DOI] [PubMed] [Google Scholar]
- 47.Brambilla G, Martelli A. Update on genotoxicity and carcinogenicity testing of 472 marketed pharmaceuticals. Mutat Res. 2009;681(2–3):209–29. doi: 10.1016/j.mrrev.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 48.Conrad J, Schlemmer K, Eisenbrand G. Nitrosation of bromohexine. Drug Dev and Evaluation. 1990;16:181–97. [Google Scholar]
- 49.Lundberg JO, Weitzberg E, Gladwin MT. The nitrate nitriteenitric oxide pathway in physiology and therapeutics. Nat Rev Drug Discov. 2008;7:156–67. doi: 10.1038/nrd2466. [DOI] [PubMed] [Google Scholar]
- 50.Bottex B, Dorne JL, Carlander D, Benford D, Przyrembel H, Heppner C, Kleiner J, Cockburn A. Risk-benefit health assessment of food—food fortification and nitrate in vegetables. Trends Food Sci Tech. 2008;19:S113–S119. doi: 10.1016/j.tifs.2008.07.005. [DOI] [Google Scholar]
- 51.Narang AS, Rao VM, Farrell T, Ferrizzi D, Castoro J, Corredor C, Jain N, Varia SA, Desai DD. Stability implications of prolonged storage of PVA and PEG-based coating suspension. AAPS poster presentation, New Orleans, LA; 2010.
- 52.Chien YW, Van Nostrand P, Hurwitz AR, Shami EG. Drug–disintegrant interactions: binding of oxymorphone derivatives. J Pharm Sci. 1981;70:709–10. doi: 10.1002/jps.2600700641. [DOI] [PubMed] [Google Scholar]
- 53.Cory W, Field K, Wu-Linhares D. Is it the method or the process—separating the causes of low recovery. Drug Dev Ind Pharm. 2004;30:891–9. doi: 10.1081/DDC-200034588. [DOI] [PubMed] [Google Scholar]
- 54.Rohrs BR, Thamann TJ, Gao P, Stelzer DJ, Bergren MS, Chao RS. Tablet dissolution affected by a moisture mediated solid-state interaction between drug and disintegrant. Pharm Res. 1999;16:1850–6. doi: 10.1023/A:1018951309506. [DOI] [PubMed] [Google Scholar]
- 55.Yu H, Cornett C, Larsen J, Hansen JH. Reaction between drug substances and pharmaceutical excipients: formation of esters between cetirizine and polyols. J Pharm and Biomed Anal. 2010;53(3):745–50. doi: 10.1016/j.jpba.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 56.Fukuyama S, Kihara N, Naksashima K, Morokoshi N, Koda S, Yasuda T. Mechanism of optical isomerization of (S)-N-[1-(2-fluorophenyl)-3,4,6,7-tetrahydro-4-oxopyrrolo[3,2,1-jk][1,4]-benzodiazepine-3-yl]-1H-indole-2-carboxamide (FK480) in soft capsules containing polyethylene glycol 400 and glycerol. Pharm Res. 1994;11:1704–6. doi: 10.1023/A:1018998813296. [DOI] [PubMed] [Google Scholar]
- 57.Nishikawa M, Fuji K. Effect of autoxidation of hydrogenated castor oil containing 60 oxyethylene groups on degradation of miconazole. Chem Pharm Bull. 1991;39:2408–11. [Google Scholar]
- 58.Zografi G, Byrn SR. The effects of residual water on solid-state stability of drugs and drug products, Water Manage. Des Distrib Qual Foods. 1999. p. 397–410.
- 59.Badawy SIF. Effect of salt formation on chemical stability of an ester prodrug of a glycoprotein IIb/IIIa receptor antagonist in solid dosage forms. Int J Pharm. 2001;223:81–7. doi: 10.1016/S0378-5173(01)00726-8. [DOI] [PubMed] [Google Scholar]
- 60.Badawy SIF, Williams RC, Gilbert D. Chemical stability of an ester prodrug of a IIb/IIIa antagonist in solid dosage forms. J Pharm Sci. 1999;8:428–33. doi: 10.1021/js9803297. [DOI] [PubMed] [Google Scholar]
- 61.Gold TB, Smith SL, Digenis GA. Studies on the influence of pH and pancreatin on 13C-formaldehyde-induced gelatin cross-links using nuclear magnetic resonance. Pharm Dev Tech. 1996;1(1):21–6. doi: 10.3109/10837459609031414. [DOI] [PubMed] [Google Scholar]
- 62.Qiu Z, Stowell JG, Cao W, Morris KR, Byrn SR, Carvajal MT. Effect of milling and compression on the solid-state Maillard reaction. J Pharm Sci. 2005;94:2568–80. doi: 10.1002/jps.20448. [DOI] [PubMed] [Google Scholar]
- 63.Chen X, Stowell JG, Morris KR, Byrn SR. Quantitative study of solid-state acid–base reactions between polymorphs of flufenamic acid and magnesium oxide using X-ray powder diffraction. J Pharm Biomed Anal. 2010;51:866–74. doi: 10.1016/j.jpba.2009.09.021. [DOI] [PubMed] [Google Scholar]
- 64.Desai D, Rao VM, Guo H, Li D, Stein D, Hu F, Kiesnowski C. An active film-coating approach to enhance chemical stability of a potent drug molecule. Pharm Dev Tech. 2010;1–9. [DOI] [PubMed]
- 65.Akers M. Antioxidants in pharmaceutical products. J Parenteral Sci and Tech. 1982;36(5):222–8. [PubMed] [Google Scholar]
- 66.Won CM, Tang SY, Strohbeck CL. Photolytic and oxidative degradation of an antiemetic agent, RG 12915. Int J Pharm. 1995;121:95–105. doi: 10.1016/0378-5173(95)00014-A. [DOI] [Google Scholar]
- 67.Kaufman MJ. Applications of oxygen polarography to drug stability testing and formulation development: solution-phase oxidation of hydroxymethylglutaryl coenzyme A (HMG-CoA) reductase inhibitors. Pharm Res. 1990;7:289–92. doi: 10.1023/A:1015886415210. [DOI] [PubMed] [Google Scholar]
- 68.Higuchi T, Lachman L. Inhibition of hydrolysis of esters in solution by formation of complexes I. Stabilization of benzocaine with caffeine. J Ame Pharm Asso. 1995;44:521–6. doi: 10.1002/jps.3030440902. [DOI] [PubMed] [Google Scholar]
- 69.Lachman L, Ravin LJ, Higuchi T. Inhibition of hydrolysis of esters in solution by formation of complexes II. Stabilization of procaine with caffeine. J Am Pharm Assoc. 1956;45:290–5. doi: 10.1002/jps.3030450506. [DOI] [PubMed] [Google Scholar]
- 70.Lachman L, Higuchi T. Inhibition of hydrolysis of esters in solution by formation of complexes. I. Stabilization of tetracaine with caffeine. J Am Pharm Assoc. 1957;46:32–6. doi: 10.1002/jps.3030460109. [DOI] [PubMed] [Google Scholar]
- 71.Guttman DE. Complex formation influence on reaction rate. I. Effect of caffeine on riboflavin base-catalyzed degradation. J Am Pharm Assoc. 1962;51:1162–6. doi: 10.1002/jps.2600511211. [DOI] [PubMed] [Google Scholar]
- 72.Pirinccioglu N, Williams A. Studies of reactions within molecular complexes: alkaline hydrolysis of substituted phenyl benzoates in the presence of xanthines. J Chem Society, Perkin Transactions 2: Phys Org Chem. 1998;1:37–40. doi: 10.1039/a705499h. [DOI] [Google Scholar]
- 73.Hirayama F, Kurihara M, Uekama K. Improvement of chemical instability of prostacyclin in aqueous solution by complexation with methylated cyclodextrins. Int J Pharm. 1987;35:193–9. doi: 10.1016/0378-5173(87)90130-X. [DOI] [Google Scholar]
- 74.Oh IJ, Song HM, Hyun M, Lee KC. Effect of 2-hydroxypropyl-beta-cyclodextrin on the stability of prostaglandin E2 in solution. Int J Pharm. 1994;106:135–40. doi: 10.1016/0378-5173(94)90311-5. [DOI] [Google Scholar]
- 75.Yamamoto M, Hirayama F, Uekama K. Improvement of stability and dissolution of prostaglandin E1 by maltosyl-β-cyclodextrin in lyophilized formulation. Chem Pharm Bulletin. 1992;40:747–51. doi: 10.1248/cpb.40.747. [DOI] [PubMed] [Google Scholar]
- 76.Bekers O, Beijnen JH, Vis BJ, Suenaga A, Otagiri M, Bult A, Underberg WJM. Effect of cyclodextrin complexation on the chemical stability of doxorubicin and daunorubicin in aqueous solutions. Int J Pharm. 1991;72:123–30. doi: 10.1016/0378-5173(91)90050-X. [DOI] [Google Scholar]
- 77.Bekers O, Beijnen JH, Kempers YAG, Bult A, Underberg WJM. Effects of cyclodextrins on N-trifluoroacetyldoxorubicin-14-valerate (AD-32) stability and solubility in aqueous media. Int J Pharm. 1991;68:271–6. doi: 10.1016/0378-5173(91)90149-I. [DOI] [Google Scholar]
- 78.Gu L, Strickley RG, Chi LH, Chowhan ZT. Drug–excipient incompatibility studies of the dipeptide angiotensin-converting enzyme inhibitor, moexipril hydrochloride: dry powder vs wet granulation. Pharm Res. 1990;7:379–83. doi: 10.1023/A:1015871406549. [DOI] [PubMed] [Google Scholar]
- 79.Delonca H, Joachim J, Joachim G, Cantos A. Wet granulation of acetylsalicylic acid. Effect of pH and stabilization agents. Farmaco Ed Prat. 1975;30:89–97. [PubMed] [Google Scholar]
- 80.Harmon PA, Wuelfing WP, Harman AB, Givand JC, Shelukar SD, Reed RA. Excipient mediated oxidation by organoperoxides. Baltimore: American Association of Pharmaceutical Scientists, AAPS; 2004. [Google Scholar]
- 81.Reed RA, Harmon P, Manas D, Wasylaschuk W, Galli C, Biddell R, Bergquist PA, Hunke W, Templeton AC. The role of excipients and package components in the photostability of liquid formulations. PDA J Pharm Sci Tech. 2003;57:351–68. [PubMed] [Google Scholar]
- 82.Polizzi MA, Singhal D, Colvin J. Mechanoradical-induced degradation in a pharmaceutical blend during high-shear processing. Pharm Dev Tech. 2008;13:457–62. doi: 10.1080/10837450802328869. [DOI] [PubMed] [Google Scholar]
- 83.Narang A, Lin J, Varia S, Badawy S. Modeling drug degradation in a tablet. AAPS Annual Meeting, New Orleans; 2010.
- 84.Badawy SI, Gawronski AJ, Alvarez FJ. Application of sorption–desorption moisture transfer modeling to the study of chemical stability of a moisture sensitive drug product in different packaging configurations. Int J Pharm. 2001;223:1–13. doi: 10.1016/S0378-5173(01)00693-7. [DOI] [PubMed] [Google Scholar]
- 85.Chen Y, Li Y. A new model for predicting moisture uptake by packaged solid pharmaceuticals. Int J Pharm. 2003;255:217–25. doi: 10.1016/S0378-5173(03)00089-9. [DOI] [PubMed] [Google Scholar]
- 86.Waterman KC, Roy MC. Use of oxygen scavengers to stabilize solid pharmaceutical dosage forms: a case study. Pharm Dev Tech. 2002;7(2):227–34. doi: 10.1081/PDT-120003490. [DOI] [PubMed] [Google Scholar]
- 87.Hartauer KJ, Arbuthnot GN, Baertschi SW, Johnson RA, Luke WD, Pearson NG, Rickard EC, Tingle CA, Tsang PK, Wiens RE. Influence of peroxide impurities in povidone and crospovidone on the stability of raloxifene hydrochloride in tablets: identification and control of an oxidative degradation product. Pharm Dev Tech. 2000;5:303–10. doi: 10.1081/PDT-100100545. [DOI] [PubMed] [Google Scholar]
- 88.Buhler V, Filges U, Schneider T. Stabilized polyvinylpyrrolidone formulation. BASF A.G., Germany. WO; 2000. p. 17.
- 89.Fereira PJ, Desjardin MA, Rohloff CM, Berry SA, Zlatkova-Karaslavova ES. Non-aqueous formulations containing biodegradable polymers and methionine and solvents for removing peroxides and reducing the oxidative degradation of drugs. Durect Corp., USA; 2005. p. 36. Cont-in-part of US Ser No. 814,826.
- 90.Ashraf-Khorassani M, Taylor LT, Waterman KC, Narayan P, Brannegan DR, Reid GL. Purification of pharmaceutical excipients with supercritical fluid extraction. Pharm Dev Tech. 2005;10:507–16. doi: 10.1080/10837450500299958. [DOI] [PubMed] [Google Scholar]
- 91.Kumar V, Kalonia S, et al. Removal of peroxides in polyethylene glycols by vacuum drying: implications in the stability of biotech and pharmaceutical formulations. AAPS PharmSciTech. 2006;7:62. doi: 10.1208/pt070362. [DOI] [PubMed] [Google Scholar]
- 92.Buhler V. Kollidon: polyvinylpyrrolidone excipients for the pharmaceuticals. Berlin: Springer; 2008. [Google Scholar]
- 93.Taylor N. Control excipient reactivity with packaging and aluminum lakes, in: product news, ingredients, excipients, and raw materials. PharmaTechnologist.com; 2010.
- 94.Nieuwmeyer F, van der Voort Maarschalk K, Vromans H. Lactose contaminant as steroid degradation enhancer. Pharm Res. 2008;25(11):2666–72. doi: 10.1007/s11095-008-9687-z. [DOI] [PubMed] [Google Scholar]
- 95.Ma D, Wasylaschuk WR, Beasley C, Zhao ZZ, Harmon PA, Ballard JM, Pitzenberger SM, Varga SL, Reed RA. Identification and quantitation of extractables from cellulose acetate butyrate (CAB) and estimation of their in vivo exposure levels. J Pharm Biomed Anal. 2004;35:779–88. doi: 10.1016/j.jpba.2004.03.004. [DOI] [PubMed] [Google Scholar]